

Abstract
Neuroblastoma (NB) is one of the most frequently occurring extracranial solid tumors of childhood (Maris et al., 2007 [1]; Brodeur, 2003 [2]). Probability of cure varies according to patient's age, extent of disease and tumor biology (Maris et al., 2007 [1]; Brodeur, 2003 [2]; Cohn et al., 2009 [3]). However, the etiology of this developmental tumor is unknown. Recent evidence has shown that pediatric solid tumors, including NB, harbor a paucity of recurrent genetic mutations, with a significant proportion of recurrent events converging on epigenetic mechanisms (Cheung et al., 2012 [4]; Molenaar et al., 2012 [5]; Pugh et al., 2013 [6]; Sausen et al., 2013 [7].
We have analyzed the DNA methylome of neuroblastoma using high-density microarrays (Infinium Human Methylation 450k BeadChip) to define the epigenetic landscape of this pediatric tumor and its potential clinicopathological impact. Here, we provide the detail of methods and quality control parameters of the microarray data used for the study. Methylation data has been deposited at NCBI Gene Expression Omnibus data repository, accession number GSE54719; superseries record GSE54721.
Keywords: Neuroblastoma, Embryonal tumor, High-density microarray, DNA methylation, HM450K
| Specifications | |
|---|---|
| Organism/cell line/tissue | Homo sapiens |
| Sex | Neuroblastoma samples (n = 35) were composed by 17 males and 18 females. All reference samples (n = 4) were males. |
| Sequencer or array type | Infinium Human Methylation 450k BeadChip (Illumina Inc., San Diego, CA). GPL13534 |
| Data format | Raw data is available as TXT non-normalized methylated and non-normalized unmethylated tables. |
| Experimental factors | Tumor and reference samples. |
| Experimental features | Normal fetal brain (n = 2) and adrenal gland (n = 2) tissues as well as ganglioneuroma (benign Schwannian stroma dominant neuroblastic tumor) samples (n = 2) were used as reference samples. Tumor cell content was evaluated by a pathologist. |
| Consent | Patients, parents or guardians signed an informed consent before sample collection. |
| Sample source location | Hospital Sant Joan de Déu — Barcelona, Passeig Sant Joan de Déu, 2, 08950 Esplugues de Llobregat, Barcelona, Spain. Latitude: 41.383906 | longitude: 2.101771 |
1. Direct link to deposited data
Methylation microarray data has been deposited at NCBI Gene Expression Omnibus data repository (GEO; http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE54719 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54719); on superseries record GSE54721 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE54721).
2. Experimental design
To define the DNA methylation landscape of neuroblastoma and its potential clinicopathological impact. Microarray DNA methylation data (Infinium Human Methylation 450k BeadChip) were analyzed and associated with functional/regulatory genome annotation data, transcriptional profiles and clinico-biological parameters.
3. Material and methods
3.1. Patients and samples
Thirty-five primary neuroblastoma tumors (including 6 stage 1, 9 stage 3, 6 stage 4s and 14 stage 4) obtained at the time of diagnosis from patients diagnosed and treated at Hospital Sant Joan de Déu (Barcelona, Spain) were included in the study. Normal fetal brain (n = 2) and adrenal gland (n = 2) tissues as well as ganglioneuroma samples (n = 2) were used as reference samples. Neuroblastoma risk assessment was defined by the International Neuroblastoma Staging System (INSS) [8]. Patients' clinical and biological characteristics are displayed in Table 1. Tumors were evaluated by a pathologist, only samples with more than 70% viable tumor cell content were included in the study. This study was approved by the Institutional Review Boards. Informed consent was obtained before collection of samples.
Table 1.
Patients' clinical and biological characteristics.
| Characteristics | Methylation array samples (n = 35) | ||
|---|---|---|---|
| Age, months | Median | 17.90 | |
| Range | 0.10–225.28 | ||
| INSS, n (%) | Stages 1–3 | 15 | 44.12% |
| Stage 4 | 14 | 41.18% | |
| Stage 4s | 6 | 17.65% | |
| MYCN status, n (%) | MYCN Amp | 7 | 20.59% |
| MYCN non-Amp | 28 | 82.35% | |
Genomic DNA was isolated from samples using Wizard DNA Purification Kit (Promega Biotech Ibérica, Spain), following manufactures' protocols.
3.2. DNA methylation microarrays
Genomic DNA bisulfite conversion and hybridization to Infinium Human Methylation 450k BeadChip (Illumina Inc., San Diego, CA) was performed at the Human Genotyping Unit of the Spanish National Cancer Center (CEGEN-CNIO, Madrid, Spain), as described previously [9].
3.3. Quality control
Microarray data was analyzed by minfi [10] and shinyMethyl [11] packages available through Bioconductor [12].
Single cytosine methylation values (β-values) in each sample were calculated as the ratio of the methylated signal intensity to the sum of methylated and unmethylated signals. Methylation values ranged from 0, fully unmethylated, to 1, fully methylated cytosine. Raw methylation β-value plots were performed for all samples to identify possible deviant samples (Fig. 1 & B).
Fig. 1.

A: Raw β-value density plot. B: Raw β-value bean density plot. C: Scatter plot of log median intensity of methylated (Meth) and unmethylated (Unmeth) channels.
Sample quality was assessed using the log median intensity in both the methylated and unmethylated channels (Fig. 1C).
Principal component analysis (PCA) was performed on the 20,000 most variable autosomal probes in order to analyze associations between sample type (adrenal gland, ganglioneuroma, fetal brain or neuroblastoma) and methylation levels (Fig. 2).
Fig. 2.
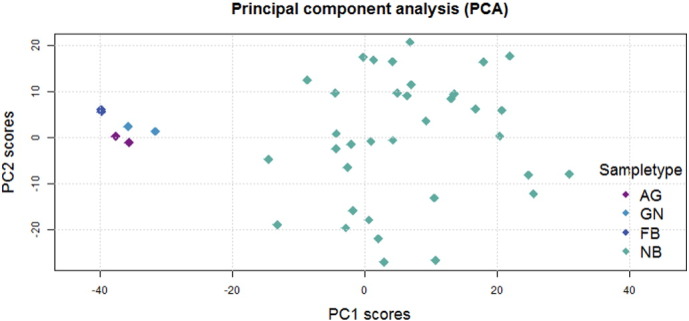
Principal component analysis. AG: adrenal gland. GN: ganglioneuroma. FB: fetal brain. NB: neuroblastoma.
3.4. Data normalization
Microarray methylation data was normalized by missMethyl package [13] using the SWAN (subset-quantile within array normalization), a within-array normalization method for Illumina Human Methylation 450k BeadChips (Fig. 3).
Fig. 3.

Density distributions of beta values before and after using SWAN.
3.5. Filtering of microarray data
An optimized pipeline with several filters developed at the IDIBAPS was used to exclude technical biases, [14], [15]. Cytosines with poor detection P-values (P > 0.01) and those with sex-specific DNA methylation (n = 6926) were removed from the initial dataset of 485,512 sites (excluding probes detecting SNPs). The remaining sites (n = 478,169) were used for downstream analyses (Fig. 4).
Fig. 4.
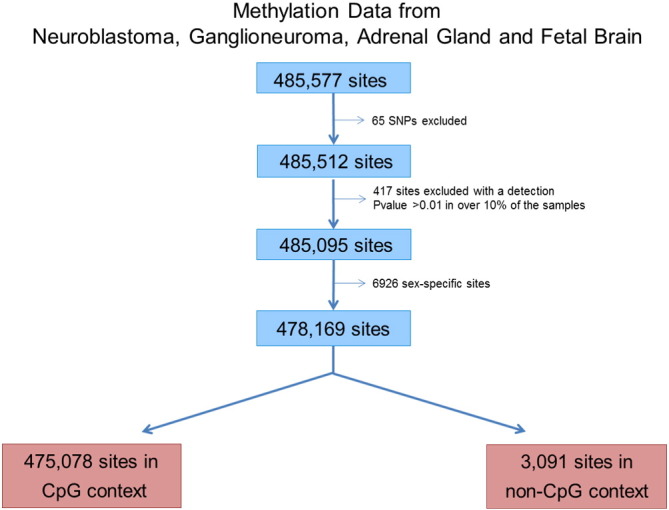
Data processing work-flow.
3.6. Bisulfite pyrosequencing
DNA methylation data were validated by bisulfite pyrosequencing as previously described [15], [16] (Fig. 5).
Fig. 5.

Technical DNA methylation validation.
(A) Scatter plot of DNA methylation levels obtained by Infinium Human Methylation 450k BeadChip (Illumina Inc., San Diego, CA) (Y-axis) and bisulfite pyrosequencing (X-axis) (n = 11). A highly significant correlation (P < 0.001, R2 = 0.978 and R2 = 0.968, respectably) between microarray and pyrosequencing data was observed. Example of pyrograms with methylated (B) and unmethylated (C) cytosines.
4. Data summary
The analysis of the DNA methylome of neuroblastoma patients revealed that: i) most DNA methylation changes take place outside promoters, ii) not only CpGs but also non-CpG sites are targeted, iii) methylation changes at these sites are associated with clinicopathological features of neuroblastoma, and iv) the identified epigenetic patterns provide new insights into the pathogenesis and clinical behavior of this pediatric tumor. The analysis of this data has been reported in our recently published study [15].
Financial disclosure
This work was supported by the NEN association (association of families and friends of patients with neuroblastoma) [to S Gómez], Hospital Sant Joan de Déu, Barcelona, Spain [BR201102 to G. Mayol] and Spanish Government [PI11/02661 and PI14/00038 Instituto de Salud Carlos III grant to C Lavarino and Ramon y Cajal Grant to JI Martín-Subero].
Acknowledgments
The authors acknowledge G. Garcia-Castellví for the fund raising and the “Biobanc de l'Hospital Infantil Sant Joan de Déu per a la Investigació” integrated in the National Network Biobanks of ISCIII for tissue samples. The authors also thank J. Ríos for statistical advice and S. Sánchez Molina for precious technical support.
References
- 1.Maris J.M., Hogarty M.D., Bagatell R., Cohn S.L. Neuroblastoma. Lancet. 2007;369(9579):2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Brodeur G.M. Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer. 2003;3(3):203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 3.Cohn S.L., Pearson A.D., London W.B. The International Neuroblastoma Risk Group (INRG) classification system: an INRG task force report. J. Clin. Oncol. 2009;27(2):289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheung N.K., Zhang J., Lu C. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307(10):1062–1071. doi: 10.1001/jama.2012.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molenaar J.J., Koster J., Zwijnenburg D.A. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature. 2012;483(7391):589–593. doi: 10.1038/nature10910. [DOI] [PubMed] [Google Scholar]
- 6.Pugh T.J., Morozova O., Attiyeh E.F. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013;45(3):279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sausen M., Leary R.J., Jones S. Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat. Genet. 2013;45(1):12–17. doi: 10.1038/ng.2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brodeur G.M., Pritchard J., Berthold F. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1993;11(8):1466–1477. doi: 10.1200/JCO.1993.11.8.1466. [DOI] [PubMed] [Google Scholar]
- 9.Bibikova M., Barnes B., Tsan C. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98(4):288–295. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Aryee M.J., Jaffe A.E., Corrada-Bravo H. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fortin J.P., Fertig E., Hansen K. shinyMethyl: interactive quality control of Illumina 450 k DNA methylation arrays in R. F1000Research. 2014;3:175. doi: 10.12688/f1000research.4680.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huber W., Carey V.J., Gentleman R. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12(2):115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maksimovic J., Gordon L., Oshlack A. SWAN: subset-quantile within array normalization for Illumina Infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13(6):R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kulis M., Heath S., Bibikova M. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet. 2012;44(11):1236–1242. doi: 10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 15.Gomez S., Castellano G., Mayol G. DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics. 2015:1–17. doi: 10.2217/epi.15.49. [DOI] [PubMed] [Google Scholar]
- 16.Mayol G., Martin-Subero J.I., Rios J. DNA hypomethylation affects cancer-related biological functions and genes relevant in neuroblastoma pathogenesis. PLoS One. 2012;7(11):e48401. doi: 10.1371/journal.pone.0048401. [DOI] [PMC free article] [PubMed] [Google Scholar]