

Summary
We present ChromATin, a quantitative high-resolution imaging approach for investigating chromatin organization in complex tissues. This method combines analysis of epigenetic modifications by immunostaining, localization of specific DNA sequences by FISH, and high-resolution segregation of nuclear compartments using array tomography (AT) imaging. We then apply this approach to understand how the genome is organized in the mammalian brain using female Rett Syndrome mice, which are a mosaic of normal and Mecp2-null cells. Side-by-side comparisons within the same field reveal distinct heterochromatin territories in wild type neurons that are altered in Mecp2-null nuclei. Mutant neurons exhibit increased chromatin compaction and a striking redistribution of the H4K20me3 histone modification into pericentromeric heterochromatin, a territory occupied normally by MeCP2. These events are not observed in every neuronal cell type, highlighting ChromATin as a powerful in situ method for examining cell type-specific differences in chromatin architecture in complex tissues.

Introduction
The organization of chromatin within the nucleus plays an important role in the regulation of gene expression (Bickmore and van Steensel, 2013; Politz et al., 2013). Although high-throughput sequencing strategies have revolutionized chromatin research by enabling genome wide analysis of chromatin interactions (Dixon et al., 2012; Lieberman-Aiden et al., 2009), fluorescent in situ hybridization (FISH) remains a powerful tool in studying the organization of chromosomal territories (Cremer and Cremer, 2010). New high-resolution imaging technologies promise to advance our understanding of how chromatin is packaged in the nucleus for appropriate gene expression (Ricci et al., 2015; Smeets et al., 2014).
New methods for examining chromatin architecture are needed. The two most widely used strategies, chromosome conformation capture (C-method) and FISH, each have their own strengths and weaknesses. Although C-methods offer base pair resolution and, in the case of HiC, genome wide analysis of chromatin, this method is most often performed on pooled cell populations, which might obscure cell type-specific differences that exist in complex tissues. On the other hand, FISH is an ideal method for analysis of different cell types in tissue, but probes are typically limited to a small number of genetic loci. Interestingly, these methods are not always in agreement with regard to chromatin organization. For example, analysis of the HoxD locus in mutant embryonic stem cells shows an open chromatin structure using FISH and a closed structure using 5C (Williamson et al., 2014). A potential source of these differences is that C-methods may involve fixation of relatively large, cross-linked chromatin domains, detecting cytological co-localization rather than direct molecular interaction (Belmont, 2014; Gavrilov et al., 2013). Also, to associate C-method and FISH results with chromatin modifications requires a separate analysis using different experimental conditions. For these reasons, we sought to develop a quantitative, high-resolution imaging method for investigating chromatin organization in complex tissues. This method would combine analysis of epigenetic modifications by immunostaining, localization of specific DNA sequences by FISH, and high-resolution segregation of nuclear compartments using an advanced imaging technique. We have adapted the array tomography (AT) imaging method for this purpose.
AT is a high-resolution imaging method developed for the reconstruction and analysis of neuronal circuitry in the brain (Micheva and Smith, 2007). The enhanced resolution is achieved by generating ultra thin serial sections of the specimen, followed by image acquisition and alignment. Acrylic sections can be stripped repeatedly allowing for multiple rounds of imaging. This multiplexed staining approach increases the amount of molecular information that can be derived from a tissue volume (Micheva et al., 2010). FISH methods have not been reported for AT, and developing this capability would increase the utility of the approach for localizing DNA sequences or expressed RNAs. Our motivation in developing this method was to gain a deeper understanding of how the genome is organized in the mammalian brain, a tissue with an extreme variety of cell types. To this end, we tested AT for examining neuronal chromatin in mice lacking the DNA binding protein, MeCP2. Mutations in MECP2 give rise to the neurological disorder, Rett Syndrome (RTT) (Amir et al., 1999). MeCP2 is expressed to high levels in neurons, and binds globally to methyl- and hydroxymethyl- cytosine within different dinucleotide contexts (Guo et al., 2014; Lewis et al., 1992; Mellén et al., 2012). Mecp2 is an X-linked gene (Quaderi et al., 1994), and cells in female RTT patients and mouse models are mosaic for loss of MeCP2 due to dosage compensation in mammals (Adler et al., 1995). This mosaicism provides an ideal experimental context wherein neurons with normal chromatin architecture are adjacent to Mecp2-null neurons. While in vitro studies suggest that MeCP2 may regulate higher order chromatin structure, it is not known how these findings impact chromatin organization in vivo. Further, several additional models for its function, including gene repression and activation, have also been proposed (Lyst and Bird, 2015).
Using AT imaging, we quantify large-scale chromatin changes in symptomatic adult female RTT mouse brain. We detect a significant increase in chromatin compaction in two types of Mecp2-null hippocampal neurons together with a striking redistribution of the H4K20me3 histone modification into pericentromeric heterochromatin. In contrast, we do not detect these changes in cerebellar granule cells. We observe a spectrum of chromatin condensation states among cells in the nervous system providing a potential mechanism to explain cell type-specific differences in gene expression upon loss of MeCP2.
In summary, we show that AT is an ideal tool for investigating chromatin architecture in complex tissues where cellular heterogeneity may confound methods that sample populations of cells. Multiplexed detection of epigenetic modifications and genomic sequences combines with the resolving power of the AT imaging method to permit quantitative analysis within defined nuclear compartments.
Results
Quantitative Analysis of Chromatin Organization in Neurons using AT
We initially chose hippocampal CA1 pyramidal neurons for our investigation due to their location within a well-defined layer in the hippocampus (Figure 1A). Neuronal nuclei, as in other cell types, can be visualized using the fluorescent DNA intercalator, DAPI (Wilson et al., 1990). To determine the utility of AT for analyzing chromatin architecture, we exploited the mosaic nature of female RTT mouse brain expressing a knock-in Mecp2-EGFP gene fusion (Lyst et al., 2013). Mosaicism is ideal for imaging comparisons because fixation, embedding, staining, and imaging steps are equivalent for the WT and mutant populations of neurons under investigation. As predicted, due to the X-linked nature of Mecp2 and random X inactivation, the ratio of WT (GFP-positive) to mutant cells was approximately 1:1 (Figure 1B).
Figure 1. Quantitative Analysis of Chromatin Architecture In Hippocampal Pyramidal Neurons.

(A) DAPI stained nuclei in a 200 nm hippocampal section from a symptomatic female RTT mouse (Mecp2-egfpB./+). The image was stitched from multiple fields acquired using a 25X objective. Scale bar, 100 μm. The white rectangle encloses the portion of the CA1 pyramidal cell layer used for high-resolution imaging. (B) Volume rendering of 59 serial sections (200 nm thick) through the hippocampal pyramidal cell layer. DAPI labels nuclei, and an antibody to GFP was used to identify cells expressing MeCP2-GFP. Note mosaic expression of Mecp2-egfp gene due to random Xi. Scale bar, 10 μm. (C-F) Nucleus from a WT pyramidal neuron. (C) Volume rendering of the nucleus viewed along the x-y axis. Scale bar, 1 μm. (D) Nucleus viewed along the y-z axis. (E) 3D surfaces (red) used to isolate heterochromatin for quantification. (F) A single 3D surface (blue) encloses the entire contents of the nucleus. (G-I) Analysis of WT and Mecp2-null pyramidal neurons. (G) Scatter plot showing the percentage of total nuclear DAPI pixel intensity located within the heterochromatin threshold (mean ± SD). (WT, 22.5 ± 2.5 %, n = 51 nuclei from 3 mice), (null, 27.1 ± 2.2 %, n = 55 nuclei from 3 mice). Unpaired t test, p < 0.0001. Xi chromosome values were subtracted from total heterochromatin. (H) Scatter plot showing the density of DAPI pixel intensity within the heterochromatin threshold (mean ± SD). Density is expressed as pixel intensity (×103) per μm3. (WT, 50.7 ± 2.8), (null, 54.4 ± 2.9). Unpaired t test, p < 0.0001. (I) Scatter plot showing heterochromatin volume (mean ± SD). (WT, 8.0 ± 1.0 μm3), (null, 9.0 ± 0.9 μm3). Unpaired t test, p < 0.0001. See also Figure S1 and Movie S1.
We found that 200 nm sections allowed for full volume reconstruction of an appropriate number of nuclei for analysis. Figure 1C shows 3D reconstruction for a WT nucleus visualized along the x-y axis, while Figure 1D shows equivalent resolution along the z-axis. Heterochromatic foci in neuronal nuclei exhibit a wide range of sizes and numbers. We therefore established a threshold based upon pixel intensity to quantify heterochromatin intensity and volume. A representative surface rendering of the heterochromatic foci enclosed by our threshold is shown in Figure 1E. A second 3D surface enclosing the entire nucleus was used to determine total nuclear volume and DAPI pixel intensity (Figure 1F). The structures enclosed by these surfaces are best visualized in Movie S1.
Using AT, we found that the total amount of nuclear DAPI did not differ between WT and Mecp2-null neurons (Figure S1A). However, loss of MeCP2 resulted in a 20% increase in the amount of DNA packaged within the heterochromatin threshold (Figure 1G). The increase in chromatin compaction was detected as both an increase in DAPI density within heterochromatin (Figure 1H), as well as an increase in heterochromatin volume (Figure 1I). These results indicate that in Mecp2-null neurons there is a redistribution of DAPI-labeled DNA into more densely compact heterochromatin, and the increase in volume is not due to “unraveling” of chromatin within heterochromatic foci.
Previous studies found that nuclear diameters in RTT neurons were smaller than WT neurons (Li et al., 2013; Stuss et al., 2013; Yazdani et al., 2012). In agreement with these reports, AT analysis of nuclear volume revealed a modest (~5%) decrease in mutant neurons compared to WT (Figure S1B). Interestingly, we found a strong negative correlation when we plotted heterochromatin content versus nuclear volume for CA1 pyramidal neurons regardless of MeCP2 status (Figure S1C). This is consistent with previous reports that show an expansion of nuclear size when chromatin is decondensed (Mazumder et al., 2008; Shen et al., 1995).
Resolving Spatial Organization of Histone Modifications with Multiplexed Immunostaining
The other notable advantage of AT over other imaging methods is the ability to perform multiple rounds of imaging on the same sections using a variety of detection reagents. The observation of more compact chromatin in Mecp2-null neurons led us to examine whether we could detect any changes in heterochromatin-associated histone modifications. In Figure 2, we show the results of multiplexed immunostaining using antibodies against five different histone modifications, as well as for DAPI, MeCP2-GFP, and Conconavalin A (ConA). Immunostaining for H3K9me2, H3K9me3, H3K27me3, and H4K20me3 show expected patterns in terms of their association with DAPI labeled heterochromatin, indicating that our cytological criterion for heterochromatin is consistent with previously published studies (Figure 2D-H).
Figure 2. Multiplexed Immunostaining for Histone Modifications.

Panels A-I represent fluorescence images acquired using the same 200 nm section through the hippocampal pyramidal cell layer. (A) DAPI staining of CA1 pyramidal neuron nuclei. Right, two WT nuclei (see panel I). Left, two mutant nuclei. Scale bar, 2 μm. (B) Binding of the lectin Concanavalin A (ConA) to membrane glycoproteins. (C) Merge of ConA (red) with DAPI (green). (D) Immunostaining for H3K9me2. (E) Immunostaining for H3K9me3. (F) Immunostaining for H3K27me3. The two intense H3K27me3 clusters represent the Xi. These clusters are not visible in the two lower nuclei because this image is of a single 200 nm section. (G) Immunostaining for H4K20me2. (H) Immunostaining for H4K20me3. (I) Immunostaining for the MeCP2-GFP fusion protein. (J) DAPI staining of a nucleus from a WT neuron. Panels J-L represent fluorescence images acquired from the same 200 nm section from another mouse. Scale bar, 1 μm. (K) Heatmap representation of DAPI pixel intensity for the section shown in Figure 2J. The two adjacent heterochromatin clusters exhibit different density profiles. Bar shows heatmap index. (L) Merged immunostaining for MeCP2-GFP (green) and H3K27me3 (red). MeCP2-GFP is enriched in the heterochromatin cluster with higher DAPI pixel intensity in Figure 2K, while H3K27me3 enriched heterochromatin has lower DAPI pixel intensity. (M) Scatter plot showing Xi DAPI pixel density (mean ± SD). Density is expressed as pixel intensity (×103) per μm3. (WT, 42.0 ± 2.5, n=51), (null, 43.0 ± 3.3, n=55). Unpaired t test, p = 0.078. (N) Scatter plot showing the percentage of MeCP2-GFP (mean ± SD) localized within the heterochromatin threshold in WT neurons. (30.4 ± 3.3 %, n=51). See also Movie S2.
We always observed a single intense cluster of H3K27me3 in female cells that represents the Xi (Figure 2F), and MeCP2-GFP was generally excluded from this heterochromatin compartment (Fig. 2J-L). These exclusive heterochromatin territories are best appreciated by viewing the full 3D reconstruction (Movie S2). Using H3K27me3 immunostaining as a guide, we isolated this compartment (green surface in Movie S1) from total heterochromatin. Consistent with the observation that MeCP2 is excluded from this territory, we did not observe a significant difference in DAPI density for the Xi chromosome between WT and mutant neurons (Figure 2M). Further, the density of DAPI within the Xi was less than the density within other heterochromatin compartments (~80%, compare with Figure 1H). This less condensed heterochromatin state for Xi has been described previously by light and electron microscopy (Rego et al., 2008).
In addition to quantifying DAPI intensity, the established heterochromatin threshold can be applied to all the imaging channels acquired in an experiment. For example, although it appears upon visual examination that most of the MeCP2 is localized to heterochromatic foci, low intensity clusters are highly abundant in euchromatin and nucleoplasm, resulting in heterochromatin MeCP2 content being ~30% of total MeCP2 in a pyramidal cell nucleus (Figure 2N).
The H4K20me3 Modification Defines a Unique Heterochromatin Territory
AT imaging revealed an unexpected alteration in the spatial organization of H4K20me3 when WT and Mecp2-null nuclei were compared. In neurons deficient for MeCP2, H4K20me3 displayed a staining pattern that was almost identical to that of DAPI (compare figures 3A-D, right nuclei), consistent with previous reports that H4K20me3 is associated with pericentromeric heterochromatin (Schotta et al., 2004). Strikingly, this extensive overlap with heterochromatic foci was not observed in WT neurons (Figure 3A-D, left nuclei). In WT neurons, MeCP2 clustered intensely in heterochromatic foci, while the H4K20me3 intensity was reduced in these regions (arrowheads, compare fig. 3D and F left nuclei), instead occupying a territory peripheral to the dense heterochromatic foci (arrows, Figure 3D and 3F). These H4K20me3 rich territories are well within the heterochromatin threshold, but they display reduced DAPI intensity when compared to pericentromeric heterochromatin (arrow, Figure 3B). The segregation of H4K20me3, H3K27me3, and MeCP2 enriched heterochromatin territories is easily observed using AT (Movie S3). We performed confocal microscopy on brain sections to confirm that the H4K20me3 territories were not an artifact of the AT procedure. Examination of CA1 pyramidal neurons from female heterozygotes or wild type male mice shows that the H4K20me3 territories can be detected using standard microscopy methods albeit at lower resolution (Figure S2).
Figure 3. The H4K20me3 Modification is Redistributed Spatially Upon Loss of MeCP2.

Panels A-F represent fluorescence images acquired using the same 200 nm section through the hippocampal pyramidal cell layer. (A) DAPI staining. Scale bar, 1 μm. (B) Relative pixel intensity of DAPI staining. Heat map index is located in the lower right corner. Arrow indicates lower intensity heterochromatin still within threshold value. Arrowhead points to high intensity heterochromatin. (C) Immunostaining for MeCP2-GFP. Left, WT nucleus; Right, Mecp2-null nucleus. (D) Immunostaining for H4K20me3. Arrows and arrowhead as in Fig. 3B. (E) Immunostaining for H4K20me2. (F) Merged image for MeCP2-GFP and H4K20me3. In the WT nucleus, MeCP2-GFP binding dominates in pericentromeric heterochromatin (arrowhead) while H4K20me3 localizes to the heterochromatin region with lower DAPI density (arrow). (G) Scatter plot showing percentage of total nuclear H4K20me3 within heterochromatin in pyramidal neurons (mean ± SD). There is a significant redistribution of H4K20me3 into heterochromatin after loss of MeCP2 (WT, 18.5 ± 3.0, n = 36 nuclei from 3 mice), (null, 30.5 ± 3.5, n = 41 nuclei from three mice). Unpaired t test, p < 0.0001. (H) Scatter plot showing the percentage of total nuclear H4K20me2 within heterochromatin (mean ± SD) for pyramidal neurons. (WT, 10.8 ± 2.1, n = 27 nuclei from 2 mice), (null, 11.0 ± 2.2, n = 31 nuclei from two mice). Unpaired t test, p = 0.72. See also Figure S2, Figure S3, and Movie S3.
We used the heterochromatin threshold to quantify the extent of this altered localization and detected a robust (65%) redistribution of H4K20me3 into dense heterochromatin upon loss of MeCP2 (Figure 3G). This spatial redistribution occurred in conjunction with a modest (11%) increase in the total nuclear amount of H4K20me3 (see Figure 4F). In contrast to H4K20me3, we did not observe a significant change in the spatial organization of H4K20me2 with loss of MeCP2 (Figure 3H), indicating specificity in spatial organization of different histone marks in mutant nuclei.
Figure 4. The H4K20me3 Modification Expands Into Pericentromeric Heterochromatin in Mecp2-Null Nuclei.

Panels A-D represent fluorescence images taken from a volume rendering through the hippocampal pyramidal cell layer. (A) DAPI staining. Scale bar, 1 μm. (B) Immunostaining for MeCP2-GFP identifies left neuron as WT and right neuron as Mecp2-null. (C) Immunostaining for H4K20me3. (D) Merged image showing H4K20me3 distribution (red) relative to major satellite FISH signal (green). In the WT neuron, H4K20me3 enriched regions are peripheral to the major satellite heterochromatin territory. (E) Scatter plot showing the percentage of total nuclear H4K20me3 within the major satellite threshold (mean ± SD). There is a significant redistribution of H4K20me3 into pericentromeric heterochromatin after loss of MeCP2 (WT, 11.8 ± 1.8, n = 19 nuclei from 3 mice), (null, 23.7 ± 4.4, n = 19 nuclei from three mice). Unpaired t test, p < 0.0001. (F) The relative amount of H4K20me3 within the nucleus (mean ± SD) is compared between WT and null neurons. Intensity units represent total integrated intensity (×106) with mean intensity normalized for WT pyramidal neurons. (WT, 1.00 ± 0.01, n = 46 nuclei from 3 animals), (null, 1.11 ± 0.02, n = 50 nuclei from three mice). Unpaired t test, p < 0.0001. See also Movie S4.
We also examined the spatial distribution of H3K9me3, another heterochromatin-associated histone modification. Unlike the case for H4K20me3, no heterochromatin territories were enriched exclusively for this modification. In fact, H3K9me3 appeared to be present throughout all of the heterochromatin territories that we observed. Although it was difficult to detect by eye, AT analysis showed that there was still a significant (26%) redistribution of H3K9me3 into densely packed heterochromatin in Mecp2-null nuclei (Figure S3A). This occurred in conjunction with a 10% increase in total nuclear H3K9me3 levels (Figure S3C). We also detected a slight redistribution of H3K9me2 into dense heterochromatin (Figure S3B).
AT FISH Shows Expansion of the H4K20me3 Modification Into Pericentromeric Heterochromatin in Mecp2-Null Nuclei
To map chromosomal territories more precisely in the nucleus, we used peptide nucleic acid probes specific for both major and minor satellite sequences to localize pericentromeric and centromeric heterochromatin, respectively (Movie S4). We performed one round of immunostaining followed by FISH on the same hippocampal sections to determine the relative positions of H4K20me3, MeCP2-GFP, and the major and minor satellite sequences. As expected, the major satellite probe localized with the most condensed heterochromatic foci (Figure 4A and D). As predicted on the basis of our DAPI staining, the H4K20me3 rich territory was adjacent to the major satellite territory in WT nuclei (Figure 4D, left nuclei). In neurons lacking MeCP2, this segregation was lost, and H4K20me3 was enriched significantly in the major satellite territory (Figure 4D, right nuclei). We created a 3D threshold surrounding the major satellite FISH signal and measured the intensity of H4K20me3 immunostaining within the major satellite zone relative to the total nuclear intensity. We detected a robust (100%) redistribution of H4K20me3 into pericentromeric heterochromatin upon loss of MeCP2 (Figure 4E). This spatial reorganization occurred in conjunction with a modest increase in the total nuclear amount of the modification (Figure 4F).
Changes in Chromatin Architecture Upon Loss of MeCP2 are Cell Type-Specific
To determine if the changes in chromatin architecture we observed for CA1 pyramidal neurons were occurring in other neuronal cell types, we performed a similar analysis for hippocampal dentate granule cells as well as cerebellar granule cells. We performed two rounds of antibody staining to collect information for both H4K20me3 and H3K9me3 modifications, followed by FISH for major satellite sequences. MeCP2-GFP is abundant in dentate granule cell nuclei, with the typical strong localization in heterochromatic foci (Figure 5B). Similar to CA1 pyramidal neurons we find that loss of MeCP2 results in a significant redistribution (93%) of H4K20me3 into the major satellite territory (Figure 5D-E). Analysis for H3K9me3 showed redistribution to a lesser extent (37%, Figure S4E and G) while H3K9me2 levels show a slight decrease in the major satellite territory (Figure S4F and H). The changes in H4K20me3 and H3K9me3 in dentate granule cells lacking MeCP2 are accompanied by a 15% increase in DAPI-labeled DNA packaged into densely packed heterochromatin (Figure 5F).
Figure 5. H4K20me3 Redistribution in Mecp2-Null Nuclei is a Cell Type Specific Event.

Panels A-D represent fluorescence images taken from a volume rendering through the suprapyramidal dentate granule cell layer. (A) DAPI staining. Scale bar, 1 μm. (B) Immunostaining for MeCP2-GFP identifies left neuron as WT and right neuron as Mecp2-null. (C) Immunostaining for H4K20me3. (D) Merged image showing H4K20me3 distribution (red) relative to major satellite FISH signal (green). (E) Scatter plot showing the percentage of total nuclear H4K20me3 within the major satellite threshold (mean ± SD). There is a significant redistribution of H4K20me3 into pericentromeric heterochromatin after loss of MeCP2 (WT, 14.0 ± 1.8, n = 15 nuclei from two mice), (null, 27.0 ± 2.1, n = 15 nuclei from two mice). Unpaired t test, p < 0.0001. (F) Scatter plot showing the percentage of total nuclear DAPI pixel intensity located within the heterochromatin threshold (mean ± SD). (WT, 24.7 ± 2.7), (null, 28.5 ± 1.8). Unpaired t test, p < 0.0001. Panels G-J represent fluorescence images taken from a volume rendering through the cerebellar granule cell layer. (G) DAPI staining. Scale bar, 1 μm. (H) Immunostaining for MeCP2-GFP distinguishes between WT and MeCP2-null granule cells. (I) Immunostaining for H4K20me3. (J) Merged image showing H4K20me3 distribution (red) relative to major satellite FISH signal (green). (K) Scatter plot showing the percentage of total nuclear H4K20me3 within the major satellite threshold (mean ± SD). (WT, 30.7 ± 2.1, n = 17 nuclei from two mice), (null, 31.2 ± 1.8, n = 17 nuclei from two mice). Unpaired t test, p = 0.4075. (L) Scatter plot showing the percentage of total nuclear DAPI pixel intensity located within the heterochromatin threshold (mean ± SD). (WT, 56.2 ± 2.5), (null, 56.9 ± 3.0). Unpaired t test, p = 0.4526. See also Figure S4, Figure S5, and Movie S5.
We next imaged chromatin organization in wild type and Mecp2-null cerebellar granule cells, which have smaller nuclei and abundant heterochromatin. While we could easily identify MeCP2-positive granule cells (Figure 5H), the expression level of MeCP2 and intensity in the major satellite territory was lower in these neurons compared to other neuronal cell types. We could not detect a change in the spatial distribution of H4K20me3 in cerebellar granule cells (Figure 5K). Consistent with their visible heterochromatic abundance, the amount of DAPI within heterochromatin was much greater for these neurons than for the hippocampal cell types we examined (Figure 5L). This was not a consequence of the brain region per se, because neighboring cell types in the cerebellum expressed much higher levels of MeCP2 than granule cells and exhibited the alterations in H4K20me3 organization that we observed for Mecp2-null hippocampal neurons. For example, cerebellar Purkinje neurons have extremely decondensed chromatin, and major satellite repeats are typically packaged in a massive heterochromatin cluster near the nucleolus (Figure S5A). In WT Purkinje neurons, MeCP2 clusters intensely in the major satellite territory, and H4K20me3 is predominantly localized to an adjacent heterochromatin territory (Figure S5B). In a Purkinje neuron that lacks MeCP2, H4K20me3 spreads into the major satellite territory (Figure S5C and Movie S5). Due to the large size of Purkinje nuclei, we have not acquired enough data from full Purkinje nuclei to quantify this change.
AT Analysis of Transcriptional Activity
We used antibodies against phosphorylated Serine5 on the carboxy terminal domain (CTD) of RNA polymerase II (RNAPII) to quantify transcriptional activity within single nuclei (Egloff and Murphy, 2008). As predicted, the signal for this antibody was excluded from highly condensed heterochromatin (Figure 6A-C). Further, analysis of the reconstructed nuclei revealed a strong negative correlation between Phospho Ser5 CTD levels and nuclear heterochromatin content (Figure 6D). We also compared total nuclear levels of Phospho Ser5 CTD between WT and mutant neurons in female RTT hippocampus. This analysis showed a slight but significant reduction in Phospho Ser5 CTD in mutant neurons (Figure 6E). Antibodies against the N-terminus of RNAPII did not work in AT, so we could not determine whether the reduction in Phospho Ser5 CTD was a result of lower total levels of RNAPII or is indeed a reflection of reduced transcriptional initiation in Mecp2-null nuclei. These experiments show that, upon the development of improved reagents, AT imaging can be effectively used to quantify transcriptional activity in different cell types.
Figure 6. AT Analysis of Transcriptional Activity.
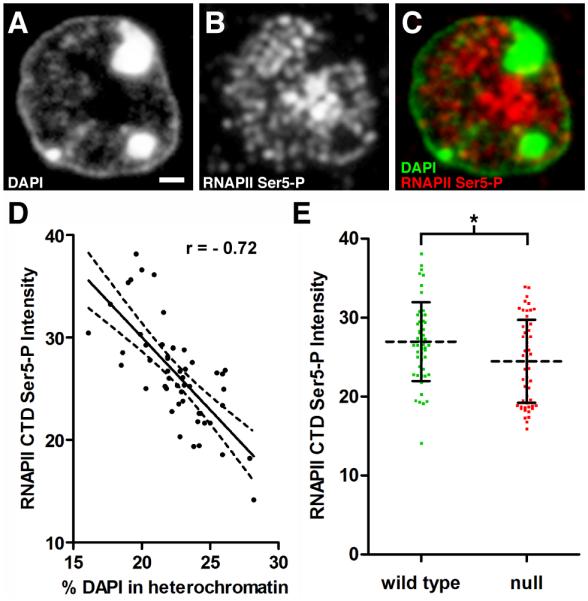
Panels A-C represent fluorescence images acquired using the same 200 nm section through a pyramidal neuron. (A) DAPI staining. Scale bar, 1 μm. (B) Immunostaining with antibodies directed against the phosphorylated CTD (Ser5) of RNA polymerase II (RNAPII Ser5-P) to detect the active polymerase. (C) Merged image of DAPI and RNAPII Ser5-P. Note that RNAPII Ser5-P is excluded from heterochromatic foci. (D) Total integrated pixel intensity (×105) for RNAPII Ser5-P from WT nuclei was plotted versus heterochromatin content. RNAPII Ser5-P levels are negatively correlated with increasing heterochromatin content (n = 51 nuclei from 3 mice, Pearson r = − 0.72, p < 0.0001). (E) Scatter plot comparing RNAPII Ser5-P levels in WT and Mecp2-null pyramidal neurons. Intensity units represent total integrated pixel intensity (×105) with mean intensity normalized for WT pyramidal neurons. (WT, 27.0 ± 5.0, n = 51 nuclei from 3 animals), (null, 24.5 ± 5.2, n = 55 nuclei from three animals). Unpaired t test, p = 0.013.
Discussion
Here we describe an adaptation and expansion of AT for the investigation of chromatin organization. The advantages that make AT a useful tool for resolving synaptic junctions in the brain also make it an ideal tool for resolving nuclear compartments. Ultrathin sectioning improves resolution along the z-axis while also eliminating signal degradation due to poor depth penetrance of staining reagents (Micheva and Smith, 2007). We have generated high-resolution, three-dimensional reconstructions of nuclei to show how AT can be used to quantify a variety of parameters of nucleus and chromatin structure.
A powerful feature of AT is the ability to acquire multiple rounds of imaging information. This provides a way to survey the molecular composition of specific compartments within the nucleus. We performed up to five rounds of imaging on one set of serial sections, but up to nine rounds have been reported with AT (Micheva and Smith, 2007). Multiplexed immunostaining permits a candidate screening approach in addition to targeted studies. In our case, we did not initiate the experiments with a predetermined hypothesis about the spatial organization of H4K20me3 and how loss of MeCP2 may alter this organization. We performed an initial screen of antibodies for histone modifications, and the results led us to focus on modifications that mark distinct heterochromatin territories. For example, H4K20me3 and H3K27me3 rich regions in WT neurons were adjacent to, but clearly segregated from MeCP2-bound heterochromatin (Movie S3). While we were able to detect the segregated distribution of H4K20me3 in CA1 pyramidal neurons by conventional confocal microscopy (Figure S2), AT imaging provided much greater resolution of these structures. The importance of examining chromatin organization in the brain is underscored by our finding that dissociated hippocampal neurons did not show distinct H4K20me3 territories adjacent to pericentromeric heterochromatin in culture (J. Sinnamon, unpublished data).
By developing FISH conditions for AT, we have added another dimension to the method that considerably broadens its applications. There has recently been great interest in relating spatial organization of genes with chromatin modifications by combining HiC analysis with ChIP (Dixon et al., 2012; Rao et al., 2014). The development of an in situ detection method for chromatin organization fills a need for investigators who work in systems with extreme cellular diversity where anatomy is an integral part of the information desired.
We challenged the AT imaging method by using it on brains from symptomatic RTT female mice at 4-6 months of age. Not only does the mammalian brain contain a staggering numbering of cell types, but the mosaic expression of MeCP2 provided a tightly controlled experimental system for comparing wild type and null cells. Using these methods, AT has provided in vivo evidence supporting a role for MeCP2 in regulating chromatin architecture in neurons. We found that chromatin becomes more densely packed upon loss of MeCP2. Along with the compaction, we see a dramatic redistribution of H4K20me3 into pericentromeric heterochromatin. Consistent with the finding of more condensed chromatin, we observed a slight reduction of active RNAPII in mutant nuclei. The ability to assess large-scale chromatin organization, histone modifications, and transcriptional activity within the same nucleus in its native environment highlights the usefulness of AT as tool for chromatin research.
What could explain the increase in chromatin compaction when MeCP2 is absent in neurons? The redistribution of H4K20me3 into pericentromeric chromatin accompanied the increased DNA compaction in Mecp2-null neurons. Given this result, it is of note that nucleosomal arrays reconstituted with H4K20me3 modified histones form locally condensed, oligomeric structures (Lu et al., 2008). Another potential mechanism is the interplay between MeCP2 and other chromatin architectural proteins, such as histone H1, which may promote higher-order aggregation of chromatin fibers when MeCP2 is absent. Previous studies have shown that MeCP2 competes with H1 for binding to nucleosomal DNA (Ghosh et al., 2010; Nan et al., 1997), and levels of H1 increase in the absence of MeCP2 (Skene et al., 2010). The presence of other chromatin architectural proteins in nuclei may explain why our results appear to be in opposition to in vitro studies that show nucleosome compaction in the presence of purified MeCP2 (Baker et al., 2013; Georgel et al., 2003). Structural studies need to be performed to compare how chromatin fibers with repressive histone modifications are packaged when bound by MeCP2 or H1. Finally, we cannot exclude the possibility that the chromatin changes are indirect and downstream effects of gene expression.
We observed similar alterations in chromatin for both hippocampal CA1 pyramidal neurons and dentate granule cells, but we did not detect changes for cerebellar granule cells. This difference between cell types is consistent with studies showing that loss of MeCP2 results in different gene expression profiles depending on neuronal type (Mellén et al., 2012; Sugino et al., 2014). It has also been shown that neurons differ with respect to levels of MeCP2 expression (Chao et al., 2010). Our general observation is that MeCP2 expression scales with nuclear size, and cerebellar granule cells express the lowest amount of MeCP2 of the cell types examined in this study (data not shown). The lack of changes in H4K20me3 distribution in cerebellar granule cells agrees with results from a previous study that did not find differences in histone modifications in Mecp2-null retinal neurons (Song et al., 2014). These results stress the importance of cell sorting or in situ approaches when studying gene expression in the brain.
Taken together with previous studies, our AT study suggests two levels of MeCP2 regulation of gene expression programs. At one level, MeCP2 prevents higher-order chromatin aggregation either by preventing H4K20me3 modification or by out-competing H1 binding at methylated nucleosomal linker DNA. At another level, MeCP2 functions as a transcriptional regulator, either through recruitment of co-repressors (Lyst et al., 2013; Nan et al., 1998) or via recruitment of co-activators (Chahrour et al., 2008). This second function may be more subject to regulation by signaling pathways, with the phosphorylation state of MeCP2 dynamically regulating association with its co-factors (Ebert et al., 2013; Lyst et al., 2013). This dual functionality would allow MeCP2 to maintain a segregated chromatin fiber that allows for long-lived neurons to adapt their gene expression programs to events including synaptic activity or injury. In the absence of MeCP2, more compact packaging of these sequences may restrict such flexibility, and this would agree with a previous model suggesting that MeCP2 functions as a facilitator of transcriptional activation (Mellén et al., 2012).
The increase in DNA compaction in mutant nuclei raises the possibility that genes required for neuronal function may become inappropriately relocated to a more repressive compartment and thus less subject to regulation by signaling pathways. In future studies, AT imaging can be combined with emerging FISH strategies (Beliveau et al., 2015; Boyle et al., 2011) to localize MeCP2-regulated genes in the RTT brain.
Experimental Procedure
Experimental Animals
Animal procedures were approved by Oregon Health and Science University Institutional Animal Care and Use Committee regulations and licenses. Mice were housed with littermates on a 12:12 h light/dark cycle. Both Mecp2tm3.1Bird (catalog #014610; Mecp2EGFP; (Lyst et al., 2013)) and Mecp2tm1.1Bird (catalog #003890; Mecp2Bnull; (Guy et al., 2001)) mice were obtained from Jackson Laboratory and were maintained on a C57BL/6 background. In order to yield heterozygote females carrying a germline Mecp2 null-mutation and a Mecp2-EGFP allele (Mecp2Bnull/EGFP), Mecp2EGFP/y male mice were crossed to heterozygous Mecp2Bnull/+ mice. Genotyping for Mecp2Bnull and Mecp2EGFP alleles were conducted as described previously (Lioy et al., 2011; Lyst et al., 2013).
Tissue Preparation
The AT procedure was performed using the published protocol with modifications (Micheva and Smith, 2007). Five month old heterozygous RTT female mice were evaluated for symptoms using the observational scale (Guy et al., 2007). Mice were anaesthetized by intraperitoneal injection of Avertin (2,2,2-tribromoethanol) and perfused transcardially with 4% depolymerized paraformaldehyde in PBS. The hippocampus was dissected, and slices of hippocampus were fixed and embedded using microwave irradiation (PELCO BioWave with ColdSpot; Ted Pella, Inc., Redding, CA). All sectioning was performed by the Array Tomography Core at the SOM Beckman Center's Cell Sciences Imaging Facility (Jon Mulholland, Director). Serial sections were cut to a thickness of 200 nm.
Immunofluorescence Procedure
Ribbons of sections were circled with an ImmEdge™ Pen (Vector Laboratories) to allow for small staining volumes. The sections were pretreated with 50 mM glycine in PBS + 0.05% Tween-20, washed with PBS, and blocked with 3% BSA in PBS (PBS-B) for 15 minutes prior to application of primary antibodies. Primary antibodies were incubated with the sections overnight at 4°C in PBS-B. Sections were washed with PBS, and secondary antibodies were incubated with sections at room temperature for 2 hours in PBS-B. The sections were washed with PBS and a water rinse before mounting with SlowFade Gold antifade with DAPI (Invitrogen, Carlsbad, CA).
To remove bound antibodies prior to second round of imaging, the ribbons were exposed to two rounds of stripping buffer. The first stripping buffer was 50 mM Tris pH 6.8, 2% SDS, 50 mM DTT, and the second stripping buffer was Restore PLUS Western Blot Stripping Buffer (Thermo Scientific, Rockford, IL). Both buffers were incubated with sections for 10 minutes at 42°C with very mild agitation. Stripped sections were washed with water and PBS before initiating another round of immunostaining.
Antibodies
Histone antibodies were prescreened using the Antibody Validation Database to limit testing to antibodies that have proven specificity records (Egelhofer et al., 2011). Each antibody was tested independently for AT application, and we continued to use only those that localized exclusively to the nucleus. Commercially sourced primary antibodies used in this study: H4K20me3, clone 6F8-D9 (Abcam, ab78517), H3K9me2 (Abcam, ab1220), H3K9me3 (Diagenode, MAb-146-050), H3K27me3 (Cell Signaling, #9733), GFP (Abcam, ab13970), RNA polymerase II clone 4H8 (Abcam, ab5408), MAP2 (Abcam, ab5392). Monoclonal antibody specific for H4K20me2 was a gift from Hiroshi Kimura. Secondary antibodies used in this study from Biotium (Hayward, CA): CF488A Goat Anti-Mouse IgG2a (20256), CF488A Goat Anti-Chicken IgY, CF647 Goat Anti-Mouse IgG. Secondary antibodies and lectins used from Life Technologies (Carlsbad, CA): Alexa Fluor 488 conjugated Concanavilin A (C11252), Alexa Fluor 555 Goat Anti-Mouse IgG2a (A21137), Alexa Fluor 555 Goat Anti-Rabbit (A21429), Alexa Fluor 647 Goat Anti-Chicken (A21449).
Fluorescent in situ hybridization
Standard protocols for FISH proved difficult to use with AT due to reduced adherence of the plastic sections to the coverslip when using formamide and elevated temperatures. We circumvented this problem by using ethylene carbonate as a solvent for melting DNA strands for hybridization (Matthiesen and Hansen, 2012), allowing for lower hybridization temperature. The following peptide nucleic acid (PNA) probes were used for FISH experiments: Cy3 labeled PNA probe against minor satellite sequence (CENPB-Cy3) (PNA Bio,Thousand Oaks, CA). The FITC labeled major satellite PNA probe was a kind gift from Peter Lansdorp (Falconer et al., 2010). After antibody stripping, the sections were washed with 1xSSC. Sections were incubated with 0.1 mg/ml pepsin in 10 mM HCl for 2 minutes at 37°C followed by washing with TE, pH 8.8. Sections were incubated with 200 μg/ml RNAse A in 1xSSC for 30 minutes at 37°C and washed with 1xSSC. The hybridization buffer was similar to the previously described protocol with some modifications (15% ethylene carbonate, 10% dextran sulfate, 600 mM NaCl, 10 mM sodium citrate, pH 6.2, 1 × Denhardt’s, and 0.1% Tween-20). After a 30 minute prehybridization incubation at 50°C, the sections were incubated with PNA probes for 2 hours at 50°C. The samples were washed with 1x SSC at 50°C, with 0.2x SSC at 50°C, and rinsed with water before mounting in SlowFade Gold.
Fluorescence Microscopy and Image Processing
Sections were imaged on a Zeiss Axio Observer.Z1 inverted microscope with motorized stage. Fluorescence imaging was performed using custom filter sets and a Lumencor Spectra light engine. Images were acquired using a Zeiss 63X/1.4 NA Plan Apochromat objective and either AxioCam MRm or Axiocam 506 mono digital camera. Image stacks were created in Fiji, and the DAPI image stacks were aligned using the MultiStackReg plugin. The alignment transform for the DAPI alignment was applied to image stacks for other fluorescent channels imaged in the first round. A detailed manual describing the AT alignment procedure is provided here: http://nisms.stanford.edu/UsingOurServices/pdf/ArrayTomographyVolumeReconstructio n_v1.4.pdf
Three Dimensional Analysis
Visualization and quantification of reconstructed nuclei was performed using Imaris software (Bitplane , Switzerland). A more detailed description of the analysis is included in Extended Experimental Procedures.
Supplementary Material
Acknowledgments
We thank Ibanri Phanwar-Wood at the Stanford Cell Sciences Imaging Facility for exceptional sectioning, Peter Lansdorp at ERIBA for the major satellite PNA, Hiroshi Kimura at the Tokyo Institute of Technology for providing antibodies, Andrea Ansari for technical assistance, Lori Vaskalis for scientific illustration, and Aurelie Snyder in The Jungers Center Advanced Light Microscopy Core for Imaris instruction (P30 NS061800). MWL thanks Kristina Micheva, Gordon Wang, Forrest Collman, and Brad Busse for advice on the array tomography procedure. Funding was provided generously by grants from Rett Syndrome Research Trust and NIH (HD056503 and NS022518) to GM. MWL received financial support from Fund for Science and MDA255543. GM is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adler DA, Quaderi NA, Brown SD, Chapman VM, Moore J, Tate P, Disteche CM. The X-linked methylated DNA binding protein, Mecp2, is subject to X inactivation in the mouse. Mamm. Genome. 1995;6:491–492. doi: 10.1007/BF00356163. [DOI] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- Baker SA, Chen L, Wilkins AD, Yu P, Lichtarge O, Zoghbi HY. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell. 2013;152:984–996. doi: 10.1016/j.cell.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beliveau BJ, Boettiger AN, Avendaño MS, Jungmann R, McCole RB, Joyce EF, Kim-Kiselak C, Bantignies F, Fonseka CY, Erceg J, et al. Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat Commun. 2015;6:7147. doi: 10.1038/ncomms8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmont AS. Large-scale chromatin organization: the good, the surprising, and the still perplexing. Curr. Opin. Cell Biol. 2014;26:69–78. doi: 10.1016/j.ceb.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickmore WA, van Steensel B. Genome architecture: domain organization of interphase chromosomes. Cell. 2013;152:1270–1284. doi: 10.1016/j.cell.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Boyle S, Rodesch MJ, Halvensleben HA, Jeddeloh JA, Bickmore WA. Fluorescence in situ hybridization with high-complexity repeat-free oligonucleotide probes generated by massively parallel synthesis. Chromosome Res. 2011;19:901–909. doi: 10.1007/s10577-011-9245-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H-T, Chen H, Samaco RC, Xue M, Chahrour M, Yoo J, Neul JL, Gong S, Lu H-C, Heintz N, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer T, Cremer M. Chromosome territories. Cold Spring Harb Perspect Biol. 2010;2:a003889–a003889. doi: 10.1101/cshperspect.a003889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Gabel HW, Robinson ND, Kastan NR, Hu LS, Cohen S, Navarro AJ, Lyst MJ, Ekiert R, Bird AP, et al. Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature. 2013;499:341–345. doi: 10.1038/nature12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelhofer TA, Minoda A, Klugman S, Lee K, Kolasinska-Zwierz P, Alekseyenko AA, Cheung M-S, Day DS, Gadel S, Gorchakov AA, et al. An assessment of histone-modification antibody quality. Nat. Struct. Mol. Biol. 2011;18:91–93. doi: 10.1038/nsmb.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Falconer E, Chavez EA, Henderson A, Poon SSS, McKinney S, Brown L, Huntsman DG, Lansdorp PM. Identification of sister chromatids by DNA template strand sequences. Nature. 2010;463:93–97. doi: 10.1038/nature08644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilov AA, Gushchanskaya ES, Strelkova O, Zhironkina O, Kireev II, Iarovaia OV, Razin SV. Disclosure of a structural milieu for the proximity ligation reveals the elusive nature of an active chromatin hub. Nucleic Acids Res. 2013;41:3563–3575. doi: 10.1093/nar/gkt067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC. Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J. Biol. Chem. 2003;278:32181–32188. doi: 10.1074/jbc.M305308200. [DOI] [PubMed] [Google Scholar]
- Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol. Cell. Biol. 2010;30:4656–4670. doi: 10.1128/MCB.00379-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014;17:215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315:1143–1147. doi: 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- Li Y, Wang H, Muffat J, Cheng AW, Orlando DA, Lovén J, Kwok S-M, Feldman DA, Bateup HS, Gao Q, et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 2013;13:446–458. doi: 10.1016/j.stem.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, Hirrlinger PG, Kirchhoff F, Bissonnette JM, Ballas N, et al. A role for glia in the progression of Rett's syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Simon MD, Chodaparambil JV, Hansen JC, Shokat KM, Luger K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol. 2008;15:1122–1124. doi: 10.1038/nsmb.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet. 2015;16:261–275. doi: 10.1038/nrg3897. [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 2013;16:898–902. doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthiesen SH, Hansen CM. Fast and non-toxic in situ hybridization without blocking of repetitive sequences. PLoS ONE. 2012;7:e40675. doi: 10.1371/journal.pone.0040675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumder A, Roopa T, Basu A, Mahadevan L, Shivashankar GV. Dynamics of chromatin decondensation reveals the structural integrity of a mechanically prestressed nucleus. Biophys. J. 2008;95:3028–3035. doi: 10.1529/biophysj.108.132274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Smith SJ. Array tomography: a new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron. 2007;55:25–36. doi: 10.1016/j.neuron.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Weiler NC, O'Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- Politz JCR, Scalzo D, Groudine M. Something silent this way forms: the functional organization of the repressive nuclear compartment. Annu. Rev. Cell Dev. Biol. 2013;29:241–270. doi: 10.1146/annurev-cellbio-101512-122317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaderi NA, Meehan RR, Tate PH, Cross SH, Bird AP, Chatterjee A, Herman GE, Brown SD. Genetic and physical mapping of a gene encoding a methyl CpG binding protein, Mecp2, to the mouse X chromosome. Genomics. 1994;22:648–651. doi: 10.1006/geno.1994.1442. [DOI] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rego A, Sinclair PB, Tao W, Kireev I, Belmont AS. The facultative heterochromatin of the inactive X chromosome has a distinctive condensed ultrastructure. J. Cell. Sci. 2008;121:1119–1127. doi: 10.1242/jcs.026104. [DOI] [PubMed] [Google Scholar]
- Ricci MA, Manzo C, García-Parajo MF, Lakadamyali M, Cosma MP. Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell. 2015;160:1145–1158. doi: 10.1016/j.cell.2015.01.054. [DOI] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Yu L, Weir JW, Gorovsky MA. Linker histones are not essential and affect chromatin condensation in vivo. Cell. 1995;82:47–56. doi: 10.1016/0092-8674(95)90051-9. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Illingworth RS, Webb S, Kerr ARW, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histoneoctamer levels and globally alters the chromatin state. Mol. Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets D, Markaki Y, Schmid VJ, Kraus F, Tattermusch A, Cerase A, Sterr M, Fiedler S, Demmerle J, Popken J, et al. Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenetics Chromatin. 2014;7:8. doi: 10.1186/1756-8935-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Feodorova Y, Guy J, Peichl L, Jost KL, Kimura H, Cardoso MC, Bird A, Leonhardt H, Joffe B, et al. DNA methylation reader MECP2: cell type- and differentiation stage-specific protein distribution. Epigenetics Chromatin. 2014;7:17. doi: 10.1186/1756-8935-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuss DP, Cheema M, Ng MK, Martinez de Paz A, Williamson B, Missiaen K, Cosman JD, McPhee D, Esteller M, Hendzel M, et al. Impaired in vivo binding of MeCP2 to chromatin in the absence of its DNA methyl-binding domain. Nucleic Acids Res. 2013;41:4888–4900. doi: 10.1093/nar/gkt213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugino K, Hempel CM, Okaty BW, Arnson HA, Kato S, Dani VS, Nelson SB. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J. Neurosci. 2014;34:12877–12883. doi: 10.1523/JNEUROSCI.2674-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson I, Berlivet S, Eskeland R, Boyle S, Illingworth RS, Paquette D, Dostie J, Bickmore WA. Spatial genome organization: contrasting views from chromosome conformation capture and fluorescence in situ hybridization. Genes Dev. 2014;28:2778–2791. doi: 10.1101/gad.251694.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WD, Tanious FA, Barton HJ, Jones RL, Fox K, Wydra RL, Strekowski L. DNA sequence dependent binding modes of 4',6-diamidino-2-phenylindole (DAPI) Biochemistry. 1990;29:8452–8461. doi: 10.1021/bi00488a036. [DOI] [PubMed] [Google Scholar]
- Yazdani M, Deogracias R, Guy J, Poot RA, Bird A, Barde Y-A. Disease modeling using embryonic stem cells: MeCP2 regulates nuclear size and RNA synthesis in neurons. Stem Cells. 2012;30:2128–2139. doi: 10.1002/stem.1180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.