

Abstract
Advent of Next Generation Sequencing has led to possibilities of de novo transcriptome assembly of organisms without availability of complete genome sequence. Among various sequencing platforms available, Illumina is the most widely used platform based on data quality, quantity and cost. Various de novo transcriptome assemblers are also available today for construction of de novo transcriptome.
In this study, we aimed at obtaining an ameliorated de novo transcriptome assembly with sequence reads obtained from Illumina platform and assembled using Trinity Assembler. We found that, primary transcriptome assembly obtained as a result of Trinity can be ameliorated on the basis of transcript length, coverage, and depth and protein homology. Our approach to ameliorate is reproducible and could enhance the sensitivity and specificity of the assembled transcriptome which could be critical for validation of the assembled transcripts and for planning various downstream biological assays.
Keywords: De novo transcriptome assembly, Trinity, Illumina
| Specifications | |
|---|---|
| Bioproject accession | PRJNA244590 |
| Organism | Helicoverpa armigera Hübner, 1809 |
| Common name | Cotton bollworm |
| Sex | – |
| Project data type | Transcriptome or gene expression |
| Platform | Illumina HiSeq 2000 |
| Data format | SRA |
| Library details | Strategy: RNA-seq |
| Source: Transcriptomic | |
| Selection: cDNA | |
| Layout: Paired | |
| Experimental factors | The data consist of 10 RNA-seq cDNA libraries. Helicoverpa armigera (Hübner) strains were cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis. Sensory organs i.e. taste organs in adults (male and female) and in larvae also. |
| Experimental features | Transcriptome survey for identifying genes relevant to chemoreception |
| Consent | – |
| Sample source location | CSIRO Ecosystem Sciences, Black Mountain, Canberra ACT 2601, Australia |
| Link | ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP041/SRP041166 |
1. Data files
| Accession | Title | Source name | Organism | Treatment |
|---|---|---|---|---|
| SRR1238087 | Mixed-feeding 5th instar-antennae | SRX516834 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1238089 | Mixed-feeding 5th instar-mouthpart | SRX516871 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1238090 | Male-adult-tarsus | SRX516872 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1239328 | Female-adult-tarsus | SRX518085 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1239329 | Mixed feeding 5th instar fat body | SRX518086 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1239330 | Female adult abdomen | SRX518087 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1239331 | Female adult head wei | SRX518088 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1239333 | Female adult head wei DSN | SRX518089 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1239334 | Male adult head wei | SRX518090 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
| SRR1239335 | Male adult head wei DSN | SRX518091 | Helicoverpa armigera | Cultured on artificial diet containing the Cry1Ac protoxin of Bacillus thuringiensis |
2. Material and methods
Deep sequencing based whole transcriptome data for reanalysis was obtained from NCBI SRA (Sequence Read Archive) with the link ftp://ftp-trace.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByStudy/sra/SRP/SRP041/SRP041166 [1]. Raw data was obtained in SRA format and further converted to FASTq format using SRA Tool kit (version 2.4) (http://www.ncbi.nlm.nih.gov/Traces/sra/) [2].
2.1. Raw data summarization
Paired End FASTq files were subjected to standard quality control using NGSQC Tool Kit [3] with the following command:
perl IlluQC_PRLL.pl -c 10 -pe SRR1238087_R1.fastq SRR1238087_R2.fastq Adapter.txt A –o “Output Folder Name”.
For each paired end data 10 core threads of processing with 2.4 GHz speed with default memory allocation were provided.
2.2. Transcriptome assembly
All the 10 HQ filtered paired end libraries were subjected to pooled de-novo transcriptome assembly as followed in the original manuscript [1]. Evaluation of multiple assemblers for de novo transcriptome assembly was already done and results are available [4], [5], [6]. For this study we chose to go with De brijn graph based Trinity Assembler [7] based on the criteria of a) default K-mer, b) less memory foot print, c) optimized for Illumina paired end data, d) reproducibility and e) configurable for all computing capacities. The following command was used to initiate the pooled assembly using Trinity.
Trinity\
-
–
seqType fq\
-
–
JM 600G\
-
–
left/data/Projects/RNASeq/Pooled_Reads/R1.fastq\
-
–
right/data/Projects/RNASeq/Pooled_Reads/R2.fastq\
-
–
CPU 50\
-
–
min_contig_length 200\
-
–
output/data/Projects/RNASeq/Pooled_Reads/Helicoverpa\
-
–
min_kmer_cov 2\
-
–
bflyHeapSpaceMax 50G\
-
–
bflyHeapSpaceInit 10G
For the pooled assembly 50 core threads of processing with 2.4 GHz speed and a maximum Heap Space of 50 GB were allotted.
2.3. Assembly validation
Since de novo transcriptome assemblers are capable of producing in fragmented/mis-assembly, validation of the assembled transcriptome is done by mapping back the HQ filtered reads to the ESTs. Bowtie [8] was used to map the HQ filtered reads from each library to the assembled transcriptome using the following parameters.
perl TRINITY_HOME/util/align_and_estimate_abundance.pl \
-
–
transcripts TrinityMergedAssembly.fasta \
-
–
seqType fq \
-
–
left SRR1238087_R1.fastq_filtered \
-
–
right SRR1238087_R2.fastq_filtered \
-
–
est_method RSEM \
-
–
aln_method bowtie \
-
–
thread_count 10
2.4. Transcript quantitation, coverage and depth analysis
Assembly validated .bam (Binary Sequence Alignment/Map) file was processed using bedtools [9] and samtools [10] for quantitation (read count estimation) for each transcript in a library and also to calculate the total coverage and average depth of the transcriptome in each library.
For quantitation the following parameters/command was used.
samtools idxstats SRR1238087.bowtie.csorted.bam > SRR1238087.bowtie.csorted.bam.idxstats
For calculating each transcript coverage and its average depth corresponding bedGraph file was generated using the following the parameters/command.
genomeCoverageBed -ibam SRR1238087.bowtie.csorted.bam -bga > SRR1238087.bedgraph
From the resultant bedGraph file, the following formulae were used to calculate coverage and average depth.
2.5. Analysis of transcriptome integrity
While doing merged assembly multiple transcripts might arise due to errors in assembly. In our approach, we performed transcriptome integrity analysis based on read count, coverage and average depth on an intra- and inter-library specific manner. Correlation coefficient graphs were plotted to understand the variations between the libraries that could be indicative of whole transcriptome integrity. Isotig analysis of validated transcriptome based on length was also done to estimate the transcriptome integrity.
2.6. Transcript annotation
Homology based annotation for each transcript was done against NCBI nrdb (Dec 2014) protein database using Blastx. Annotation and statistical ranking of the results were done using Blast2GO [11]. Also domain level annotation was performed using the Online InterProScan tool [7] RunIprScan-1.1.0 (http://michaelrthon.com/runiprscan/).
Blastx and Blast2GO parameters used are
e-Value <= 10 − e4
Similarity ≥ 35%
Annotation cutoff ≥ 55
GO weight cutoff ≥ 5.
2.7. Normalization and expression profiling
Primary advantage of using NGS based transcriptome profiling is to identify sample/condition specific expressed transcripts which is not easy with earlier hybridization methods. Transcripts with a read count ≥ 10 in any one of the library was considered to be as likely expressed. A sub .bam file was created from the master .bam file based on the above criteria. The sub .bam file was subjected to normalization and expression profiling using RSEM software [12]. The following parameters/commands were used to normalize each library.
rsem-prepare-reference\
--no-polyA\
--no-bowtie\
ValidTranscripts.fasta\
Harmigera_RSEM
/data/Program/rsem-1.2.12/rsem-calculate-expression\
--paired-end\
-p 8\
--bam\
SRR1238087.bowtie.csorted.bam\
Harmigera_RSEM\
SRR1238087
RSEM software provides an output for each library with expected normalized read count, TPM (tags per million) and FPKM (fragments per kilobase per million). Log to the base 2 of FPKM was considered as absolute expression or Delta CT equivalent value.
3. Results
3.1. Raw data summarization and transcriptome assembly
Quality control of individual libraries using NGSQC tool kit revealed an average of 95.85% HQ reads based on Q20 score. Total number of HQ reads on an average per library was ~ 47 million, indicating significant amount of reads to proceed with transcriptome assembly (Table 1, Fig. 1). HQ paired reads from all the libraries were merged and provided as input to Trinity Assembler V2014. Transcriptome assembly performed with parameters as outlined resulted in assembly of 74,966 putative transcripts with an overall size of 78.61 Mb. N50 of the assembled transcriptome was 2.12 kb. Transcript length distribution analysis revealed 51.79% of the transcript length less than 500 bases. This is a typical observation of most of the de novo transcriptome assemblies using Illumina short read deep sequencing approach [13], [14], [15] (Table 2, Fig. 2).
Table 1.
Distribution of reads based on quality score from each library indicating percentage of high quality and low quality reads.
| Sample ID | HQ reads | Low quality reads |
|---|---|---|
| SRR1238087 | 47,523,826 (99.69%) | 148,556 (0.31%) |
| SRR1238089 | 49,202,920 (99.68%) | 160,166 (0.32%) |
| SRR1238090 | 49,791,084 (99.68%) | 160,610 (0.32%) |
| SRR1239328 | 46,376,028 (99.76%) | 113,622 (0.24%) |
| SRR1239329 | 48,642,138 (93.75%) | 3,244,266 (6.25%) |
| SRR1239330 | 31,475,588 (91.96%) | 2,753,084 (8.04%) |
| SRR1239331 | 66,399,420 (94.23%) | 4,065,716 (5.77%) |
| SRR1239333 | 32,605,304 (93.83%) | 2,142,264 (6.17%) |
| SRR1239334 | 56,840,276 (91.69%) | 5,151,868 (8.31%) |
| SRR1239335 | 42,943,308 (94.27%) | 2,612,200 (5.73%) |
Fig. 1.

Histogram representation of high quality and low quality reads from each individual library.
Table 2.
Statistics of merged de novo transcriptome sequence assembly using Illumina paired end reads using Trinity Assembler 2014 release.
| Number of transcripts | 74,966 |
| Transcriptome size (Mb) | 78.61 |
| Mean (bp) | 1049 |
| Stdev (bp) | 1319 |
| Median (bp) | 472 |
| Smallest (bp) | 201 |
| Largest (bp) | 29,186 |
| N50 length (bp) | 2123 |
Fig. 2.

Histogram representation of abundance of putative assembled transcripts based on their length range.
3.2. Validation of putative assembled transcriptome and quantitation
A merged de novo assembly is expected to provide representative transcriptome of transcripts from individual libraries. This would be evident from mapping the reads from each library to the putative transcriptome to validate the build. In our approach too, we mapped/aligned the reads back to the putative transcripts from the assembly to understand if there is any library specific bias (enrichment or depletion). Copy number of each transcript from each library was also measured to understand any copy number specific bias that could arise due to upstream sample preparation artifacts. We observed on an average of 72% of HQ reads from each library mapped to the putative transcriptome (Table 3, Fig. 3).
Table 3.
Alignment statistics indicative of reads aligned to the assembled transcriptome as a result of standard parameters used in Trinity Assembler.
| Sample ID | Aligned reads | Unaligned reads |
|---|---|---|
| SRR1238087 | 34,972,638 (73.36%) | 12,551,188 (26.33%) |
| SRR1238089 | 36,915,066 (74.78%) | 12,448,020 (25.22%) |
| SRR1238090 | 37,346,712 (74.77%) | 12,604,982 (25.23%) |
| SRR1239328 | 33,406,742 (71.86%) | 13,082,908 (28.14%) |
| SRR1239329 | 32,489,850 (62.62%) | 19,396,554 (37.38%) |
| SRR1239330 | 23,811,380 (69.57%) | 10,417,292 (30.43%) |
| SRR1239331 | 53,009,538 (75.23%) | 17,455,598 (24.77%) |
| SRR1239333 | 25,688,662 (73.93%) | 9,058,906 (26.07%) |
| SRR1239334 | 45,313,564 (73.1%) | 16,678,580 (26.9%) |
| SRR1239335 | 34,384,314 (75.48%) | 11,171,194 (24.52%) |
Fig. 3.

HQ read alignment to putative transcriptome.
3.3. Analysis of transcriptome integrity and refinement
HQ aligned reads were subjected to integrity analysis with reference to coverage and average depth ratio in each library. Coverage was calculated as percentage of the transcript length supported by aligned reads and average depth was calculated as number of bases supporting each nucleotide in a transcript. We found that a total of 37,930 transcripts (50.59%) were covered at ≥ 70% with an average depth of 5 × in one or more of the libraries (Table 4). Further, we subjected the 37,930 transcripts to length and annotation analysis to establish the integrity and refinement. We observed the minimal transcript length of 201 bases and maximum to be 29.18 kb with an average length of 1.19 kb. A total of 15,197 out of 37,930 transcripts got assigned to a protein based on homology at protein level (Fig. 4). Comparative analysis of assembled and ameliorated transcriptome on the basis of length, coverage, depth and annotation showed clear improvement in the assembly process (Fig. 5). Isotig analysis of assembled transcriptome and ameliorated transcriptome also showed significant improvement in the quality of the transcriptome build (Fig. 6).
Table 4.
Matrix representation of depth vs coverage of individual libraries with highlight on transcripts with ≥ 70% coverage and ≥ 5 × depth.
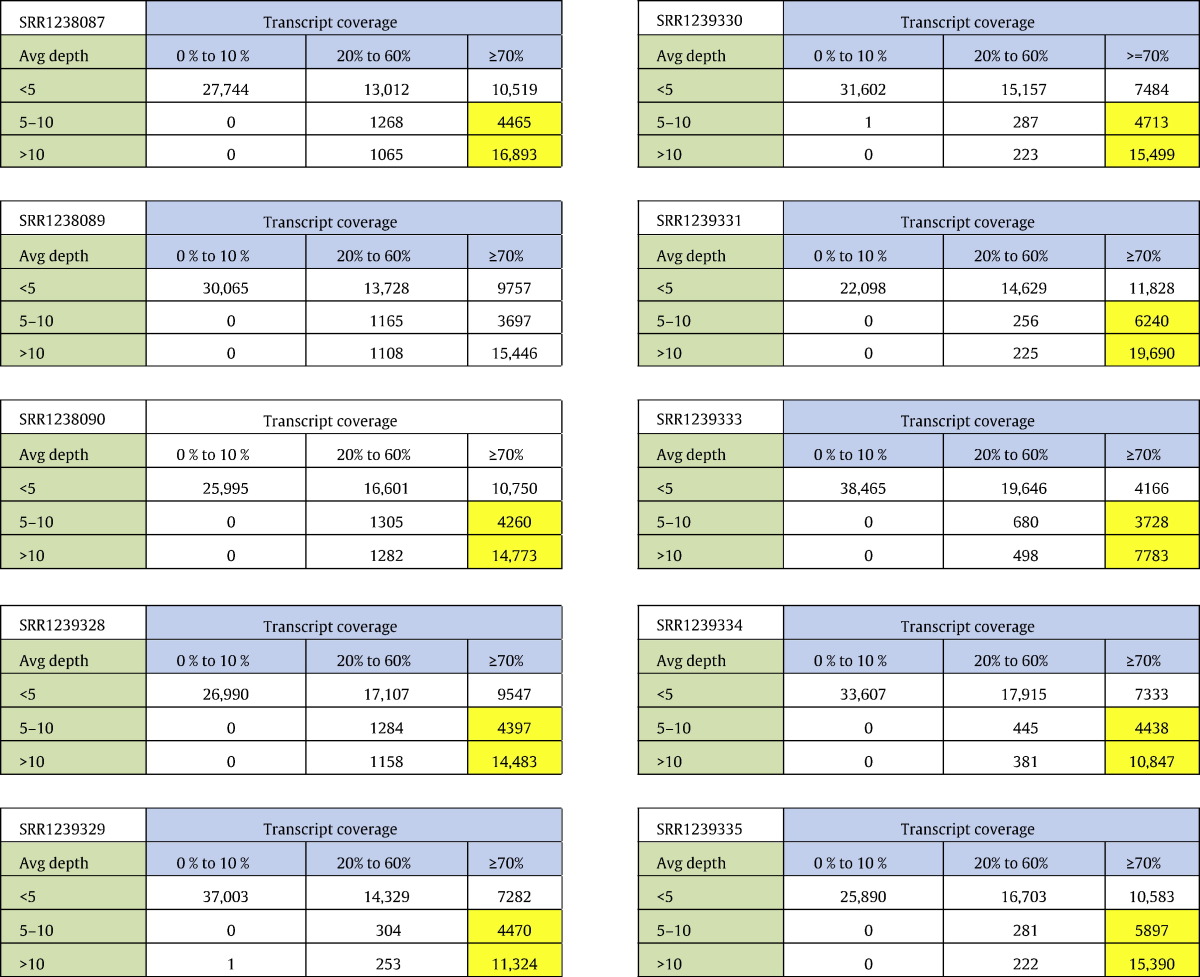
Fig. 4.

a — Distribution of assembled transcripts with reference to length and their annotation with NRDB representing noise of smaller transcripts.
b — Distribution of ameliorated transcripts with reference to length and their annotation with NRDB shows nice correlation by reducing noise of smaller length transcripts.
Fig. 5.

Species distribution of assembled transcriptome and ameliorated transcriptome [Top 10].
Fig. 6.

Isotig analysis with respect to assembled transcriptome and ameliorated transcriptome.
3.4. Expression profiling, Gene Ontology and Pathway enrichment of ameliorated transcriptome
Normalization of assembled transcriptome and ameliorated transcriptome was done as per the discussed method. Box plot representation of both the transcriptome showed significant difference in the ameliorated global expression profile in comparison to assembled transcriptome (Fig. 7). Ameliorated transcriptome was subjected to Gene Ontology and Pathway analysis as discussed in the methods to identify key enriched gene ontology categories and pathways. Top 10 GO categories were found to represent essential biological processes (Fig. 8). Complete assembled and annotated transcriptome along with transcript length, read count, depth and coverage is provided along with the transcriptome sequence (Supplementary File 1, Supplementary File 2).
Fig. 7.
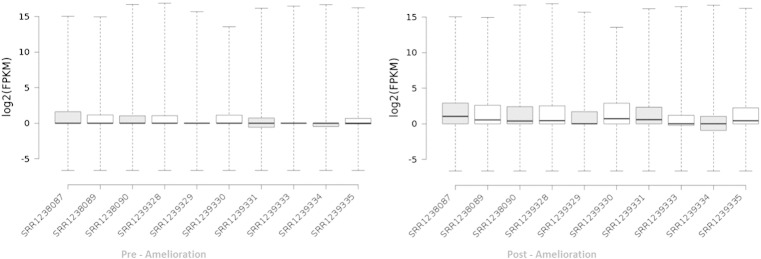
Global expression profiling of ameliorated transcriptome (post-amelioration) in comparison to complete expression profile (pre-amelioration) represented in Box Plot.
Fig. 8.

Top 10 GO categories were found to represent essential biological processes.
4. Discussion
Next Generation Sequencing based gene expression studies enable faster and cheaper data generation. Illumina is the widely used sequencing platform for whole transcriptome studies. Since the advent of novel sequencing methodologies, de novo transcriptome sequencing is the method of choice for conducting spatial, temporal and condition specific gene expression profiling in both non-model and model organisms.
With hundreds of de novo transcriptomes published with majority using Illumina sequencing platform, the integrity and resolution of the assembled transcriptome remain un-addressed. The choice of the platform, assembler and sample size and study design largely determines the sensitivity and specificity of the assembled transcriptome. The most important step in de novo RNA-seq analysis is the assembly of the sequencer generated short reads into full-length transcripts. Among the well-known, publicly available software's for de novo transcriptome sequence assembly are: Trinity, Velvet-Oases, SOAPdenovo-trans assembler and the Trans-ABySS. Trans-ABySS, SOAPdenovo-trans and Velvet-Oases are extensions of the pre-developed genome assembler programs. The Illumina based Trinity Assembler is the most widely used tool that was developed primarily for de novo RNA-seq data assembly.
In case of a multiple sample study involving analysis of the differentially expressed genes it is recommended to combine all the reads from independent samples and obtain a merged assembly [16]. Thus obtained merged assembly has a representation of all the transcripts in the given set of samples allowing for a true differential expression analysis. Although Trinity is among the most efficient tools for reconstructing transcripts in the absence of a reference sequence yet number of limitations has been encountered with this assembler. The first among them is the lack of reproducibility. Second are the high rate of false positives in the assembly ranging from 20 to 30% and the presence of large number of fusion transcripts as well as partial transcripts. Third, the number of obtained transcripts is too high compared to the expected number in the particular organism in the study. As a result the number of annotated transcripts is observed to be very hugely different in every experiment in the range of 40–90%. The high degree of variability in the results is evidenced by a low validation score.
In this study we attempted to benchmark various parameters when taken into consideration, could result in enhancing the sensitivity and specificity of the assembled transcriptome, considering Illumina as the sequencing platform of choice and Trinity as the assembler of choice.
The following are the supplementary data related to this article.
Spreadsheet with list of 74,966 transcripts with information about transcript length, read count, coverage, average depth and annotation.
FASTA file of assembled transcriptome (74,966 transcripts).
References
- 1.Liu N.Y., Xu W., Papanicolaou A., Dong S.L., Anderson A. Identification and characterization of three chemosensory receptor families in the cotton bollworm Helicoverpa armigera. BMC Genomics. 2014;15(1):597. doi: 10.1186/1471-2164-15-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leinonen R., Sugawara H., Shumway M. The sequence read archive. Nucleic Acids Res. 2010;gkq1019 doi: 10.1093/nar/gkq1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel R.K., Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2) doi: 10.1371/journal.pone.0030619. e30619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li B., Fillmore N., Bai Y., Collins M., Thomson J.A., Stewart R., Dewey C.N. Evaluation of de novo transcriptome assemblies from RNA-Seq data. Genome Biol. 2014;15(12):553. doi: 10.1186/s13059-014-0553-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chopra R., Burow G., Farmer A., Mudge J., Simpson C.E., Burow M.D. Comparisons of de novo transcriptome assemblers in diploid and polyploid species using peanut (Arachis spp.) RNA-Seq data. PLoS One. 2014;9(12):e115055. doi: 10.1371/journal.pone.0115055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Surget-Groba Y., Montoya-Burgos J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010;20(10):1432–1440. doi: 10.1101/gr.103846.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henschel R., Lieber M., Wu L.S., Nista P.M., Haas B.J., LeDuc R.D. Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging From the Extreme to the Campus and Beyond. ACM; 2012, July. Trinity RNA-Seq assembler performance optimization; p. 45. [Google Scholar]
- 8.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., ..., Durbin R. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conesa A., Götz S., García-Gómez J.M., Terol J., Talón M., Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 12.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(1):323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sreedhar R.V., Kumari P., Rupwate S.D., Rajasekharan R., Srinivasan M. Exploring triacylglycerol biosynthetic pathway in developing seeds of Chia (Salvia hispanica L.): a transcriptomic approach. PLoS One. 2015;10(4) doi: 10.1371/journal.pone.0123580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar M., Gantasala N.P., Roychowdhury T., Thakur P.K., Banakar P., Shukla R.N., ..., Rao U. 2014. De novo transcriptome sequencing and analysis of the cereal cyst nematode, Heterodera avenae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhardwaj A.R., Joshi G., Kukreja B., Malik V., Arora P., Pandey R., Shukla R.N., Bankar K.G., Katiyar-Agarwal S., Goel S., Jagannath A.A., Kumar A.A., Agarwal M. Global insights into high temperature and drought stress regulated genes by RNA-Seq in economically important oilseed crop Brassica juncea. BMC Plant Biol. Jan 21 2015;15:9. doi: 10.1186/s12870-014-0405-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Q.Y., Wang Y., Kong Y.M., Luo D., Li X., Hao P. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics. 2011;12(Suppl. 14):S2. doi: 10.1186/1471-2105-12-S14-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spreadsheet with list of 74,966 transcripts with information about transcript length, read count, coverage, average depth and annotation.
FASTA file of assembled transcriptome (74,966 transcripts).