

Abstract
The discovery of highly recurrent mutations in melanoma, such as BRAFV600E, completely changed the clinical management including therapy of melanoma patients. In the era of Personalized Medicine targeted melanoma therapies showed high response rates, currently evidenced by BRAF inhibitors or immune-stimulating therapies. In addition to genetic biomarkers, epigenetic knowledge in melanoma has undergone a major step forward in recent years. In particular, epigenetics is unveiling new perspectives to fight this disease, providing an encouraging number of DNA methylation based biomarkers that will likely improve patient stratification for prognosis and treatment. In this regard, putative targetable biomarkers or those with predictive value for the outcome of currently applied therapies are promising tools for future precision oncology strategies. In addition, the progress made in genetic and epigenetic profiling technologies and their reconfiguration to real-time clinical screening approaches makes personalized medicine in melanoma an achievable objective in upcoming years.
Keywords: Melanoma, DNA methylation, biomarkers, target therapy, chemoresistance, dynamic rewiring, translational science
Epigenetics in melanoma: new biomarkers based on DNA methylation
Melanoma is one of the solid tumors less epigenetically interrogated. However, huge efforts have been made in the last decade to improve the knowledge we had regarding this aggressive and multi-resistant tumor type. Clinical outcome of melanoma patients justifies this scientific involvement given the pronounce survival difference between localized and metastatic disease (98% and 16-62% 5-year survival, respectively) (1). In addition, approximately 15% of diagnostic melanomas are in fact benign lesions (false positive rate) and 17% of diagnostic benign lesions are malignant melanomas (false negative rate). Hence, this setting provided enough stimuli to derive resources to improve the clinical management and diagnosis of melanoma patients.
Epigenetic inactivation of particular tumor suppressor genes has been extensively implicated in tumor initiation, promotion and progression (2-6). It was the first epigenetic alteration studied in melanoma occurring preferentially by the specific DNA methylation of promoter regions. Promoters are CG enriched portions of the genes involved in the transcription and susceptible to be silenced by the deposition of methyl groups in the cytosines closed by guanines. Figure 1 summarized first reported genes regulated by DNA methylation in melanoma: PTEN (methylated in ~60% of melanoma) (7,8), SYK (methylated in 30% of melanomas) (9), TNFSF10D and LOX (methylated in 80% and 50% of melanomas) (10), or COL1A2 (methylated in 80% of melanomas) (9). Later on, genome-wide screenings led to the discovery of multi-locus DNA-methylation signatures and new target genes associated with melanoma malignancy: MeDIP assay (11), Illumina GoldenGate Methylation Cancer Panel I (12), Illumina Infinium HumanMethylation27 BeadChips (13-16), or the Illumina Infinium HumanMethylation450 Beadchips (17-19).
Figure 1.

Illustrative image of melanoma evolution from an epigenetic point of view, highlighting the aberrant DNA methylation events occurring during the malignization of melanocytes. TSGs, tumor suppressor genes.
Even though the first discoveries were made on disrupted genes by DNA hypermethylation, epigenome-wide DNA methylation landscape of melanoma revealed a global wave of hypomethylation throughout intergenic regions, gene bodies and locations away from CpG islands (genome regions with enriched CpG sites). Actually, Guo and coworkers comparing the epigenomes of a set of 34 primary cutaneous melanoma tumors harboring BRAFV600E mutation with those of 27 BRAFWT samples, found that the 98% of the disrupted methylated CpG sites were events of loss of methylation whilst only a 2% were events of methylation gain (20). Interestingly, the authors related this particular mutation (BRAFV600E) with an event of global hypomethylation and a decrease in DNMT3A expression, one of de novo DNA methyltransferases in cells. Regarding to the effects that a global wave of hypomethylation can produce at transcriptional level, it is noteworthy the reactivation of microphthalmia-associated transcription factor (MITF) which is the master regulator of melanin production and identified as a lineage-specific oncogene in melanoma. Guo and colleagues defined the reactivation of MITF as one of the most significant up-regulated genes under the control of BRAFV600E mutation (20) and Lauss and colleagues demonstrated its regulation by DNA methylation in melanoma tumors (17). Interestingly, neither the BRAFV600E mutation, which is also present in at least 20% of normal nevi, or the MITF expression are sufficient to drive the melanoma progression, but their roles in melanoma tumorigenesis are undisputed. Firstly, MITF introduces complex transcriptional changes in cells, triggering new gene pathways affecting cell proliferation, pigmentation synthesis, endosome trafficking or drug resistance (21,22). Secondly, BRAFV600E mutation throughout its hyperactivity reprograms the proliferative cell machinery but the bright side is that patients become susceptible to specific BRAF-inhibitor treatments (23). Responsiveness to BRAF-inhibitors has been one of the quickest and most potent feedbacks found into the clinics in the last decade but the patients also acquired resistance to the treatment in short-term which makes more difficult to deal with the malignancy. Interestingly, new mechanisms of resistance depending on epigenetic aberrations have been already described. Among them, we highlight the reactivation of the epidermal growth factor receptor (EGFR) by the demethylation of its own regulatory DNA elements located upstream and downstream of the transcription start site or the reactivation of a cryptic variant of the TBC1D16 gene (19,24,25). Particularly, Vizoso and colleagues describe in melanoma the modulation of EGFR receptor by an imbalance of the endosome/lysosome cell machinery. They explain how the epigenetic modulation of EGFR by TBC1D16 contributes to the dynamic rewiring of survival pathways in melanoma to evade target therapy-associated cell death (19).
Systematic characterization of epigenetically disrupted genes in melanoma has provided new insights regarding to the progression of the malignancy or the acquisition of resistance in melanoma patients. In this line, some genes have been reported as good candidates to be used as melanoma biomarkers (e.g., CDH11, CLDN11, MAPK13) and others have shown correlation with disease-free survival and overall survival (e.g., PTEN and HOXD9 genes). However, it would be desirable to identify novel target genes epigenetically silenced or reactivated with an extra clinical load, improving the stratification capacity of known histopathological parameters such as the Breslow index, the mitotic index, or ulceration. At this respect, the most valuable DNA methylation-associated biomarkers would be (I) those intimately related to the metastatic stage but being also present in low grade primary melanomas with bad prognosis; (II) those able to re-stratify traditional markers such as tumor thickness and ulceration; and (III) those that provide information about targeted cancer therapy-associated response in melanoma patients. Thus, melanoma could be molecularly stratified and personalized treatments could be applied improving the melanoma patients survival. For this purpose, it is essential to do a right selection of the patients’ cohort according with all these clinical prognostic factors. Finally, the best biomarkers would be those also susceptible to be directly targetable by small drug molecules.
Resistance to antineoplastic agents
Resistance to cancer therapy is a multifactorial process related not only to the kind of neoplasia and the tumor genotype and heterogeneity but also to own patients’ features. Actually, drugs are uptaken, processed and metabolized in a different way for the same tumour type in patients with different genetic and epigenetic background, altering both effectiveness and toxicity of treatments. Moreover, some neoplasias, such as melanoma, are refractory to particular therapies due to molecular characteristics that are decisive in their carcinogenesis process (26,27). Antineoplastic agent’s administration exerts selective pressure on those clones with more appropriate alterations to survive, making them refractory to treatment (intrinsic or primary resistance). In addition, cancer cells frequently can adapt to and evade the drug effects, either by developing new favourable molecular mechanisms or by activating alternative compensatory pathways that can bypass the effects of the therapy (acquired or secondary resistance) (28,29).
General mechanisms of cell chemoresistance
In order to comprehend mechanisms implicated in chemoresistance is important to know pathways triggered by anticancer drugs (30,31). Mechanisms of resistance to chemotherapy (Figure 2) have widely been associated with alterations in drug uptake, not only at level of cell capacity to assimilate the drug but also in the detoxification processes (32). It is important noteworthy the influence of tumour intrinsic factors such as size or low vascularisation, and pharmacokinetic drug specific parameters. As well, therapeutic target interaction is also crucial for its effectiveness. This interface activates damage recognition machinery and cell gatekeepers that triggered cell cycle arrest and DNA repair systems and, subsequently, mechanisms of cell death. Alterations that benefit cell survival have been related with lack of drug efficacy. Hence, resistance phenomenon appears associated with alterations that led to the cell bypass this blockage and remain viable (33). Apart from the cell kinetic parameters, tumour microenvironment factors have to be taken into account. Angiogenesis, lymphangiogenesis, tumour-associated fibroblasts (TAFs) and immune system exert a significant influence on the anticancer activity of these agents (34-36).
Figure 2.

Representation of drug resistance associated cell mechanisms that can take place against the anticancer therapy. Besides, tumor microenvironment associated processes also affects tumor cell drug responsiveness.
Chemoresistance in melanoma
Multidrug-resistance
The impact of vesicle trafficking in melanocytes
Melanoma is a highly aggressive form of skin cancer, with an average survival of 6-10 months in advanced cases who receives conventional treatment (37,38). Despite the unquestionable clinical benefit of the new recently approved targeted therapies, conventional chemotherapy continues being an option of treatment, both combined with immunotherapy strategies at first-line treatment and after targeted therapy progression (27,39). The notorious unresponsiveness of melanoma cells to current chemotherapy has been widely observed, presenting an intrinsic multi-drug resistance (MDR) phenotype, often related to over-expression of ABC-transporters family of proteins, responsible for extrusion of lots of substances chemically different (32,40). Particularly, some studies described the influence of ATP-binding cassette sub-family B member 5 (ABCB5) over-expressions in melanoma chemoresistance, proposing it as potential therapeutic target, even in clinical trials (40-43). Apart from this, other mechanisms involved in chemotherapy uptake have been associated with melanoma multi-drug refractoriness. Vesicle trafficking are crucial during melanogenesis and processes of autophagy. In this sense, Chen et al. described an in vitro association between drug accumulation and melanogenesis phases, suggesting the importance of this process in chemoresistance (40). Autophagy and endoplasmatic-reticulum (ER) stress both also play roles although their real impact on melanoma is still unclear. Corazzari et al. reported a chronic ER stress status directly increasing basal cell autophagy, which finally results in higher levels of drug resistance in melanomas BRAFV600E (44). Likewise, the effect of vesicle trafficking in chemoresistance has also been comprehensively demonstrated using in vitro models. Huang et al. showed the effect of the vacuolar protein sorting 33 homologs A (VPS33A) and the cappuccino homolog protein (CNO) down-regulation, implicated in vesicles formation during melanosomes maturation, increasing sensitivity to cisplatin and dacarbazine. The authors also suggested the inhibition of receptor-mediated signalling for melanosome formation as potential therapeutic strategy of chemoresistance reversion (45).
Novel pathways
Combination treatments between conventional chemotherapy and new agents that targeted downstream drug-associated pathways could be a good strategy to alleviate chemoresistance in melanoma cells (46,47). In this sense, Kreiseder et al. suggested the cytoskeletal linker protein a-catulin (CTNNAL1) as a potential targetable candidate, demonstrating its role in the upregulation of epithelial-to-mesenchymal (EMT) genes, nuclear factor kappa (NF-κB) and MAPK pathway, and chemosensitization of melanoma cells with elevated basal-expression of the gene upon CTNNAL1 depletion (48). Elevated levels of other proteins involved in EMT process, such as thrombospondin 1 (THBS1), has been related to chemoresistance, giving support to its inhibition as a potential therapeutic strategy (49). A comparable effect was observed by other authors when it was targeted particular proteins, direct or indirectly implicated in apoptosis (47,50,51). Hence, Liu et al. reported the therapeutic potential of targeting myeloid cell leukemia 1 (Mcl-1), an anti-apoptotic protein belonging to the Bcl-2 family, widely expressed in melanoma, which contributes to chemoresistance (50). Additionally, it has been observed a decreased cell apoptosis against cisplatin caused by the overexpression of aldolase A (ALDOA) and angiopoietin-like 4 (ANGPTL4) (51). At this regard, TBC1D16 could also be another good candidate as mentioned before.
Epigenetic mechanisms of resistance acquisition to alkylating agents
Alkylating agents have been the unique family of cytotoxic drugs that have demonstrated some efficacy in melanoma patients, being only effective in around 10-20% of cases in monotherapy (37,38). They trigger cell death by binding to DNA, mainly introducing in O6 position of guanine aberrant alkylations (methylation or chloroethylation). Cell damage is recognized by cell mechanisms such as the mismatch repair (MMR) machinery, activating DNA repair to counteract the drug effects (52). DNA-repair enzyme O6-methyl-guanine-DNA methyltransferase (MGMT) is responsible for reversing the alkylation and avoiding the formation of lethal base-pair cross-links in DNA (Figure 3). Its inactivation by hypermethylation in the promoter of the gene in relation with a better overall survival and disease-free survival in gliomas treated by alkylating agents was described at first time by Esteller et al. The authors observed that this alteration was present in a 40% of gliomas and that it was an independent prognostic factor (2). Later, this finding was observed in other tumour types, as glioblastoma, melanoma and colorectal cancer (52-54). In melanoma context, it was described an in vitro reactivation of MGMT previous to fotemustine resistance acquisition and a better tolerance to temozolamide treatment in MGMT-methylated tumours from melanoma patients (55). Apart from methylation, SNPs in the sequence of the gene could also affect to MGMT expression levels and, consequently, to response to the treatment (56,57). Additionally, the levels of other proteins involved in damage recognition and DNA repair have been linked with resistance to alkylating agents. Naumann et al. observed an in vitro refractory MGMT-independent effect of methylating agents in melanoma cell lines caused by a loss of expression of MSH2 and MSH6, responsible for the DNA damage recognition. However, these MMR proteins downregulation did not affect cell sensitivity to chloroethylating agents, indicating a partial cross-resistance between alkylating agents subfamilies (58).
Figure 3.
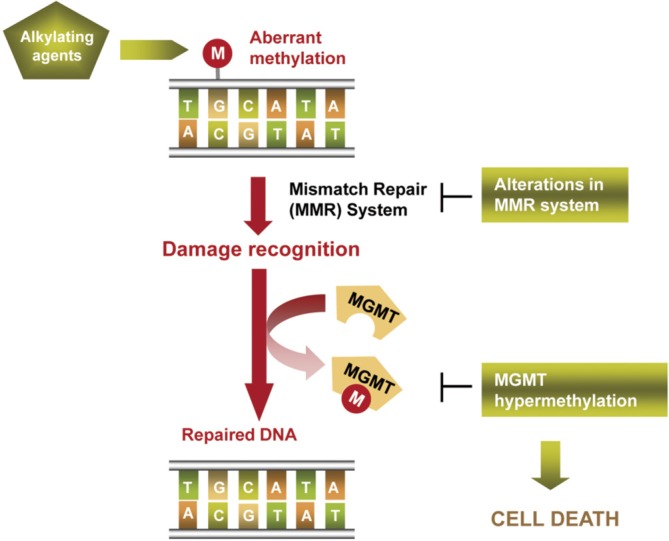
Scheme of alkylating agents’ mechanism of action and the role of MGMT in repairing drug-associated DNA damage. MGMT inactivating alterations enhance drug activity avoiding tumor cell viability by cell death activation. MGMT, methyl-guanine-DNA methyltransferase.
Epigenetic perspectives in tailored oncology
The implementation of personalized medicine has revolutionized the current strategies in oncology. Human tissue samples remain the gold standard resource to identify biomarkers that could be of utility in cancer clinical care (59). Paraffin-embedded tissue (FFPE) is the most widely used method for tissue preparation after surgical excision to preserve histology, whereas the application of diagnostic assays based on frozen tissue is and will stay behind limited due to its unwieldy collection process in the hospital setting. Tumor nucleic acids can be degraded upon formalin fixation and this can limit researchers’ ability to perform high-throughput analyses. However, advances in technology mean that the analyses are becoming more feasible (59). In the last years, a substantial number of publications have reported the study of DNA methylation in FFPE and other kind of stored samples of readily accessible in the hospital environment (52,60-62). Moreover, DNA methylation analyses can be performed using a wide range of technical approaches, not only at small-scale, as MSP and pyrosequencing, but also new-generation strategies, such as microarray platforms or WBGS (63,64). Even though most of them are ready to use into the clinical setting, the ground-breaking technology of NGS has just been adapted to diagnostic requirements. Interestingly, Moran et al. demonstrated the suitability of Infinium 450K DNA methylation microarray platform to analyze DNA samples coming from FFPE tissue, obtaining similar results as comparing with frozen tissue counterparts (62). As well, it has been reported the opportunity to perform epigenetic studies in a considerable range of biological samples obtained at non-invasive manner. For instance, based on the rational approach that tumour cells are continuously shed into the colonic lumen and mixed with stool, Carmona et al. confirmed the recognition of epigenetic biomarkers in this kind of samples (60). In the same way, it has been detected DNA methylation in urine, specially highlighting GSTP1 methylation as a potential predictive factor of disease aggressiveness in prostate cancer (61). Indeed, epigenetic biomarkers are capable to be detected in liquid biopsies, based on detection of DNA methylation in circulating tumour-DNA (ctDNA) from tumour or circulating tumour cells (CTC) (65,66). It is worth to emphasize the relevance of studying molecular biomarkers in liquid biopsies, since ctDNA approach let to monitor genetic and epigenetic markers, in easy-getting samples at non-invasive manner, along disease follow-up and treatment exposure, in order to notice cancer recurrence and secondary resistance phenomenon (67,68). An example is the use of hypomethylation in ctDNA for monitoring the tumours follow-up in hepatocellular carcinoma (69). Furthermore, deciphering the precise spatial orientation of stored FFPE tumor blocks using the routine clinical annotation to reconstruct intratumour heterogeneity can be difficult (70,71). ctDNA is more amenable to serial sampling and presumably represents cancer genomes and epigenomes from multiple metastatic sites (72,73). Nevertheless and despite the introduction of more suitable and optimized tools as regards histopathologic and molecular analyses, the supply of appropriate samples is still a limiting issue.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
Financial Disclosure: A Martinez-Cardús was supported by Red Temática de Investigación Cooperativa en Cáncer (RTICC) and Fundación Privada Olga Torres. M Vizoso was supported by a Formacion de Profesorado Universitario fellowship from the Spanish Ministry of Education. JL Manzano was supported by Fondo de Investigación Sanitaria (FIS) - Instituto de Salud Carlos III (ISCIII).
References
- 1.American Cancer Society. Cancer Facts & Figures, 2014. Atlanta: American Cancer Society; 2014. [Google Scholar]
- 2.Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med 2000;343:1350-4. [DOI] [PubMed] [Google Scholar]
- 3.Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst 2000;92:564-9. [DOI] [PubMed] [Google Scholar]
- 4.Esteller M. Epigenetics in cancer. N Engl J Med 2008;358:1148-59. [DOI] [PubMed] [Google Scholar]
- 5.Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol 2005;45:629-56. [DOI] [PubMed] [Google Scholar]
- 6.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 2003;349:2042-54. [DOI] [PubMed] [Google Scholar]
- 7.Mirmohammadsadegh A, Marini A, Nambiar S, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res 2006;66:6546-52. [DOI] [PubMed] [Google Scholar]
- 8.Lahtz C, Stranzenbach R, Fiedler E, et al. Methylation of PTEN as a prognostic factor in malignant melanoma of the skin. J Invest Dermatol 2010;130:620-2. [DOI] [PubMed] [Google Scholar]
- 9.Muthusamy V, Duraisamy S, Bradbury CM, et al. Epigenetic silencing of novel tumor suppressors in malignant melanoma. Cancer Res 2006;66:11187-93. [DOI] [PubMed] [Google Scholar]
- 10.Liu S, Ren S, Howell P, et al. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res 2008;21:545-58. [DOI] [PubMed] [Google Scholar]
- 11.Koga Y, Pelizzola M, Cheng E, et al. Genome-wide screen of promoter methylation identifies novel markers in melanoma. Genome Res 2009;19:1462-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conway K, Edmiston SN, Khondker ZS, et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi. Pigment Cell Melanoma Res 2011;24:352-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonazzi VF, Nancarrow DJ, Stark MS, et al. Cross-platform array screening identifies COL1A2, THBS1, TNFRSF10D and UCHL1 as genes frequently silenced by methylation in melanoma. PLoS One 2011;6:e26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sigalotti L, Covre A, Fratta E, et al. Whole genome methylation profiles as independent markers of survival in stage IIIC melanoma patients. J Transl Med 2012;10:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao L, Smit MA, van den Oord JJ, et al. Genome-wide promoter methylation analysis identifies epigenetic silencing of MAPK13 in primary cutaneous melanoma. Pigment Cell Melanoma Res 2013;26:542-54. [DOI] [PubMed] [Google Scholar]
- 16.Gao L, van den Hurk K, Moerkerk PT, et al. Promoter CpG island hypermethylation in dysplastic nevus and melanoma: CLDN11 as an epigenetic biomarker for malignancy. J Invest Dermatol 2014;134:2957-66. [DOI] [PubMed] [Google Scholar]
- 17.Lauss M, Haq R, Cirenajwis H, et al. Genome-Wide DNA Methylation Analysis in Melanoma Reveals the Importance of CpG Methylation in MITF Regulation. J Invest Dermatol 2015;135:1820-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marzese DM, Scolyer RA, Huynh JL, et al. Epigenome-wide DNA methylation landscape of melanoma progression to brain metastasis reveals aberrations on homeobox D cluster associated with prognosis. Hum Mol Genet 2014;23:226-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vizoso M, Ferreira HJ, Lopez-Serra P, et al. Epigenetic activation of a cryptic TBC1D16 transcript enhances melanoma progression by targeting EGFR. Nat Med 2015;21:741-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo X, Xu Y, Zhao Z. In-depth genomic data analyses revealed complex transcriptional and epigenetic dysregulations of BRAF (V600E) in melanoma. Mol Cancer 2015;14:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ploper D, De Robertis EM. The MITF family of transcription factors: Role in endolysosomal biogenesis, Wnt signaling, and oncogenesis. Pharmacol Res 2015;99:36-43. [DOI] [PubMed] [Google Scholar]
- 22.Ji Z, Erin Chen Y, Kumar R, et al. MITF Modulates Therapeutic Resistance through EGFR Signaling. J Invest Dermatol 2015;135:1863-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 2011;29:3085-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Huang SK, Marzese DM, et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J Invest Dermatol 2015;135:532-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goueli BS, Powell MB, Finger EC, et al. TBC1D16 is a Rab4A GTPase activating protein that regulates receptor recycling and EGF receptor signaling. Proc Natl Acad Sci USA 2012;109:15787-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrone L, Hersey P. The chemoresistance of human malignant melanoma: an update. Melanoma Res 1999;9:51-8. [DOI] [PubMed] [Google Scholar]
- 27.Menzies AM, Long GV. Systemic treatment for BRAF-mutant melanoma: where do we go next? Lancet Oncol 2014;15:e371-81. [DOI] [PubMed] [Google Scholar]
- 28.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005;5:275-84. [DOI] [PubMed] [Google Scholar]
- 29.Sun C, Wang L, Huang S, et al. Reversible and adaptive resistance to BRAF (V600E) inhibition in melanoma. Nature 2014;508:118-22. [DOI] [PubMed] [Google Scholar]
- 30.Perona R, Sánchez-Pérez I. Control of oncogenesis and cancer therapy resistance. Br J Cancer 2004;90:573-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duffy MJ, Crown J. Companion biomarkers: paving the pathway to personalized treatment for cancer. Clin Chem 2013;59:1447-56. [DOI] [PubMed] [Google Scholar]
- 32.Ozben T. Mechanisms and strategies to overcome multiple drug resistance in cancer. FEBS Lett 2006;580:2903-9. [DOI] [PubMed] [Google Scholar]
- 33.Vadlapatla RK, Vadlapudi AD, Pal D, et al. Mechanisms of drug resistance in cancer chemotherapy: coordinated role and regulation of efflux transporters andmetabolizing enzymes. Curr Pharm Des 2013;19:7126-40. [DOI] [PubMed] [Google Scholar]
- 34.Neesse A, Krug S, Gress TM, et al. Emerging concepts in pancreatic cancer medicine: targeting the tumor stroma. Onco Targets Ther 2013;7:33-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madorsky Rowdo FP, Baron A, Urrutia M, et al. Immunotherapy in Cancer: A Combat between Tumors and the Immune System; You Win Some, You Lose Some. Front Immunol 2015;6:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung HJ, Mahalingam M. Angiogenesis, vasculogenic mimicry and vascular invasion in cutaneous malignant melanoma-implications for therapeutic strategies and targeted therapies. Expert Rev Anticancer Ther 2014;14:621-39. [DOI] [PubMed] [Google Scholar]
- 37.Middleton MR, Grob JJ, Aaronson N, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol 2000;18:158-66. Erratum in: J Clin Oncol 2000;18:158-66. [DOI] [PubMed] [Google Scholar]
- 38.Avril MF, Aamdal S, Grob JJ, et al. Fotemustine compared with dacarbazine in patients with disseminated malignant melanoma: a phase III study. J Clin Oncol 2004;22:1118-25. [DOI] [PubMed] [Google Scholar]
- 39.Stadler S, Weina K, Gebhardt C, et al. New therapeutic options for advanced non-resectable malignant melanoma. Adv Med Sci 2015;60:83-8. [DOI] [PubMed] [Google Scholar]
- 40.Chen D, Lin Q, Box N, et al. SKI knockdown inhibits human melanoma tumour growth in vivo. Pigment Cell Melanoma Res 2009;22:761-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mimeault M, Batra SK. Novel biomarkers and therapeutic targets for optimizing the therapeutic management of melanomas. World J Clin Oncol 2012;3:32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Czyz M, Koprowska K, Sztiller-Sikorska M. Parthenolide reduces the frequency of ABCB5-positive cells and clonogenic capacity of melanoma cells from anchorage independent melanospheres. Cancer Biol Ther 2013;14:135-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature 2008;451:345-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corazzari M, Rapino F, Ciccosanti F, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ 2015;22:946-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang ZM, Chinen M, Chang PJ, et al. Targeting protein-trafficking pathways alters melanoma treatment sensitivity. Proc Natl Acad Sci USA 2012;109:553-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Erfle H, Pashayeva K, Harder N, et al. Targeting mitosis-regulating genes in cisplatin-sensitive and -resistant melanoma cells: A live-cell RNAi screen displays differential nucleus-derived phenotypes. Biotechnol J 2015;10:1467-77. [DOI] [PubMed] [Google Scholar]
- 47.Kramer D, Schön M, Bayerlová M, et al. A pro-apoptotic function of iASPP by stabilizing p300 and CBP through inhibition of BRMS1 E3 ubiquitin ligase activity. Cell Death Dis 2015;6:e1634. [DOI] [PMC free article] [PubMed]
- 48.Kreiseder B, Holper-Schichl YM, Muellauer B, et al. Alpha-catulin contributes to drug-resistance of melanoma by activating NF-κB and AP-1. PLoS One 2015;10:e0119402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jayachandran A, Anaka M, Prithviraj P, et al. Thrombospondin 1 promotes an aggressive phenotype through epithelial-to-mesenchymal transition in human melanoma. Oncotarget 2014;5:5782-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu Y, Xie M, Song T, et al. A novel BH3 mimetic efficiently induces apoptosis in melanoma cells through direct binding to anti-apoptotic Bcl-2 family proteins, including phosphorylated Mcl-1. Pigment Cell Melanoma Res 2015;28:161-70. [DOI] [PubMed] [Google Scholar]
- 51.Sun Y, Long J, Zhou Y. Angiopoietin-like 4 promotes melanoma cell invasion and survival through aldolase A. Oncol Lett 2014;8:211-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Christmann M, Verbeek B, Roos WP, et al. O(6)-Methylguanine-DNA methyltransferase (MGMT) in normal tissues and tumors: enzyme activity, promoter methylation and immunohistochemistry. Biochim Biophys Acta 2011;1816:179-90. [DOI] [PubMed]
- 53.Hegi ME, Liu L, Herman JG, et al. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J Clin Oncol 2008;26:4189-99. [DOI] [PubMed] [Google Scholar]
- 54.Amatu A, Sartore-Bianchi A, Moutinho C, et al. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin Cancer Res 2013;19:2265-72. [DOI] [PubMed] [Google Scholar]
- 55.Christmann M, Pick M, Lage H, et al. Acquired resistance of melanoma cells to the antineoplastic agent fotemustine is caused by reactivation of the DNA repair gene MGMT. Int J Cancer 2001;92:123-9. [PubMed] [Google Scholar]
- 56.Hassel JC, Sucker A, Edler L, et al. MGMT gene promoter methylation correlates with tolerance of temozolomide treatment in melanoma but not with clinical outcome. Br J Cancer 2010;103:820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma S, Egyházi S, Ueno T, et al. O6-methylguanine-DNA-methyltransferase expression and gene polymorphisms in relation to chemotherapeutic response in metastatic melanoma. Br J Cancer 2003;89:1517-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Naumann SC, Roos WP, Jöst E, et al. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and p53. Br J Cancer 2009;100:322-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dietel M, Jöhrens K, Laffert M, et al. Predictive molecular pathology and its role in targeted cancer therapy: a review focussing on clinical relevance. Cancer Gene Ther 2013;20:211-21. [DOI] [PubMed] [Google Scholar]
- 60.Carmona FJ, Azuara D, Berenguer-Llergo A, et al. DNA methylation biomarkers for noninvasive diagnosis of colorectal cancer. Cancer Prev Res (Phila) 2013:6;656-65. [DOI] [PubMed] [Google Scholar]
- 61.Ploussard G, de la Taill A. Urine biomarkers in prostate cancer. Nat Rev Urol 2010;7:101-9. [DOI] [PubMed] [Google Scholar]
- 62.Moran S, Vizoso M, Martinez-Cardús A, et al. DNA Methylation profiling on Formalin-Fixed Paraffin-Embedded samples using Infinium HumanMethylation 450 BeadChips. Epigenetics 2014;9:829-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moutinho C, Martinez-Cardús A, Santos C, et al. Epigenetic inactivation of the BRCA1 interactor SRBC and resistance to oxaliplatin in colorectal cancer. J Natl Cancer Inst 2014;106:djt322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stevens M, Cheng JB, Li D, et al. Estimating absolute methylation levels at single-CpG resolution from methylation enrichment and restriction enzyme sequencing methods. Genome Res 2013;23:1541-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Diaz LA, Jr, Bardelli A. Liquid biopsies: genotyping circulating tumour DNA. J Clin Oncol 2014;32:579-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.De Mattos-Arruda L, Cortes J, Santarpia L, et al. Circulating tumour cells and cell-free DNA as tools for managing breast cancer. Nat Rev Clin Oncol 2013;10:377-89. [DOI] [PubMed] [Google Scholar]
- 67.Crowley E, Di Nicolantonio F, Loupakis F, et al. Liquid biopsy: monitoring cancer-genetics in the blood. Nat Rev Clin Oncol 2013;10:472-84. [DOI] [PubMed] [Google Scholar]
- 68.Murtaza M, Dawson SJ, Tsui DW, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013;497:108-12. [DOI] [PubMed] [Google Scholar]
- 69.Dawson SJ, Rosenfeld N, Caldas C. Circulating tumour DNA to monitor metastatic breast cancer. N Engl J Med 2013;369:93-4. [DOI] [PubMed] [Google Scholar]
- 70.Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bedard PL, Hansen AR, Ratain MJ, et al. Tumour heterogeneity in the clinic. Nature 2013;501:355-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Davis M, Pragani R, Popovici-Muller J, et al. ML309: A potent inhibitor of R132H mutant IDH1 capable of reducing 2-hydroxyglutarate production in U87 MG glioblastoma cells. Probe Reports from the NIH Molecular Libraries Program [Internet] 2013. [PubMed] [Google Scholar]
- 73.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene 2009;28:1653-68. [DOI] [PubMed] [Google Scholar]