

Summary
Tumour necrosis factor (TNF)‐α, a proinflammatory cytokine central to many autoimmune diseases, has been implicated in the depigmentation process in vitiligo. We review its role in vitiligo by exploring its pro‐ and anti‐inflammatory properties and examine the effects of blocking its actions with TNF‐α antagonist therapeutics in reports available in the literature. We found that TNF‐α inhibition halts disease progression in patients with progressive vitiligo but that, paradoxically, treatment can be associated with de novo vitiligo development in some patients when used for other autoimmune conditions, particularly when using adalimumab and infliximab. These studies reinforce the importance of stating appropriate outcomes measures, as most pilot trials propose to measure repigmentation, whereas halting depigmentation is commonly overlooked as a measure of success. We conclude that TNF‐α inhibition has proven useful for patients with progressive vitiligo, where TNF‐α inhibition is able to quash cytotoxic T‐cell‐mediated melanocyte destruction. However, a lingering concern for initiating de novo disease will likely prevent more widespread application of TNF inhibitors to treat vitiligo.
Short abstract
What's already known about this topic?
Increased levels of tumour necrosis factor (TNF)‐α are present in active vitiligo lesions.
Upon TNF‐α inhibition in vitiligo, anti‐TNF‐α agents have been labelled as ineffective, as repigmentation was not achieved.
What does this study add?
In patients with progressive disease, TNF‐α inhibition may provide a means to halt depigmentation in vitiligo.
A dual role for tumour necrosis factor‐α in vitiligo
Tumour necrosis factor (TNF)‐α, also known as cachectin, is a polypeptide hormone1 that plays a role in inflammatory, infectious and autoimmune processes in human disease.2, 3 The TNF gene is located on chromosome 6, and its production is regulated at the transcriptional and post‐transcriptional levels.4 TNF‐α exists in two forms: a transmembrane protein and a soluble protein. Both are biologically active in vivo.5
While TNF‐α is produced by a diverse array of resident tissue cells including melanocytes,6, 7, 8 the most prominent producers are migratory immune cells such as activated macrophages2 and activated T cells.9 TNF‐α binds to, and thereby acts through, receptors TNFR1 and TNFR2. TNFR1 receptors are expressed by a wide variety of cell types10, 11 and mediate apoptosis through a caspase 3‐dependent pathway.12 The cytoplasmic domain of TNF receptor I has a death domain, which recruits TNF receptor‐associated death domain (TRADD), Fas‐associated death domain (FADD) and FADD‐like interleukin‐1β‐converting enzyme (FLICE). This leads to caspase‐3 activation, which then induces apoptosis by prompting degradation of multiple proteins.12
TNFR2 receptors are far more rare, and expression is limited to fewer cell types including lymphocytes.13 TNFR2 does not contain a death domain and therefore cannot mediate apoptosis.14 Instead, through TNFR2 receptors, TNF‐α can activate and stimulate the proliferation of T cells.14 TNF depletion in the presence of specific TNFR2 agonists may be an ideal way to treat autoimmune disease, as TNFR2 agonism results in selective regulatory T cell (Treg) activation.15 Interestingly, melanocytes can express both TNFR1 and TNFR2 receptors in response to stress.16
TNF‐α has been implicated in the pathophysiology of several autoimmune diseases, particularly within the fields of rheumatology, gastroenterology and dermatology. Indeed, TNF‐α levels are elevated at prime sites of inflammation in certain autoimmune diseases, including the synovial fluid of patients with rheumatoid arthritis17 and the synovium18, 19 and sacroiliac joints20 of patients with ankylosing spondylitis. In Crohn disease, TNF‐α is thought to increase intestinal epithelial permeability, thereby interrupting the gut barrier and driving inflammation.21 The proinflammatory properties of TNF‐α stem from its activation of the proinflammatory cytokines interleukin (IL)‐1 and IL‐622 and of numerous nuclear transcription factors, most importantly nuclear factor (NF)‐κB, which is involved in maintaining inflammatory responses.23
The CD4+/CD8+ ratio is markedly decreased in perilesional skin of actively depigmenting patients.24 In vitiliginous skin, perilesional T cells of the CD8+ and CD4+ subsets secrete predominantly type 1 cytokines, including TNF‐α.25 TNF‐α plays an important role in the development of cytotoxic T lymphocytes (CTLs),26 which are implicated in disease initiation in vitiligo, and it enhances activated lymphocytic expression of interferon (IFN)‐γ,27 a cytokine implicated in the depigmentation process in vitiligo.28 As patients with vitiligo exhibit reduced mRNA levels for full‐length and soluble cytotoxic T‐lymphocyte‐associated protein 4 compared with controls, and these receptors negatively regulate T‐cell function, patients are more likely to experience the excessive T‐cell responses implicated in vitiligo pathogenesis.29
Recent evidence suggests that TNF‐α might possess anti‐inflammatory properties as well. For example, TNF‐α activates and induces proliferation of Tregs in vivo.30, 31, 32 Tregs are responsible for immune homeostasis. They suppress the activity of autoreactive T cells that escape clonal deletion in the thymus, such as those reactive against melanocytes in vitiligo, dampening inflammation as needed.33 Their abundance is markedly reduced in skin of patients with vitiligo.34 Given these functional characteristics, TNF‐α potentially plays both harmful and protective roles in vitiligo, by activating the cytotoxic T cells that are detrimental to melanocytes, and stimulating Tregs, respectively.
Tumour necrosis factor‐α inhibitors: current applications in clinical practice
TNF‐α antagonists are therapeutic agents that have proven efficacious in the treatment of certain autoimmune diseases. The five main TNF‐α antagonists approved by the US Food and Drug Administration and currently used in clinical practice are infliximab, etanercept, adalimumab, golimumab and certolizumab pegol.
Adalimumab and golimumab are anti‐TNF monoclonal antibodies with a human variable region and a human IgG1 constant region. Infliximab is a humanized anti‐TNF monoclonal antibody, containing a mouse variable region and a human IgG1 constant region. All three of these agents act as decoy receptors to bind TNF‐α so the cytokine cannot bind and act through its true receptors.35
Etanercept is a fusion protein of human TNFR2 and a truncated human IgG constant region. It contains duplicate domains of the human TNFR2 receptor, which is located on the lymphocyte surface and promotes T‐cell activation.36 This allows competitive binding of TNF‐α, thereby preventing binding of TNF‐α to true TNFRs on cell surfaces.37 Etanercept was found to bind soluble TNF‐α with a greater affinity than infliximab or adalimumab, although all three agents bound transmembrane TNF‐α with similar affinities.38
Finally, certolizumab is a humanized, anti‐TNF‐α monoclonal antibody covalently attached to polyethylene glycol chains (PEGylated), which binds TNF‐α with great affinity. Unlike other TNF‐α inhibitors, certolizumab does not contain an Fc region.39 Pegylation provides a longer half‐life for the drug by preventing its clearance from the plasma, thereby allowing for less frequent dosing.40 All agents inhibit both soluble and membrane‐bound TNF, but have different structures and may have different mechanisms of action.41, 42, 43 These TNF‐α antagonists have proven successful in treating ankylosing spondylitis,44, 45 rheumatoid arthritis,46, 47 psoriasis,48, 49, 50, 51 psoriatic arthritis52, 53 and Crohn disease,54, 55 supporting the pathological role of TNF‐α in these diseases.
Tumour necrosis factor‐α, melanocytes and vitiligo: the rationale for tumour necrosis factor‐α antagonists in the treatment of vitiligo
Vitiligo is a cutaneous disease presenting with a loss of epidermal melanocytes.56 Vitiligo is a multifactorial disease, but the Vitiligo European Task Force reached a consensus that autoimmunity plays at least some role in every type of vitiligo.57 T‐cell‐mediated autoimmunity in particular plays a prominent role in disease progression, as skin‐infiltrating cytotoxic T lymphocytes are reactive with melanocytes.58 The ultimate destruction of melanocytes may be due, in part, to an imbalance in the local cytokine environment. An abundance of proinflammatory cytokines such as TNF‐α has been reported in vitiligo‐affected skin as compared with nonlesional skin.59, 60, 61
Multiple triggers, including infectious agents, might precipitate such cytokine imbalances. In fact, recent evidence suggests that viral infections might trigger melanocyte secretion of proinflammatory cytokines including TNF‐α, which ultimately results in disease development.62 TNF‐α was proposed as a contributor to depigmentation in vitiligo after discovery of increased TNF‐α levels in lesional vitiligo skin, particularly in patients with active vitiligo.59, 60, 63 In fact, tissue levels of TNF‐α directly correlate with vitiligo disease activity.64
Data for TNF‐α serum levels are mixed, yet the largest study to date demonstrated that serum TNF‐α levels are increased in patients with vitiligo compared with healthy controls, and serum TNF‐α levels are higher in patients with generalized vitiligo than in those with localized disease.65, 66, 67 It is likely that localized TNF‐α plays an active role in vitiligo pathogenesis by activating CTLs within the skin, whereas serum TNF‐α may be of some use as a biomarker of vitiligo activity in addition to the more clearly established vitiligo‐associated cytokine, IFN‐γ.68, 69
In vivo, TNF‐α inhibits melanocyte proliferation and decreases melanocyte tyrosinase activity in a dose‐dependent manner, and is thus expected to interfere with repigmentation.70 The disappearance of differentiated melanocytes is a feature of vitiligo skin56 in progressive disease, and hopes for repigmentation are based on the availability of stem cells that may be stimulated to migrate, differentiate and repigment the lesions. TNF‐α can inhibit melanocyte stem cell differentiation, thereby further inhibiting repigmentation.71 TNF‐α has also been shown to promote melanocyte apoptosis in vitro.72 These functional data and findings provided a rationale for employing TNF‐α inhibition via the use of infliximab, etanercept and/or adalimumab for the purpose of halting depigmentation and supporting repigmentation in vitiligo.
When considering inhibition of TNF receptors rather than depleting the cytokine itself, it must be taken into account that lymphotoxin‐α (LTA), also known as TNF‐β, binds to receptors TNFR1 and TNFR2 as well.7 As increased expression of (genetic variants of) LTA has been associated with vitiligo susceptibility,73 receptor inhibition may offer additional advantages over inhibiting TNF‐α alone. The lack of uniformly efficacious therapeutics for vitiligo has provided ample incentive for utilizing TNF‐α antagonists in a clinical setting. In order to understand further the aetiology of vitiligo and to move towards development of more effective therapeutics, it is important to reflect on the data obtained to date.
Mixed results for tumour necrosis factor‐α antagonists in the clinic
Three clinical pilot studies have been reported and are summarized in Table 1. The largest pilot study was performed with six patients with widespread, progressive vitiligo, who were randomized to treatment with infliximab, etanercept or adalimumab. Dosage was based on recommended guidelines for psoriasis. These patients had not received recent treatment for their vitiligo, and vitiligo lesions were assessed via digital photography. One patient treated with infliximab experienced further disease progression and may have required higher treatment doses to halt disease, but 83% of patients displayed disease stabilization lasting up to 6 months after cessation of therapy.71
Table 1.
Patients treated with tumour necrosis factor (TNF)‐α antagonists for vitiligo
| Age (years)/sex | Disease treated | Vitiligo disease activity | Treatment regimen | TNF‐α antagonist dosing | Results | Report type | References |
|---|---|---|---|---|---|---|---|
| 15–38/5 M, 1 F | Vitiligo | Progressive | Untreated for ≥ 3 months, then ETA (n = 2), IFX (n = 2), ADA (n = 2) | IFX: 5 mg kg−1 IV at weeks 0, 2, 6, then q8 wk. ETA: 50 mg SC 2 ×/wk. ADA: 80 mg SC at week 0, 40 mg SC at week 1, then 40 mg SC q2 wk | One worsened, five stabilized | PS | 71 |
| 24–34/4 M | Vitiligo | Progressive | Untreated for 2 months, then ETA | 50 mg SC 1 ×/wk for 12 weeks, then 25 mg 1 ×/wk for 4 weeks | Stabilized | PS | 74 |
| 42/F, 46/F | Vitiligo | Progressive | Continued NB‐UVB and topical calcineurin inhibitors, added ETA | 50 mg 2 ×/wk, then 50 mg 1 ×/wk for ≥ 1 year | Disease stabilization and repigmentation | PS | 64 |
M, male; F, female; ETA, etanercept; IFX, infliximab; ADA, adalimumab; NB‐UVB, narrowband ultraviolet B; IV, intravenously; SC, subcutaneously; PS, pilot study; q2 wk, every 2 weeks; 2 ×/wk, two times per week.
In another pilot trial, four men with previously untreated, progressive vitiligo underwent therapy with etanercept. All patients experienced disease stabilization during their 16‐week course of treatment, but post‐treatment follow‐up was not documented.74 In the smallest pilot study two patients with progressive, recalcitrant vitiligo were administered etanercept, while continuing all previous vitiligo therapies. Both subjects experienced repigmentation.64 Finally, one of us (A.B.G.) conducted a pilot study looking at the safety and efficacy of etanercept in treating vitiligo. The study did not meet the primary end point, which was the achievement of ≥ 50% repigmentation at 6 months of treatment. However, it is unknown whether the subjects experienced stabilization or worsening of their disease control with etanercept, and other parameters were not measured (K.C. Webb, R. Tung, L.S. Winterfield, A.B. Gottlieb, J.M. Eby, S.W. Henning, I.C. Le Poole, unpublished results). In aggregate, 11 of 12 treated subjects experienced improvement in their vitiligo disease activity, from progressive to stable disease, or even repigmentation.
Additionally, there are several case reports detailing the effects of anti‐TNF‐α agents on established vitiligo, as well as the emergence of de novo vitiligo upon initiation of a TNF‐α antagonist to treat another autoimmune condition (Table 2). Four patients with long‐standing vitiligo underwent treatment with a TNF‐α antagonist for another indication (ankylosing spondylitis, psoriasis and seronegative inflammatory arthritis). Three patients noted improvement in their vitiligo,75, 76, 77 while one patient noted rapid, marked worsening of his vitiligo after starting therapy.78 In the latter case, the response of his ankylosing spondylitis is not reported, and the patient experienced partial repigmentation after stopping adalimumab.78
Table 2.
Patients treated with tumour necrosis factor (TNF)‐α antagonists for nonvitiligo conditions
| Age (years)/sex | Disease treated | Vitiligo disease activity | TNF‐α antagonist | TNF‐α antagonist dosing and duration | Results | Report type | References |
|---|---|---|---|---|---|---|---|
| 24/M | AS | Progressive | IFX | 350 mg IV at weeks 0, 2, 6 then q8 wk for 10 months | AS improved. Stabilization of vitiligo and repigmentation by 6 months | CR | 75 |
| 65/M | Plaque psoriasis | Progressive | ETA | 50 mg 2 ×/wk for 12 weeks then 25 mg 2 ×/wk for 12 weeks | Psoriasis improved. Stabilization of vitiligo and repigmentation at 24 weeks | CR | 76 |
| 76/F | SIA | Progressive | ETA | Weekly | Repigmentation after 2 months | PP/CR | 77 |
| 57/M | AS | Stable | ADA | 40 mg SC q2 wk | Worsening of vitiligo after 3 months. Partial repigmentation after ADA stopped and adjuvant therapies | CR | 78 |
| 61/M | RA | NA | IFX | 3 mg kg−1 IV every 2 months | Developed vitiligo at 6 months. Partial repigmentation with adjuvant therapies | CR | 79 |
| 51/M | UC | NA | IFX | 5 mg kg−1 at weeks 0, 2, 6 | UC improved. Developed vitiligo at 6 weeks. Partial repigmentation with stopping IFX and adjuvant vitiligo therapies | CR | 80 |
| 55/M | PsA | NA | IFX | 5 mg kg−1 IV q6 wk for 2 years, then 10 mg kg−1 q6 wk for 12 weeks | Developed vitiligo within 2 weeks after IFX dose increased. Partial repigmentation after IFX stopped | LE/CR | 81 |
| 54/F | CD | NA | ADA | 160 mg at week 0, 80 mg at week 2, then 40 mg q2 wk | CD improved. Developed vitiligo after 8 months | CR | 82 |
| 60/M | PRP | NA | IFX | 5 mg kg−1 IV then 10 mg kg−1 IV q5–6 wk for 27 months | PRP improved. Developed vitiligo after 27 months. Near complete repigmentation after IFX stopped and adjuvant vitiligo therapies | CR | 83 |
| 33/M | AS | NA | ADA | 40 mg q2 wk | AS improved. Developed vitiligo after 18 months | CR | 84 |
| 46/F | RA | NA | IFX | q8 wk for 8 months | Developed vitiligo after 2 months | CR | 85 |
| Eight patients, mean age 38·2 ± 11·5 | Psoriasis, CD, UC, RA, AS, PsA | NA | 7 ADA, 1 IFX | Unknown dose and duration | Developed vitiligo after mean treatment time of 17·4 ± 15·8 months | CS | 86 |
| One patient | RA | NA | IFX | Unknown dose and duration | Developed vitiligo | PC | 87 |
| 71/M | CD | NA | IFX | Unknown dose and duration | Developed vitiligo after 7 months | PC | 88 |
| 44/M | Behçet disease | NA | ADA | Unknown dose and duration | Developed vitiligo after 6 months | PC | 89 |
M, male; F, female; AS, ankylosing spondylitis; SIA, seronegative inflammatory arthritis; RA, rheumatoid arthritis; UC, ulcerative colitis; PsA, psoriatic arthritis; CD, Crohn disease; PRP, pityriasis rubra pilaris; NA, not applicable; IFX, infliximab; ETA, etanercept; ADA, adalimumab; IV, intravenously; SC, subcutaneously; q2 wk, every 2 weeks; 2 ×/wk, two times per week; CR, case report; PP, poster presentation; LE, letter to the editor; CS, case series; PC, prospective cohort.
There are also 18 reported patients who developed de novo vitiligo after initiating therapy with a TNF‐α antagonist for nonvitiligo conditions. Seven of these patients are detailed in case reports,79, 80, 81, 82, 83, 84, 85 with an additional eight patients in one case series86 and three reported in observational studies.87, 88, 89 In two observational studies looking at adverse cutaneous events that developed during TNF‐α antagonist treatment for rheumatological conditions, one of 5437 patients developed vitiligo in one study,89 and one of 435 patients developed vitiligo in another.87 A third prospective observational study exclusively reported patients who developed adverse cutaneous events while undergoing TNF‐α antagonist therapy. In this report, one of the 41 patients developed vitiligo.88 Finally, one case series reports eight patients who developed vitiligo when treated with an anti‐TNF‐α agent for a rheumatological, dermatological or gastrointestinal ailment.86 Among patients who developed de novo vitiligo, 10 patients were treated with adalimumab and eight were treated with infliximab.
In sum, 14 of 16 patients with established vitiligo experienced improvement in their vitiligo control when treated with a TNF‐α antagonist for either vitiligo or another autoimmune condition. Compiling the data from all studies reported here, a total of only 18 among 5928 patients treated with a TNF‐α antagonist for a nonvitiligo condition developed de novo vitiligo while on therapy. Nevertheless, a concern for activating vitiligo in treated patients cannot be overlooked.
Effects of tumour necrosis factor‐α inhibitors on vitiligo vary by subgroup
Although the number of reported subjects is low, the results for TNF‐α inhibition in the treatment of progressive vitiligo in patients without other autoimmune conditions are actually very promising. Indeed, in the three pilot studies, TNF‐α antagonists proved effective in halting disease progression in most patients with progressive vitiligo. These encouraging findings are likely not confounded by adjunctive vitiligo treatments, as the subjects had either been off all other vitiligo therapies during the months prior to and during the study,71, 74 or the patients' vitiligo had already proven refractory to therapies continued during the study (narrowband ultraviolet B and topical calcineurin inhibitors).64
TNF‐α inhibitors have been found to increase T‐cell activity in the periphery while decreasing localized (tissue) T‐cell activity, as evidenced by downregulation of inflammatory genes in target tissues.90 Unfortunately, no data are currently available to support reduced cutaneous CD8+ T‐cell infiltration in response to treatment with TNF‐α inhibitors in vitiligo. However, numerous studies describe the effects of TNF‐α inhibitors on cutaneous T‐cell levels in psoriasis,91, 92 another disease in which T cells (mainly of the CD4+ subtype) drive disease pathology.93 Decreased CD4+ and CD8+ T‐cell levels were found in psoriatic plaques following treatment with etanercept as compared with untreated skin,92 and skin‐homing CD8+ lymphocytes in psoriatic skin expressed markedly inhibited activation and increased susceptibility to apoptosis following exposure to infliximab.91 Meanwhile, there was no difference in absolute peripheral lymphocyte numbers following anti‐TNF‐α therapy.92 If these agents have comparable effects on CD8+ T‐cell numbers and activation in vitiliginous skin, this could explain why patients with progressive vitiligo experienced halting of their depigmentation when treated with TNF‐α inhibitors.
The duration of treatment with TNF‐α inhibitors required to halt depigmentation cannot be definitively concluded due to the lack of uniformly reported treatment durations in the reported studies. However, subjects showed stabilization of disease or repigmentation by the fourth month of treatment in two of the pilot studies.64, 74 In the four patients with established vitiligo treated with an anti‐TNF‐α agent for a nonvitiligo condition, it is interesting that the three patients who experienced improvement in their vitiligo had progressive disease for 11–20 years prior to anti‐TNF‐α exposure,75, 76, 77 while the patient who had marked worsening of his vitiligo had stable disease for 20 years prior to exposure.78 These findings show that TNF‐α inhibition was helpful in patients with progressive vitiligo but harmful in the one with stable disease.
Important differences exist in the immunological profile of the microenvironment in stable vs. progressive vitiligo. In actively spreading lesions, CD8+ T cells are grouped into clusters abutting melanocytes, whereas the concentration of skin‐infiltrating CD8+ T cells is much lower in stable vitiligo,24, 58, 94 as active melanocyte destruction is not occurring in the latter case. In our review, TNF‐α inhibitors were found to halt active depigmentation, likely because these agents interfere with T‐lymphocyte activity. Moreover, whereas the activity of vitiligo impacts the extent to which the disease can be controlled with TNF‐α inhibition, the data do not support a correlation between disease duration prior to anti‐TNF‐α exposure and treatment efficacy. As for treatment regimens, etanercept at a dose of 50 mg subcutaneously once or twice weekly for at least 2 months, with or without decreasing the dose or administration frequency during the course of treatment, was the most commonly recorded successful treatment. Efficacy was demonstrated in 10 of the 14 patients with established vitiligo who experienced improvement in their vitiligo control.
Paradoxical effects have been observed with the use of TNF‐α inhibitor therapeutics,95 but we found that most patients with active vitiligo improved with these agents. Furthermore, reports of vitiligo as a cutaneous adverse event of TNF‐α inhibitor therapy are sparse, and the majority of patients treated do not develop vitiligo. Indeed, a mere 18 of the 5928 reported patients treated with TNF‐α antagonists for nonvitiligo conditions developed de novo vitiligo. However, the fact that patients developed vitiligo when exposed to these agents suggests that TNF‐α might have been conferring a protective effect against depigmentation prior to its blockade. A possible explanation for this is provided below and is detailed in Figure 1. Interestingly, two patients who had been taking a stable dose of an anti‐TNF‐α agent did not develop vitiligo until their dose was doubled. In both cases the agent was infliximab.81, 83 Indeed, although it is low, the risk of developing vitiligo as a cutaneous adverse event during anti‐TNF‐α therapy does appear greater with the use of adalimumab or infliximab than with etanercept.
Figure 1.
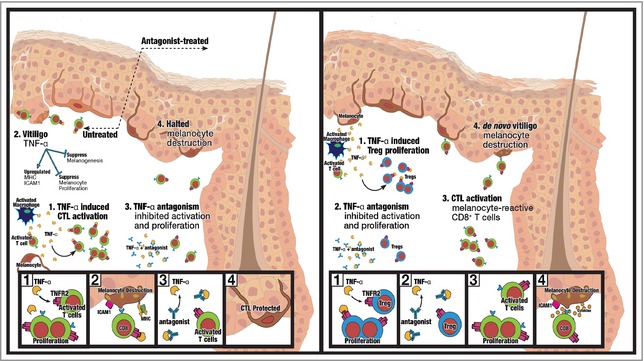
Effects of tumour necrosis factor (TNF)‐α inhibition in patients with progressive vitiligo (left) and in patients without vitiligo (right). In patients with progressive vitiligo (left), successful halting of depigmentation is observed with the use of TNF‐α inhibitors. In the skin of patients with progressive vitiligo, there is a surplus of cytotoxic T lymphocytes (CTLs) and a relative deficiency of regulatory T cells (Tregs) to suppress cytotoxic effects. With the addition of TNF‐α inhibitors, there is preferential inhibition of the effects of TNF‐α on CD8+ T‐cell activation and proliferation, resulting in reduced melanocyte destruction. In patients without vitiligo, a surplus of CTLs in the skin is not observed, and Treg numbers are normal. Thus, inhibiting TNF‐α instead preferentially inhibits the ability of TNF‐α to promote the activation and proliferation of Tregs, resulting in removal of the skin's innate protection against depigmentation and de novo vitiligo development (right). On the left, in progressive vitiligo, TNF‐α induces CTL activation and proliferation (Box 1), and upregulates expression of melanocyte cell‐surface molecules that result ultimately in melanocyte destruction (Box 2). With TNF‐α inhibitor therapy, CTL activation and proliferation is inhibited (Box 3), resulting in melanocyte survival and halted depigmentation (Box 4). On the right, in patients without vitiligo, an increase in cutaneous self‐reactive CTLs is not observed. Instead, immunological homeostasis exists, with a normal amount of Tregs suppressing effects of any self‐reactive CD8+ T cells. TNF‐α induces activation and proliferation of Tregs in the skin (Box 1). When TNF‐α is inhibited, Treg activation and proliferation are inhibited (Box 2), and self‐reactive cutaneous CD8+ T cells can destroy cutaneous melanocytes (Box 3), resulting in de novo vitiligo development (Box 4). ICAM, intercellular adhesion molecule; TNFR, TNF receptor.
Reduced regulatory T‐cell activity may contribute to disease progression in the face of tumour necrosis factor‐α antagonist treatment
There exists a bipartite explanation for the unfavourable cutaneous environment in vitiligo, in that there is both an overabundance of the T cells that promote localized cutaneous inflammation (CTLs) and a shortage of the T cells that curb these inflammatory responses (Tregs). Despite stimulatory effects of the cytokine on pathological CTLs, TNF‐α knockout mice were still able to develop vitiligo in a T‐cell‐receptor transgenic model of spontaneous disease,28 suggesting that TNF‐α is not the sole player in promoting depigmentation. Given that cytotoxic T cells facilitate target cell death through either granzyme/perforin‐ or Fas/Fas ligand (FasL)‐mediated pathways,96, 97 it would follow that one of these mechanisms facilitates melanocyte death in vitiligo. However, granzyme knockout mice were unimpaired in their ability to depigment, and melanocytes were found to be insensitive to FasL‐mediated apoptosis.98 From these studies, one can conclude that neither the TNF‐α‐, Fas/FasL‐ nor granzyme‐mediated apoptotic mechanism is solely responsible for T‐cell‐mediated cytotoxicity. This would explain, in part, why depleting TNF‐α alone can be insufficient to interfere with CTL function and depigmentation.
Instead, it is more likely that other inflammatory cytokines, such as IFN‐γ, and other factors in the local microenvironment are the chief culprits in depigmentation. In fact, IFN‐γ and TNF‐α are observed to promote synergistically the inflammatory response, which is accredited to the cooperative interaction between the transcription factors signal transducer and activator of transcription‐1 and NF‐κB.99 Furthermore, IFN‐γ, TNF‐α and TNF‐β induced the expression of intercellular adhesion molecule (ICAM)1 on the surface of melanocytes,100 which promotes leucocyte recruitment and T‐cell activation.101
Interestingly, melanocytes from perilesional vitiligo skin and vitiligo lesions have upregulated ICAM1 levels,102 suggesting a potentially significant role of these molecules in vitiligo disease. Indeed, genetic variants of IFN‐γ and ICAM1 were shown to affect disease onset and progression of generalized vitiligo.103 Melanocytes were found to be capable of antigen presentation via major histocompatibility complex (MHC) II molecules, and these MHC II–antigen complexes are recognized by CD4+ cells.104 Expression of the costimulatory molecule ICAM1 on the melanocyte cell surface correlated with T‐cell responses to antigens presented by melanocytes.104 Together, this may afford a partial explanation for vitiligo pathogenesis in that melanocytes with greater ICAM1 cell‐surface expression experience increased T‐cell recognition, which ultimately results in increased cytotoxic T‐cell‐mediated melanocyte destruction.
Ample support exists for the role of IFN‐γ in vitiligo. For example, IFN‐γ knockdown68 and knockout28, 105 mice experienced impaired depigmentation. The latter publication also provides insight into the detrimental effects observed following TNF‐α inhibition in some patients. In IFN‐γ knockout mice, depigmentation is still observed when Tregs are depleted, and IFN‐γ knockout mice exhibited an increase in Tregs. This suggests that the mechanism by which IFN‐γ can induce depigmentation is explained, at least in part, by suppressing Treg development.106, 107
Further supporting the importance of Tregs in vitiligo is the finding that upon adoptive transfer of Tregs, disease remission was measured in a mouse model of vitiligo.28 Knowing that TNF‐α activates Tregs in vivo 32 and will support Treg development,30, 31, 32, 108 it follows that depleting TNF‐α can lead to decreased Treg production and activation, resulting in even less skin homing of Tregs in patients with vitiligo, which is problematic and relatively specific to vitiligo.34 Inhibiting TNF‐α may also reduce the number of available Tregs in the skin of patients without vitiligo, permitting cytotoxic T lymphocytes the opportunity to exert an unchecked inflammatory response within the epidermis without suppression by Tregs, and tipping the scale in favour of depigmentation. This explanation may account for the unexpected worsening of pre‐existing vitiligo and de novo vitiligo development observed in some patients treated with anti‐TNF‐α agents. It is then probable that such local Treg deficiency can be overcome by recruiting Tregs to the skin by means of topical CCL22 administration.109 Once developed for human use, such treatment may be helpful in preventing the adverse development of vitiligo in patients treated with TNF‐α inhibitors for other conditions.
Conclusions and future directions
Upon review, TNF‐α appears to have both pro‐ and anti‐inflammatory properties (Box 3), within the skin, possibly due to its ability to stimulate both cytotoxic T cells and Tregs. Although blocking TNF‐α was effective in halting disease progression and even in promoting repigmentation in nearly every patient with progressive vitiligo, 18 of 5928 patients developed de novo vitiligo when treated for another autoimmune condition. Until such side‐effects can be fully averted, anti‐TNF‐α treatment is unlikely to gain much traction in the treatment of vitiligo. An important observation is that halting depigmentation was not necessarily followed by promoting repigmentation. Indeed, in future clinical trials to treat vitiligo it will be of the utmost importance to consider both parameters when evaluating treatment efficacy, so that a positive outcome is not inadvertently overlooked. Given the complex nature of vitiligo development, it is possible that a combination of treatments will prove most efficacious for vitiligo treatment. To prevent vitiligo from developing as a side‐effect of anti‐TNF‐α treatment in patients with other autoimmune conditions, it may be important simultaneously to promote Treg infiltration into the skin.
TNF‐α inhibitors have actually provided significant benefit by halting disease that might otherwise be progressive. For this reason, failure to repigment should not necessarily be considered a treatment failure. Furthermore, etanercept should be considered the preferred anti‐TNF‐α agent to employ in patients at risk for developing vitiligo, in order to mitigate the risk of this adverse event during therapy for other conditions. In all, TNF‐α inhibitors have offered benefit in patients with progressing vitiligo, while unexpected observations made during clinical trials for these agents have provided new insight into the differential involvement of TNF‐α in autoimmune disease.
Funding sources
National Institutes of Health grant R01AR057643 to I.C.L.P.
Conflicts of interest
None declared.
References
- 1. Wakefield PE, James WD, Samlaska CP, Meltzer MS. Tumor necrosis factor. J Am Acad Dermatol 1991; 24:675–85. [DOI] [PubMed] [Google Scholar]
- 2. Vassali P. The pathophysiology of tumour necrosis factor. Annu Rev Immunol 1992; 10:411–52. [DOI] [PubMed] [Google Scholar]
- 3. Aggarwal BB. Signaling pathways of the TNF superfamily: a double‐edged sword. Nat Rev Immunol 2003; 3:745–56. [DOI] [PubMed] [Google Scholar]
- 4. Spriggs DR, Deutsch S, Kufe DW. Genomic structure, induction, and production of TNF‐alpha. Immunol Ser 1992; 56:3–34. [PubMed] [Google Scholar]
- 5. Black RA, Rauch CT, Kozlosky CT et al A metalloproteinase disintegrin that releases tumour‐necrosis factor‐alpha from cells. Nature 1997; 385:729–33. [DOI] [PubMed] [Google Scholar]
- 6. Krüger‐Krasagakes S, Krasagakis K, Garbe C et al Production of cytokines by human melanoma cells and melanocytes In: Skin Cancer: Basic Science, Clinical Research and Treatment (Garbe C, Schmitz S, Orfanos CE, Dummer R, eds). Berlin, Heidelberg: Springer, 1995; 155–68. [DOI] [PubMed] [Google Scholar]
- 7. Bradley JR. TNF‐mediated inflammatory disease. J Pathol 2008; 214:149–60. [DOI] [PubMed] [Google Scholar]
- 8. Tam I, Stepien K. Secretion of proinflammatory cytokines by normal human melanocytes in response to lipopolysaccharide. Acta Biochim Pol 2011; 58:507–11. [PubMed] [Google Scholar]
- 9. Kriegler MC, Perez K, DeFay I et al A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF. Cell 1988; 53:45–53. [DOI] [PubMed] [Google Scholar]
- 10. MacEwan DJ. TNF receptor subtype signaling: differences and cellular consequences. Cell Signal 2002; 14:477–92. [DOI] [PubMed] [Google Scholar]
- 11. Vandenabeele P, Declercq W, Beyaerte R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol 1995; 5:392–9. [DOI] [PubMed] [Google Scholar]
- 12. Nagata S, Golstein P. The Fas death factor. Science 1995; 267:1449–56. [DOI] [PubMed] [Google Scholar]
- 13. McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation 2008; 5:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Faustman D, Davis M. TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov 2010; 9:482–93. [DOI] [PubMed] [Google Scholar]
- 15. Faustman DL, Davis M. TNF receptor 2 and disease: autoimmunity and regenerative medicine. Front Immunol 2013; 4:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kroll TM, Bommiasamy H, Boissy RE et al 4‐Tertiary butyl phenol exposure sensitizes human melanocytes to dendritic cell‐mediated killing: relevance to vitiligo. J Invest Dermatol 2005; 124:798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saxne T, Palladino MA, Heinegård D et al Detection of tumor necrosis factor α but not tumor necrosis factor β in rheumatoid arthritis synovial fluid and serum. Arthritis Rheum 1989; 31:1041–5. [DOI] [PubMed] [Google Scholar]
- 18. Grom AA, Murray KJ, Luyrink L et al Patterns of expression of tumor necrosis factor alpha, tumor necrosis factor beta, and their receptors in synovia of patients with juvenile rheumatoid arthritis and juvenile spondylarthropathy. Arthritis Rheum 1996; 39:1703–10. [DOI] [PubMed] [Google Scholar]
- 19. Cañete JD, Llena J, Collado A et al Comparative cytokine gene expression in synovial tissue of early rheumatoid arthritis and seronegative spondyloarthropathies. Br J Rheumatol 1997; 36:38–42. [DOI] [PubMed] [Google Scholar]
- 20. Braun J, Bollow M, Neure L et al Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis Rheum 1995; 38:499–505. [DOI] [PubMed] [Google Scholar]
- 21. Suenaert P, Bulteel V, Lemmens L et al Anti‐tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol 2002; 97:2000–4. [DOI] [PubMed] [Google Scholar]
- 22. Tracey D, Klareskog L, Saso EH et al Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 2008; 117:244–79. [DOI] [PubMed] [Google Scholar]
- 23. Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev 2008; 19:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Le Poole IC, van den Wijngaard RM, Westerhof W, Das PK. Presence of T cells and macrophages in inflammatory vitiligo skin parallels melanocyte disappearance. Am J Pathol 1996; 148:1219–28. [PMC free article] [PubMed] [Google Scholar]
- 25. Wańkowicz‐Kalińska A, van den Wijngaard RM, Tigges BJ et al Immunopolarization of CD4+ and CD8+ T cells to type‐1‐like is associated with melanocyte loss in human vitiligo. Lab Invest 2003; 83:683–95. [DOI] [PubMed] [Google Scholar]
- 26. Ranges GE, Figari IS, Espevik T, Palladino MA Jr. Inhibition of cytotoxic T cell development by transforming growth factor β and reversal by recombinant tumor necrosis factor α. J Exp Med 1987; 166:191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scheurich P, Thoma B, Ricer U, Pfizenmaier K. Immunoregulatory activity of recombinant human tumor necrosis factor (TNF)‐alpha: induction of TNF receptors on human T cells and TNF‐alpha‐mediated enhancement of T cell responses. J Immunol 1987; 138:1786–90. [PubMed] [Google Scholar]
- 28. Chatterjee S, Eby JM, Al‐Khami AA et al A quantitative increase in regulatory T cells controls development of vitiligo. J Invest Dermatol 2014; 134:1285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dwivedi M, Laddha NC, Imran M et al Cytotoxic T‐lymphocyte‐associated antigen‐4 (CTLA‐4) in isolated vitiligo: a genotype–phenotype correlation. Pigment Cell Melanoma Res 2011; 24:737–40. [DOI] [PubMed] [Google Scholar]
- 30. Grinberg‐Bleyer Y, Saadoun D, Baeyens A et al Pathogenic T cells have a paradoxical protective effect in murine autoimmune diabetes by boosting Tregs. J Clin Invest 2010; 120:4558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Biton J, Semerano L, Delavallée L et al Interplay between TNF and regulatory T cells in a TNF‐driven murine model of arthritis. J Immunol 2011; 186:3899–910. [DOI] [PubMed] [Google Scholar]
- 32. Biton J, Boissier MC, Bessis N. TNF‐alpha: activator or inhibitor of regulatory T cells? Joint Bone Spine 2012; 79:119–23. [DOI] [PubMed] [Google Scholar]
- 33. Sakaguchi S, Sakaguchi N, Shimizu J et al Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev 2001; 182:18–32. [DOI] [PubMed] [Google Scholar]
- 34. Klarquist J, Denman CJ, Hernandez C et al Reduced skin homing by functional Treg in vitiligo. Pigment Cell Melanoma Res 2010; 23:276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Silva LC, Origosa LC, Bernard G. Anti‐TNF agents in the treatment of immune‐mediated inflammatory diseases: mechanisms of actions and pitfalls. Immunotherapy 2010; 2:817–33. [DOI] [PubMed] [Google Scholar]
- 36. Mohler KM, Torrance DS, Smith CA et al Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol 1993; 151:1548–61. [PubMed] [Google Scholar]
- 37. Spencer‐Green G. Etanercept (Enbrel): update on therapeutic use. Ann Rheum Dis 2000; 59:i46–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kaymakcalan Z, Sakorafas P, Bose S et al Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol 2009; 131:308–16. [DOI] [PubMed] [Google Scholar]
- 39. Smith B, Ceska T, Henry A et al Detailing the novel structure of the biopharmaceutical certolizumab pegol. Am J Gastroenterol 2008; 103:S430. [Google Scholar]
- 40. Baker M, Stringer F, Stephens S. Pharmacokinetic properties of the anti‐TNF agent certolizumab pegol. Gut 2006; 55 (Suppl. V):A122. [Google Scholar]
- 41. Thalayasingam N, Isaacs JD. Anti‐TNF therapy. Best Pract Res Clin Rheumatol 2011; 25:549–67. [DOI] [PubMed] [Google Scholar]
- 42. Benucci M, Saviola G, Manfredi M et al Tumor necrosis factors blocking agents: analogies and differences. Acta Biomed 2012; 83:72–80. [PubMed] [Google Scholar]
- 43. Park W, Hrycaj P, Jeka S et al A randomized, double‐blind, multicenter, parallel‐group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT‐P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis 2013; 72:1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Braun J, Brandt J, Listing J et al Treatment of active ankylosing spondylitis with infliximab: a randomised controlled multicentre trial. Lancet 2002; 359:1187–93. [DOI] [PubMed] [Google Scholar]
- 45. Davis JC Jr, van der Heijde D, Braun J et al Recombinant human tumor necrosis factor receptor (etanercept) for treating ankylosing spondylitis: a randomized, controlled trial. Arthritis Rheum 2003; 48:3230–6. [DOI] [PubMed] [Google Scholar]
- 46. Elliott MJ, Maini RN, Feldmann M et al Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor α. Arthritis Rheum 1993; 36:1681–90. [DOI] [PubMed] [Google Scholar]
- 47. Elliot MJ, Maini RN, Feldmann M et al Randomised double‐blind comparison of chimeric monoclonal antibody to tumour necrosis factor a (cA2) versus placebo in rheumatoid arthritis. Lancet 1994; 344:1105–10. [DOI] [PubMed] [Google Scholar]
- 48. Leonardi CL, Powers JL, Matheson RT et al Etanercept as monotherapy in patients with psoriasis. N Engl J Med 2003; 349:2014–22. [DOI] [PubMed] [Google Scholar]
- 49. Gottlieb AB, Evans R, Li S et al Infliximab induction therapy for patients with severe plaque‐type psoriasis: a randomized, double‐blind, placebo‐controlled trial. J Am Acad Dermatol 2004; 51:534–42. [DOI] [PubMed] [Google Scholar]
- 50. Pitarch G, Sanchez‐Carazo JL, Mahiques L et al Treatment of psoriasis with adalimumab. Clin Exp Dermatol 2007; 32:18–22. [DOI] [PubMed] [Google Scholar]
- 51. Reich K, Ortonne JP, Gottlieb AB et al Successful treatment of moderate to severe plaque psoriasis with the PEGylated Fab′ certolizumab pegol: results of a phase II randomized, placebo‐controlled trial with a re‐treatment extension. Br J Dermatol 2012; 167:180–90. [DOI] [PubMed] [Google Scholar]
- 52. Antoni C, Dechant C, Lorenz H et al Successful treatment of severe psoriatic arthritis with infliximab. Arthritis Rheum 1999; 42:S371. [Google Scholar]
- 53. Mease PJ, Goffe BS, Metz J et al Etanercept in the treatment of psoriatic arthritis and psoriasis: a randomised trial. Lancet 2000; 356:385–90. [DOI] [PubMed] [Google Scholar]
- 54. Akobeng AK, Zachos M. Tumor necrosis factor‐alpha antibody for induction of remission in Crohn's disease. Cochrane Database Syst Rev 2004; (1):CD003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ford AC, Sandborn WJ, Khan KJ et al Efficacy of biological therapies in inflammatory bowel disease: systematic review and meta‐analysis. Am J Gastroenterol 2011; 106:644–59. [DOI] [PubMed] [Google Scholar]
- 56. Le Poole IC, van den Wijngaard RM, Westerhof W et al Presence or absence of melanocytes in vitiligo lesions: an immunohistochemical investigation. J Invest Dermatol 1993; 100:816–22. [DOI] [PubMed] [Google Scholar]
- 57. Ezzedine K, Lim HW, Suzuki T et al Reviewed classification/nomenclature of vitiligo and related issues: the Vitiligo Global Issues Consensus Conference. Pigment Cell Melanoma Res 2012; 25:E1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van den Wijngaard R, Wankowicz‐Kalinska A, Le Poole C et al Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab Invest 2000; 80:1299–309. [DOI] [PubMed] [Google Scholar]
- 59. Moretti S, Spallanzani A, Amato L et al New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res 2002; 15:87–92. [DOI] [PubMed] [Google Scholar]
- 60. Birol A, Kisa U, Kurtipek GS et al Increased tumor necrosis factor alpha (TNF‐α) and interleukin 1 alpha (IL1‐α) levels in the lesional skin of patients with nonsegmental vitiligo. Int J Dermatol 2006; 45:992–3. [DOI] [PubMed] [Google Scholar]
- 61. Moretti S, Fabbri P, Baroni G et al Keratinocyte dysfunction in vitiligo epidermis: cytokine microenvironment and correlation to keratinocyte apoptosis. Histol Histopathol 2009; 24:849–57. [DOI] [PubMed] [Google Scholar]
- 62. Wang S, Liu D, Ning W, Xu A. Cytosolic dsDNA triggers apoptosis and pro‐inflammatory cytokine production in normal human melanocytes. Exp Dermatol 2015; 24:298–300. [DOI] [PubMed] [Google Scholar]
- 63. Attwa E, Gamil H, Assaf M, Ghonemy S. Over‐expression of tumor necrosis factor‐? in vitiligo lesions after narrow‐band UVB therapy: an immunohistochemical study. Arch Dermatol Res 2012; 304:823‐30. [DOI] [PubMed] [Google Scholar]
- 64. Kim NH, Torchia D, Rouhani P et al Tumor necrosis factor‐α in vitiligo: direct correlation between tissue levels and clinical parameters. Cutan Ocul Toxicol 2011; 30:225–7. [DOI] [PubMed] [Google Scholar]
- 65. Tu CX, Gu JS, Lin XR. Increased interleukin‐6 and granulocyte–macrophage colony stimulating factor levels in the sera of patients with non‐segmental vitiligo. J Dermatol Sci 2003; 31:73–8. [DOI] [PubMed] [Google Scholar]
- 66. Laddha NC, Dwivedi M, Begum R. Increased tumor necrosis factor (TNF)‐α and its promoter polymorphisms correlate with disease progression and higher susceptibility towards vitiligo. PLoS One 2012; 7:e52298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Singh S, Singh U, Pandey SS. Serum concentration of IL‐6, IL‐2, TNF‐α, and IFN γ in vitiligo patients. Indian J Dermatol 2012; 57:12–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Harris JE, Harris TH, Weninger W et al A mouse model of vitiligo with focused epidermal depigmentation requires IFN‐γ for autoreactive CD8+ T‐cell accumulation in the skin. J Invest Dermatol 2012; 132:1869–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang L, Wei Y, Sun Y et al Interferon‐γ inhibits melanogenesis and induces apoptosis in melanocytes: a pivotal role of CD8+ cytotoxic T lymphocytes in vitiligo. Acta Derm Venereol 2015; 95:664–70. [DOI] [PubMed] [Google Scholar]
- 70. Swope VB, Abdel‐Malek Z, Kassem LM, Nordlund JJ. Interleukins 1α and 6 and tumor necrosis factor α are paracrine inhibitors of human melanocyte proliferation and melanogenesis. J Invest Dermatol 1991; 96:180–5. [DOI] [PubMed] [Google Scholar]
- 71. Alghamdi KM, Khurrum H, Taieb A, Ezzedine K. Treatment of generalized vitiligo with anti‐TNF‐α agents. J Drugs Dermatol 2012; 11:534–9. [PubMed] [Google Scholar]
- 72. Kim NH, Jeon S, Lee HJ, Lee AY. Impaired PI3K/Akt activation‐mediated NF‐κB inactivation under elevated TNF‐α is more vulnerable to apoptosis in vitiliginous keratinocytes. J Invest Dermatol 2007; 127:2612–17. [DOI] [PubMed] [Google Scholar]
- 73. Laddha NC, Dwivedi M, Gani AR et al Tumor necrosis factor B (TNFB) genetic variants and its increased expression are associated with vitiligo susceptibility. PLoS One 2013; 8:e81736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rigopoulos D, Gregoriou S, Larios G et al Etanercept in the treatment of vitiligo. Dermatology 2007; 215:84–5. [DOI] [PubMed] [Google Scholar]
- 75. Simon JA, Burgos‐Vargas R. Vitiligo improvement in a patient with ankylosing spondylitis treated with infliximab. Dermatology 2008; 216:234–5. [DOI] [PubMed] [Google Scholar]
- 76. Campanati A, Guiliodori K, Ganzetti G et al A patient with psoriasis and vitiligo treated with etanercept. Am J Clin Dermatol 2010; 11 (Suppl. 1):S46–8. [DOI] [PubMed] [Google Scholar]
- 77. Tolaymat L, Sluzevich J. Repigmentation of chronic generalized vitiligo following etanercept therapy for seronegative inflammatory arthritis. J Am Acad Dermatol 2010; 62 (Suppl. 1):AB121. [Google Scholar]
- 78. Maruthappu T, Leandro M, Morris SD. Deterioration of vitiligo and new onset of halo naevi observed in two patients receiving adalimumab. Dermatol Ther 2013; 26:370–2. [DOI] [PubMed] [Google Scholar]
- 79. Ramirez‐Hernandez M, Marras C, Martinez‐Escribano JA. Infliximab‐induced vitiligo. Dermatology 2005; 210:79–80. [DOI] [PubMed] [Google Scholar]
- 80. Ismail WA, Al‐Enzy SA, Alsurayei SA, Ismail AE. Vitiligo in a patient receiving infliximab for refractory ulcerative colitis. Arab J Gastroenterol 2011; 12:109–11. [DOI] [PubMed] [Google Scholar]
- 81. Lahita RG, Vernace MA. Vasculitis, vitiligo, thyroiditis, and altered hormone levels after anti‐tumor necrosis factor therapy. J Rheumatol 2011; 38:579–80. [DOI] [PubMed] [Google Scholar]
- 82. Posada C, Flórez A, Batalla A et al Vitiligo during treatment of Crohn's disease with adalimumab: adverse effect or co‐occurrence? Case Rep Dermatol 2011; 3:28–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mattox AR, Chappell JA, Hurley MY. New‐onset vitiligo during long‐term, stable infliximab treatment of pityriasis rubra pilaris. J Drugs Dermatol 2013; 12:217–19. [PubMed] [Google Scholar]
- 84. Toissirot E, Salard D, Algros MP. Aubin F [Occurrence of vitiligo in a patient with ankylosing spondylitis receiving adalimumab]. Ann Dermatol Venereol 2013; 140:801–4 (in French). [DOI] [PubMed] [Google Scholar]
- 85. Carvalho CLDB, Ortigosa LCM. Segmental vitiligo after infliximab use for rheumatoid arthritis – a case report. An Bras Dermatol 2014; 89:154–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Mery‐Bossard L, Chaby G, Maccari F et al New onset vitiligo under biologic agents: a case series. Ann Rheum Dis 2014; 73:S2. [Google Scholar]
- 87. Exarchou SA, Voulgari PV, Markatseli TE et al Immune‐mediated skin lesions in patients treated with anti‐tumour necrosis factor alpha inhibitors. Scand J Rheumatol 2009; 38:328–31. [DOI] [PubMed] [Google Scholar]
- 88. Bonnet N, Guis S, Koeppel MC et al [Cutaneous events during anti‐TNF‐α therapy: a prospective observational study of 41 cases]. Ann Dermatol Venereol 2010; 137:12–20 (in French). [DOI] [PubMed] [Google Scholar]
- 89. Hernández MV, Sanmartí R, Cañete JD et al Cutaneous adverse events during treatment of chronic inflammatory rheumatic conditions with tumor necrosis factor antagonists: study using the Spanish registry of adverse events of biological therapies in rheumatic diseases. Arthritis Care Res 2013; 65:2024–31. [DOI] [PubMed] [Google Scholar]
- 90. Bosè F, Raeli L, Garutti C et al Dual role of anti‐TNF therapy: enhancement of TCR‐mediated T cell activation in peripheral blood and inhibition of inflammation in target tissues. Clin Immunol 2011; 139:164–76. [DOI] [PubMed] [Google Scholar]
- 91. Bedini C, Nasorri G, Girolomoni O et al Antitumour necrosis factor‐α chimeric antibody (infliximab) inhibits activation of skin‐homing CD4+ and CD8+ T lymphocytes and impairs dendritic cell function. Br J Dermatol 2007; 157:249–58. [DOI] [PubMed] [Google Scholar]
- 92. Mahiques L, Pitarch G, Sánchez‐Carazo JL et al [Analysis of lymphocyte populations in psoriatic plaques following inhibition of tumour necrosis factor‐α with etanercept]. Actas Dermosifiliogr 2007; 98:539–44 (in Spanish). [PubMed] [Google Scholar]
- 93. Nickoloff BJ, Wrone‐Smith T. Injection of pre‐psoriatic skin with CD4+ T cells induces psoriasis. Am J Pathol 1999; 155:145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Benzekri L, Gauthier Y, Hamada S, Hassam B. Clinical features and histological findings are potential indicators of activity in lesions of common vitiligo. Br J Dermatol 2013; 168:265–71. [DOI] [PubMed] [Google Scholar]
- 95. Speeckaert R, Speeckaert MM, van Geel N. Why treatments do(n't) work in vitiligo: an autoinflammatory perspective. Autoimmun Rev 2015; 14:332–40. [DOI] [PubMed] [Google Scholar]
- 96. Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T‐cell cytotoxicity is mediated through perforin and Fas lytic pathway. Nature 1994; 370:650–2. [DOI] [PubMed] [Google Scholar]
- 97. Barry M, Bleackley RC. Cytotoxic T lymphocytes: all roads lead to death. Nat Rev Immunol 2002; 2:401–9. [DOI] [PubMed] [Google Scholar]
- 98. Rivoltini L, Radrizzani M, Accornero P et al Human melanoma‐reactive CD4+ and CD8+ CTL clones resist Fas ligand‐induced apoptosis and use Fas/Fas ligand‐independent mechanisms for tumor killing. J Immunol 1998; 161:1220–30. [PubMed] [Google Scholar]
- 99. Ohmori Y, Schreiber RD, Hamilton TA. Synergy between interferon‐γ and tumor necrosis factor‐α in transcriptional activation is mediated by cooperation between signal transducer and activator of transcription 1 and nuclear factor κB. J Biol Chem 1997; 272:14899–907. [DOI] [PubMed] [Google Scholar]
- 100. Yohn JJ, Critelli M, Lyons MB, Norris DA. Modulation of melanocyte intercellular adhesion molecule‐1 by immune cytokines. J Invest Dermatol 1990; 95:233–7. [DOI] [PubMed] [Google Scholar]
- 101. Hedley SJ, Metcalfe R, Gawkrodger DJ et al Vitiligo melanocytes in long‐term culture show normal constitutive and cytokine‐induced expression of intercellular adhesion molecule‐1 and major histocompatibility complex class I and class II molecules. Br J Dermatol 1998; 139:965–73. [DOI] [PubMed] [Google Scholar]
- 102. Morelli JG, Norris DA. Influence of inflammatory mediators and cytokines on human melanocyte function. J Invest Dermatol 1993; 100 (2 Suppl.):191S–5S. [PubMed] [Google Scholar]
- 103. Dwivedi M, Laddha NC, Shah K et al Involvement of interferon‐gamma genetic variants and intercellular adhesion molecule‐1 in onset and progression of generalized vitiligo. J Interferon Cytokine Res 2013; 33:646–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Le Poole IC, Mutis T, van den Wijngaard RM et al A novel, antigen‐presenting function of melanocytes and its possible relationship to hypopigmentary disorders. J Immunol 1993; 151:7284–92. [PubMed] [Google Scholar]
- 105. Gregg RK, Nichols L, Chen Y et al Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase‐specific TCR transgenic mice. J Immunol 2010; 184:1909–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Nakajima CT, Mukai N, Yamaguchi Y et al Induction of the chemokine receptor CXCR3 on TCR‐stimulated T cells: dependence on the release from persistent TCR‐triggering and requirement for IFN‐gamma stimulation. Eur J Immunol 2002; 32:1792–801. [DOI] [PubMed] [Google Scholar]
- 107. Moser B, Wolf M, Walz A, Loetscher P. Chemokines: multiple levels of leukocyte migration control. Trends Immunol 2004; 25:75–84. [DOI] [PubMed] [Google Scholar]
- 108. Chen X, Bäumel M, Männel DN et al Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+ CD25+ T regulatory cells. J Immunol 2007; 179:154–61. [DOI] [PubMed] [Google Scholar]
- 109. Eby JM, Kang HK, Tully ST et al CCL22 to activate Treg migration and suppress depigmentation in vitiligo. J Invest Dermatol 2015; 135:1574–80. [DOI] [PMC free article] [PubMed] [Google Scholar]