

Abstract
Objective
Paraneoplastic neurologic disorders (PND) are autoimmune diseases associated with cancer and ectopic expression of a neuronal antigen in a peripheral tumor. Patients with PND harbor high‐titer antibodies and T cells in their serum and cerebrospinal fluid (CSF) that are specific to the tumor antigen, and treatment with the immunosuppressant FK506 (tacrolimus) decreases CSF white blood cell counts. The objective of this study was to determine the effect of FK506 on CSF chemokine levels in PND patients.
Methods
CSF samples before and after FK506 treatment were tested by multiplex assay for the presence of 27 cytokines. Follow‐up in vitro experiments aimed to determine whether T cells secrete CXCL10 in response to cognate antigen.
Results
Here we report that PND patients harbor high levels of the chemokine CXCL10 in their CSF. CXCL10 is a cytokine that recruits CXCR3+ cells such as activated T cells, and we found that FK506 treatment specifically decreased CSF CXCL10 from among 27 cytokines tested. In vitro, CXCL10 was only produced during antigen‐specific cognate interactions between T cells and antigen‐presenting cells (APCs) when interferon‐γ (IFNγ) receptors were present on the T cell.
Interpretation
These results support a model in which antigen‐specific T cell stimulation by PND APCs triggers IFNγ, followed by CXCL10 production and further lymphocyte recruitment, suggesting that treatments targeting T cells or CXCL10 in the central nervous system (CNS) may interrupt a destructive positive feedback loop present in CNS inflammation. Ann Neurol 2015;78:619–629
Paraneoplastic neurologic disorders (PND) are characterized by an immune response against a neuronal antigen that is ectopically expressed by tumors outside the brain. This autoimmune response not only targets the tumor, but also the neurons that normally express the antigen. High‐titer antibodies to the neuronal protein are found in both the serum and cerebrospinal fluid (CSF) of patients with PND.1 However, antibodies are unlikely to be the sole cause of the neuronal destruction for those PNDs in which the target antigens are intracellular.2, 3 Moreover, PND antigen‐specific CD8+ T cells are present in the peripheral blood and CSF of patients with both paraneoplastic cerebellar degeneration (PCD) and the paraneoplastic Hu syndrome subtypes of PND.4, 5, 6
Our laboratory has used FK506 (tacrolimus) in the experimental treatment of patients with PND with the goal of decreasing the number of PND antigen‐specific T cells in the brain and attempting to arrest disease progression. FK506 reduces the number of activated T cells in the CSF of patients with PCD.4 Not all PND patients demonstrate dramatic clinical improvements with this treatment,7 perhaps in part because most patients are diagnosed and referred after the onset of the neuronal destruction that is characteristic of PND.1 FK506, a lipid‐soluble immunosuppressant used to prevent transplant rejection,8 partitions well into the central nervous system (CNS). FK506 interferes with calcineurin phosphatase activity via FK‐binding protein 12, leading to decreased activation of inflammatory genes regulated by the transcription factor NFAT,8, 9, 10 and thereby inhibits T cell function.9
Here we examined cytokine levels in the CSF of PND patients before and after treatment with FK506 after noting recurrent episodes of CSF pleocytosis following FK506 treatments in one PCD patient, suggesting that recruitment of peripheral PCD antigen‐specific T cells to the CSF resumed after treatment. Such CXCL10‐dependent recruitment of T cells into the CNS has been documented previously in mice infected with West Nile virus. In this model, neurons of infected mice generate CXCL10, which recruits CD8+ T cells into the CNS.11 Cytokines and their receptors have also been proposed to help mediate CSF entry of peripheral T cells in multiple sclerosis (MS).12 In particular, the chemokine CXCL10 is elevated in CSF during exacerbations of MS, and both CXCL10 and its receptor, CXCR3, are elevated in and colocalize with active MS lesions.13
When we examined the peripheral blood and CSF of 8 patients for the presence of 27 cytokines, CXCL10 was consistently elevated in the CSF of all patients with PND. Remarkably, CXCL10 was the only cytokine that decreased after FK506 treatment in patients, and correlated with decreases in CSF white blood cell (WBC) counts. We investigated the potential contribution of T cells in the elevation of CXCL10 levels in the CSF, because both were affected by drug treatment. We observed that T cells and antigen‐presenting cells (APCs) produce CXCL10 in an antigen‐specific fashion and that production of this chemokine by T cells required expression of the interferon‐γ receptor (IFNγR) on the T cell. These observations support a model in which a positive feedback develops when antigen‐specific T cells encounter APCs presenting target antigen in the brain, stimulating CXCL10 production, further T cell recruitment, and PND disease progression. Hence, targeting autoimmune T cells and CXCL10 together may help break this cycle, blocking further T cell homing to the CNS, suggesting a new treatment strategy for PND and allied T cell–mediated CNS disorders.
Materials and Methods
Patient Samples
Patients positive for the presence of Hu or Yo antibodies were enrolled in this Rockefeller University Institutional Review Board–approved study at the Rockefeller University Hospital (RDA‐572). Where noted, patients were given FK506 0.15 to 0.3mg/kg/day in 2 divided doses to maintain plasma levels at 5 to 20ng/ml and prednisone (60mg/day, tapered to 2.5mg/day) for 10 to 28 days. Whole blood and CSF samples were obtained before and after treatment. Peripheral blood mononuclear cells and plasma were isolated from peripheral blood by Ficoll separation. CSF WBC counts were determined at the Memorial Sloan–Kettering Cancer Center Hematology laboratory. CSF was placed on ice immediately after lumbar puncture and centrifuged immediately at 4°C to remove cells from the supernatant. Cells were resuspended in culture medium or fluorescence‐activated cell sorting (FACS) buffer. Samples used for cytokine analysis were taken from the third or fourth collection tube. CSF supernatant and plasma were stored in aliquots at −80°C until analysis.
Mouse Experiments
Female C57BL/6 and IFNγR−/− mice were obtained from Jackson Laboratory (Bar Harbor, ME) and were housed in the Rockefeller University specific pathogen‐free animal facility. CXCL10−/− mice were obtained from the laboratory of Dr A. Luster at Massachusetts General Hospital. All animal procedures were performed according to institutional animal care and use committee (IACUC)‐approved animal protocols. Mice were immunized intraperitoneally with influenza A/PR/8 (Charles River Laboratory, Wilmington, MA) 2 weeks before spleens were harvested for isolation of CD4+ and CD8+ T cells by MACS separation (Miltenyi Biotec, Auburn, CA). Dendritic cells (DCs) were generated from bone marrow cells grown in RPMI/10% fetal bovine serum (FBS) with granulocyte‐macrophage colony–stimulating factor (GM‐CSF) from J558L cell culture supernatant.14 Enzyme‐linked immunospot (Elispot) plates (Millipore, Billerica, MA) were coated with and detection was performed with anti‐IFNγ antibody matched pairs (MabTech, Mariemont, OH). Spots were counted in a blinded fashion by Zellnet Consulting (Fort Lee, NJ).
Flow Cytometry
All antibodies were purchased from BD Biosciences/Pharmingen (Franklin Lakes, NJ). Cells were resuspended in FACS buffer (1% FBS/1% pooled human serum in phosphate‐buffered saline) and stained with fluorochrome‐conjugated antibodies for 20 minutes at 4°C, washed 3 times, and analyzed on a Becton Dickinson (Franklin Lakes, NJ) FACSCaliber. Data were analyzed using FlowJo software (TreeStar, Ashland, OR).
Bead‐Based Immunoassay of Cytokines
Samples were analyzed using the Bioplex system (BioRad, Hercules, CA) 27‐plex human cytokine kit according to the manufacturer's instructions. Plasma was diluted 1:5 in the provided buffers, and CSF was assayed without dilution. All patient samples were analyzed in duplicate. Tissue culture supernatants were assayed undiluted and at 1:10. Human cytokines tested were: IL‐2, IL‐4, IL‐6, IL‐8, IL‐10, GM‐CSF, IFNγ, TNFα, IL‐1b, IL‐5, IL‐7, IL‐12(p70), IL‐13, IL‐17, G‐CSF, MCP‐1, MIP‐1β, IL‐1ra, IL‐9, IL‐15, eotaxin, FGF‐basic, IP‐10 (CXCL10), MIP‐1α, PDGF‐bb, RANTES, and VEGF. CSF CXCL10 levels were initially quantified by bead array, and confirmed by human CXCL10 enzyme‐linked immunosorbent assay (ELISA).
ELISA
CSF samples and tissue culture supernatants were tested for the presence of mouse IFNγ and IL‐17 and human and mouse CXCL10 using ELISA kits according to the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Statistics
Cytokine levels before and after FK506 treatment and the percentage of CXCR3+ cells before and after FK506 were compared using the Wilcoxon matched pairs signed rank test, with p < 0.05 considered significant. Percentages of CXCR3+ cells in blood and CSF were compared using an unpaired, 2‐tailed t test assuming unequal variances, with p < 0.05 considered significant.
Declaration of Patient and Animal Study Approvals
The patient studies were approved by the Rockefeller University Institutional Review Board and were conducted at the Rockefeller University Hospital. Written, informed consent was obtained from all patients before they enrolled in the study (RDA‐572). All experiments involving animals were conducted according to a Rockefeller University IACUC‐approved animal protocol and in accordance with the United States Public Health Service's Policy on the Humane Care and Use of Laboratory Animals.
Results
CXCL10 Is Elevated in the CSF of Patients with PND
We screened CSF samples from 8 patients with PND for the presence of 27 cytokines both before and after FK506 treatment using a multiplex bead‐based assay. Patient characteristics are outlined in Table 1. This initial bead‐based screen showed high pretreatment levels of CSF CXCL10 in these patients, so we confirmed this finding by quantifying CSF CXCL10 by ELISA. As shown in Figure 1A (dark bars), all patients with PND had elevated levels of CXCL10 (average = 2,054 pg/ml, range = 347–6,846 pg/ml). The CSF CXCL10 levels in our patients were similar to or in some cases higher than those reported previously in the literature for MS patients and were also higher than values seen in normal CSF.15 Although CSF CXCL10 elevation has been reported in other disease states,16, 17, 18 this is the first report to our knowledge that PND patients have elevated CSF CXCL10. CCL2 was also detected in PND patient CSF pretreatment, but levels were comparable to those reported for normal individuals (data not shown). Four of 8 PND patients had a <2‐fold increase in pretreatment CSF CXCL8 compared to controls (data not shown). Other cytokines tested were either not present or were below the limit of detection of the assay (listed in Materials and Methods).
Table 1.
Paraneoplastic Neurologic Disorder Patient Characteristics
| Patient ID | Age, yr | Antibody | Tumor Type | Neurological Symptoms | Time from Diagnosis, months | CSF WBC/μl | CSF Total Cells Seen (maximum of 100) | CSF Lymphocytes (subset of total cells seen) | CSF RBC/μl |
|---|---|---|---|---|---|---|---|---|---|
| 0312 | 58 | Yo | Ovarian | Cerebellar/brainstem syndrome | 8 | 2 | 38 | 36 | 13 |
| 0611 | 73 | Hu | SCLC | Sensory neuropathy | 6 | 6 | 100 | 88 | None seen |
| 0304 | 57 | Yo | Ovarian | PCD | 8 | None seen | None seen | None seen | 10 |
| 0308 | 61 | Hu | SCLC | Peripheral neuropathy/pancerebellar syndrome | 12 | 2 | 82 | 72 | 1 |
| 0508 | 60 | Yo | Unknown primary | PCD | 1 | 9 | 100 | 94 | None seen |
| 0603 | 74 | Yo | Ovarian and breast | PCD, sensory and motor neuropathy | 3 | 3 | 24 | 14 | 3 |
| 0318 | 59 | Yo | Ovarian | PCD | 5 | 2 | 10 | 10 | 2 |
| 0615 | 45 | Yo | Ovarian | PCD | 1 | 51 | 100 | 87 | 10 |
CSF = cerebrospinal fluid; PCD = paraneoplastic cerebellar degeneration; RBC = red blood cells; SCLC = small cell lung cancer; WBC = white blood cells.
Figure 1.
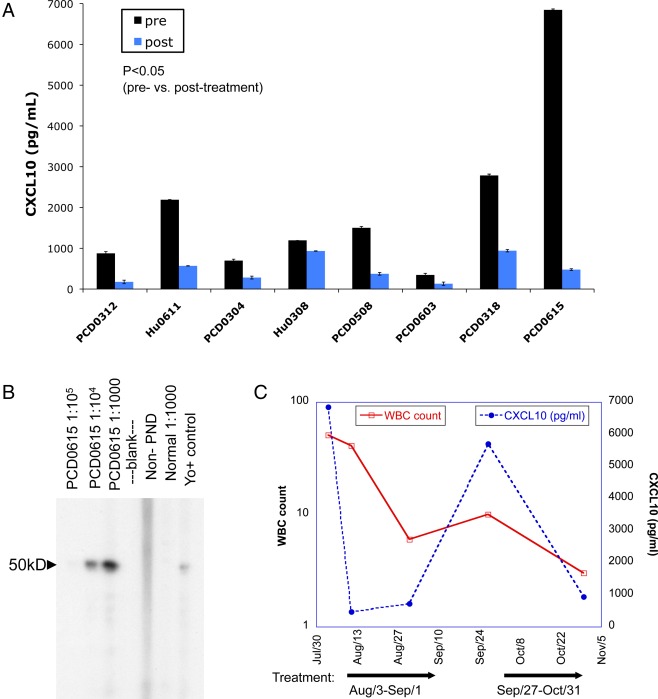
CXCL10 is elevated in paraneoplastic neurologic disorder (PND) patient cerebrospinal fluid (CSF) and is decreased after FK506 treatment. (A) Levels of CXCL10 in the CSF of 8 PND patients with the Hu syndrome or paraneoplastic cerebellar degeneration (PCD) before and after FK506 treatment, as determined by CXCL10 ELISA. Bars represent the average of duplicate wells. p = 0.011 for patients with PND pre‐ versus post‐treatment by Wilcoxon signed rank test with p < 0.05 considered significant, indicating a decrease in CXCL10 with treatment. (B) Western blot of serum from PCD patient PCD0615 recognizing cdr2 (∼50kD). (C) CSF white blood cell (WBC) counts and CXCL10 levels over time in PND patient PCD0615, who was treated with repeated courses of FK506. Arrows indicate treatment periods.
We also measured cytokine and chemokine levels in these patients after FK506 treatment. Remarkably, CXCL10 was the only CSF cytokine measured that was consistently and significantly changed after treatment with FK506 (Fig 1A, dark bars vs light bars, average fold decrease = 4.56; p = 0.011). CCL2 and CXCL8 were not significantly altered by the treatment (average fold decrease = 1.3, p = 0.401 for CCL2; average fold decrease = 0.88, p = 0.207 for CXCL8 before vs after, data not shown). Of note, IL‐17, which is an important autoimmune cytokine in experimental autoimmune encephalomyelitis (EAE) mouse models of MS, was not detected in the CSF of our patients (data not shown). However, we cannot rule out the possibility that IL‐17 might be bound to cell receptors or is otherwise inaccessible to our assay.
We studied the inflammatory response of one patient with Yo antibody‐positive PCD (previously described in detail7; see Fig 1B) over time. This patient was initially seen with relatively mild symptoms of cerebellar dysfunction that had been acutely worsening, and was treated with FK506 for 14 days, with subsequent clinical stabilization. After experiencing periods of symptom stability lasting from weeks to several months, she repeatedly developed episodes of acute symptom exacerbation. During the first two of these episodes, CSF was obtained prior to and following treatment. Analysis of her CSF revealed that during each clinical exacerbation the patient demonstrated a CSF pleocytosis that was reduced to normal or near‐normal levels following FK506 treatment, which paralleled the reduction in CXCL10 levels in her CSF (see Fig 1C). This repeated elevation in CSF WBC suggested that after their elimination from the CSF, peripheral T cells were regaining access to the CNS.
CXCR3+ T Cells Are Enriched in the CSF of PND Patients Compared to Peripheral Blood
CXCL10 is an IFNγ inducible cytokine that stimulates the production and infiltration of inflammatory T cells and other cells expressing the CXCR3 receptor into inflamed sites.19 PND patients typically have abnormally high numbers of WBCs in their CSF.1 Hence, we examined CSF of PND patients for the presence of CXCR3+ T cells. As has been reported in MS patients,13 we observed enrichment of CXCR3+ T cells in PND CSF when compared to the peripheral blood CXCR3+ T cell population (Fig 2). This population was enriched nearly 3‐fold for CD8+ T cells and 4‐fold for CD4+ T cells for CXCR3, relative to peripheral blood T cells. Furthermore, the number of CXCR3+ T cells decreased after FK506 treatment in all 3 patients for whom these data were available, ranging from a nearly 2‐fold decrease in one patient to a >7‐fold reduction in another (Table 2). These results suggest that there may be a role for a specific inflammatory pathway in patients with PND involving the cytokine CXCL10 and its receptor CXCR3 on T cells.
Figure 2.
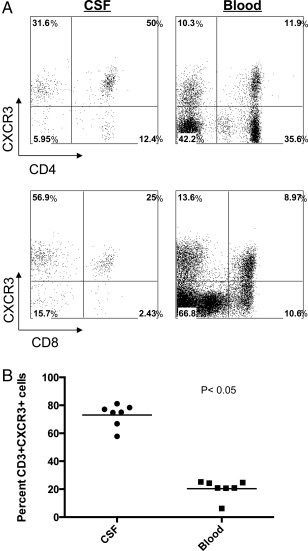
Enrichment of CXCR3+ T cells in paraneoplastic neurologic disorder (PND) patient cerebrospinal fluid (CSF). (A) Flow cytometry analysis of PND patient cells showing CXCR3+CD4+ T cells (top panels) and CXCR3+CD8+ T cells (bottom panels) in CSF and peripheral blood. (B) Percentage of CD3+/CXCR3+ T cells in CSF and peripheral blood from individual PND patients. Percentage of CSF CD3+/CXCR3+ cells mean = 73; percentage of peripheral blood CD3+/CXCR3+ cells mean = 20.4. A t test assuming unequal variance was applied to the data with p < 0.05 considered significant. p = 2e10−8 for CSF versus peripheral blood CXCR3+ cells.
Table 2.
Number of CSF CXCR3+ T Cells per Microliter
| Patient | Pre‐FK506 | Post‐FK506 |
|---|---|---|
| Hu0308 | 1.57 | 0.83 |
| PCD0318 | 2.25 | 0.75 |
| PCD0615 | 35.19 | 4.68 |
T Cells from Normal Donors Produce CXCL10 in an Antigen‐Specific Manner
Based on the parallel decreases in patient CSF T cells and CXCL10 after FK506 treatment, we sought to determine whether T cells could produce CXCL10 and thus implicate them in production of the CXCL10 detected in the PND patient CSF. Previous work by our laboratory has shown that peripheral CD8+ T cells specific for the HuD PND antigen express CXCL10.6 Due to insufficient patient CSF T cells available for direct study, we modeled this question by assessing CXCL10 production from normal donor T cells upon encounter with specific antigen presented by DCs as APCs. CD4+ or CD8+ T cells were cocultured with DCs loaded with apoptotic influenza‐infected 3T3 cells (for CD4+) or DCs directly infected with influenza virus (for CD8+). Analysis of supernatants from the cultures revealed that CXCL10 was only produced after a cognate CD4+ or CD8+ T cell–DC interaction (Fig 3A). CXCL10 secretion was coincident with IFNγ production, which was measured as a positive control (Fig 3B). These data indicate that CXCL10 is produced after an interaction between antigen‐specific T cells and an APC but not after noncognate interactions.
Figure 3.
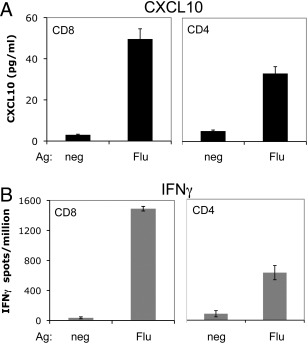
CXCL10 is produced in an antigen (Ag)‐specific fashion in cultures of human T cells and dendritic cells (DCs) from normal donors. (A) CXCL10 was measured by ELISA in supernatants from cocultures of CD8+ or CD4+ T cells and DCs presenting influenza antigen (Flu) or uninfected DCs (neg). (B) Interferon‐γ (IFNγ) Elispot assay of normal donor CD8+ and CD4+ T cells cultured with DCs presenting influenza antigen, performed in parallel with the experiment in A. Error bars represent standard deviation from the mean of triplicate wells for each condition.
Production of CXCL10 in Mouse T Cell and DC Cocultures Is Antigen Specific and Requires the IFNγR on the T Cell
To assess whether these antigen‐specific interactions led to CXCL10 production by T cells only, APCs only, or both cell types, we employed an assay similar to the human cell assay just described, but instead using cells from wild‐type or CXCL10 null mice (Fig 4 A‐D). T cells from influenza‐immunized mice were cocultured with DCs pulsed with relevant influenza nucleoprotein peptide or an irrelevant control peptide. Production of CXCL10 in wild‐type conditions was found to be antigen‐specific and paralleled production of IFNγ, as was seen with human T cells. Similar results were obtained using T cells from ovalbumin‐immunized mice (not shown). When influenza‐specific CXCL10−/− CD4+ or CD8+ T cells were cocultured with wild‐type DCs presenting antigen, the number of IFNγ‐producing cells present was similar to the number of IFNγ‐producing cells in the wild‐type T cell condition. However, when CXCL10−/− T cells were cocultured with wild‐type DCs, 3‐fold less CXCL10 was detected. In comparison, no CXCL10 was detected when CXCL10−/− T cells were cocultured with CXCL10−/− DC. Similar data were obtained using T cells from ovalbumin protein‐immunized rather than virus‐immunized CXCL10−/− mice (data not shown). Taken together, these results indicate that T cells produce the majority of CXCL10 in this assay and that they do so in an antigen‐specific manner. DCs themselves also contribute to the production of CXCL10 after encounter with a cognate T cell in this assay, because a low level of CXCL10 is still present in the CXCL10−/− T cells plus wild‐type DC condition.
Figure 4.
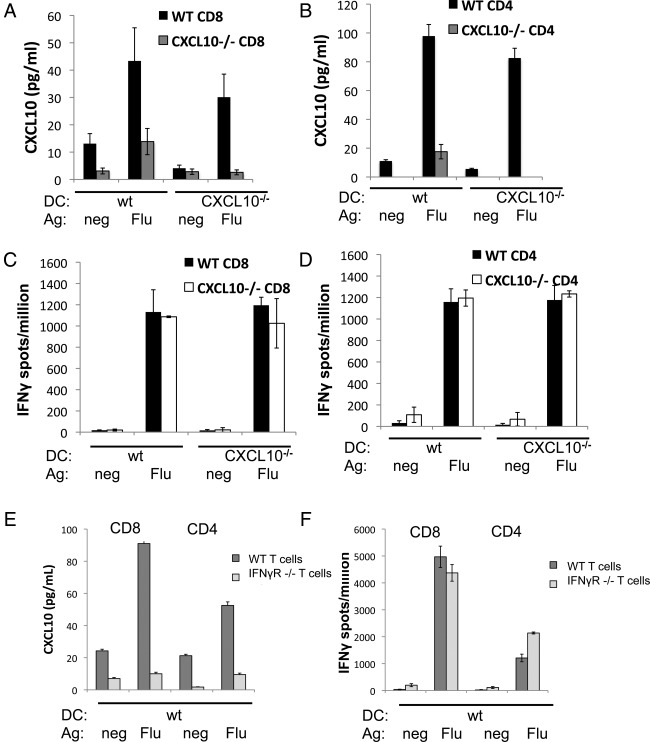
T cell interferon‐γ (IFNγ) receptor is required for antigen (Ag)‐specific production of CXCL10 in cocultures of T cells and dendritic cells (DCs). (A–D) CXCL10 (A, B) and IFNγ (C, D) were measured in cocultures of T cells from influenza‐immunized CXCL10−/− or wild‐type (wt) C57BL/6 mice with DC from CXCL10−/− or wild type mice. For CD8+ cocultures (A, C), DCs were pulsed with influenza nucleoprotein peptide (Flu) or ovalbumin peptide control (neg). For CD4+ cocultures (B, D), DCs were fed apoptotic influenza‐infected 3T3 cells (Flu) or uninfected 3T3 cells (neg). (E) CXCL10 measured in cocultures set up as in A, using T cells from influenza‐immunized IFNγR−/− mice. (F) IFNγ production was determined in parallel. Each experiment was repeated at least 3 times with similar results. CXCL10 in coculture supernatants was measured by ELISA and IFNg secretion was measured by Elispot.
DC production of CXCL10 in an antigen‐specific manner suggests the presence of a signal from T cells that have encountered their cognate antigen to the DC upon which the antigen is presented. One possible mediator of such a signal from the T cells is IFNγ, because CXCL10 is known to be IFNγ inducible20 and both cytokines were produced in an antigen‐specific manner in our cocultures. To address this possibility, we assessed CXCL10 production following antigen‐specific T cell–DC interactions using cells from mice lacking the receptor for IFNγ (IFNγR−/−). Both IFNγ and CXCL10 were produced when wild‐type T cells encountered DCs presenting influenza antigen. However, when IFNγR−/− T cells encountered wild‐type DCs presenting influenza antigen, CXCL10 was not detected despite detection of expected levels of IFNγ (Fig 4E, F). These data support the idea that the IFNγR is required on the T cell for antigen‐specific CXCL10 production.
Discussion
Our study began with a remarkable observation of a high CXCL10 concentration in the CSF of an acutely ill PCD patient that was decreased dramatically after treatment with the immunosuppressive drug FK506. A second course of FK506 treatment in the same patient resulted in a similar decrease in CSF CXCL10 (Fig 1C). We subsequently found that CXCL10 was decreased after FK506 treatment in 7 other PND patients (Fig 1A, p = 0.011). Importantly, 2 other chemokines also present in the CSF, CCL2 and CXCL8, were unaffected by the drug treatment in the same patients (p = 0.401 and p = 0.207, respectively). Moreover, the levels of 24 other cytokines (listed in Materials and Methods) were either undetectable or very similar to normal CSF, and were unaffected by FK506 treatment. The specificity of the effect of immunosuppressive therapy suggested a strong and potentially clinically relevant link to CNS inflammation in these patients that was pursued to discover a specific positive feedback loop. In this model (Fig 5), antigen‐specific T cell stimulation by APCs within the CNS triggers T cell IFNγ production, followed by CXCL10 secretion by both T cells and APCs, leading to further lymphocyte recruitment to the area of inflammation.
Figure 5.
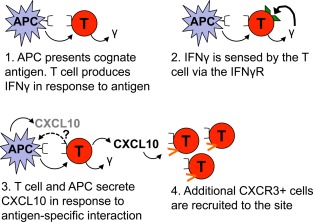
Proposed model of the interactions leading to CXCL10 production by T cells and antigen‐presenting cells (APCs). The T cell is activated by its cognate antigen presented on an APC. The T cell secretes interferon‐γ (IFNγ) in response to the antigen and senses autocrine or paracrine IFNγ via the IFNγ receptor (IFNγR). The T cell and APC secrete CXCL10 in this antigen‐specific interaction, which recruits additional CXCR3+ cells into the area. The signal that tells the APC to produce CXCL10 was not investigated in the current experiments, but may be related to expression of the IFNγR by APCs or other signals.
Many cell types in the brain are able to produce CXCL10, such as MS patient astrocytes,13 neurons,21 and microglia.22 In addition, a correlation between CSF CXCL10 levels and CSF leukocyte count has previously been observed in MS.15 Here, we hypothesized that a key source of CXCL10 in PND patient CSF might be T cells, given that both T cells and CXCL10 were elevated in CSF from PND patients (see Table 1 and Fig 1), and that we have previously seen CSF PND antigen‐specific T cell responses in these patients.5, 6, 23 In support of this idea, we found that CXCL10 can be produced by both human and mouse T cells, and that this production occurs specifically upon encounter with cognate antigen (Figs 3 and 4).
Interestingly, when we used CXCL10−/− T cells in this assay we found that a low level of CXCL10 production persisted in response to antigen, and we attribute this production to the APCs (Fig 4A, B). This result supports the possibility that the APCs are receiving antigen‐specific signals from the T cells during their interaction, inducing them to elaborate the same chemokine, a potential component of the positive feedback loop (Fig 5) that would be location specific. The additional release of CXCL10 would further increase recruitment of T cells to the local area. A recently published study demonstrated that CXCL10 production by DC is important in DC–T cell interactions during priming of T cells in the lymph node.24 It is possible that CXCL10 production by APCs is similarly important for reactivation and further recruitment of memory T cells in the brain in PND, and that CXCL10 production by responding T cells augments the response.
It has been shown previously that microglia are able to produce CXCL10 and express its receptor CXCR3.25 Our observation of CXCL10 production by antigen‐specific T cells uncovers a new, potentially clinically relevant dimension to T cell inflammation in the brain. It has been shown previously that CXCL10‐deficient mice infected with mouse hepatitis virus show decreased migration of effector T cells to the brain.19 In light of our observation that CXCL10 production is antigen specific, and given cells capable of presenting antigen within the brain,13, 26 we suggest that PND T cells encountering antigen in the CNS may be the source of the high CXCL10 levels in CSF from patients with PND.
A leading candidate for mediating the signal for CXCL10 production between the APC and the T cell was the IFNγR, because IFNγ is known to induce CXCL10 secretion.20 When T cells lacking the IFNγR were cocultured with DC, antigen‐specific production of CXCL10 was completely obliterated, whereas IFNγ production remained unchanged (Fig 4E, F). This suggests that there is a T cell–intrinsic feedback loop for CXCL10 production, and that the low level of CXCL10 produced by the DCs may require the IFNγR‐mediated T cell feedback loop as well (Fig 5). CXCL10 and the CXCR3 receptor have been proposed to work in an inflammation‐promoting loop in allergy27 and to modulate IFNγ production in EAE.28 CXCR3 has been suggested not only to play a role in T cell priming in the lymph node24 but also in the recruitment of T cells into peripheral target tissues.29 Taken together, these data suggest that the high concentration of CXCL10 present in PND patient CSF may be due to production of this chemokine by infiltrating autoimmune T cells interacting with APCs in the CNS.30 However, EAE can be induced in CXCL10−/− mice, indicating that this chemokine is not essential for the development of this MS‐like disease in mice.30
It is important to note that previous in vivo studies of the inflammatory response to CNS virus infection and chronic EAE have demonstrated that astrocytes are a major source of CXCL10.13, 31, 32 Our observations of CXCL10 production by DCs and T cells in an antigen‐specific in vitro system do not rule out the possible additional production of CXCL10 by astrocytes or other cells in patients with PND. It may even be that one cell type is responsible for CXCL10 production early during disease initiation, and other cell types such as T cells and APCs participate in CXCL10 production to propagate the disease state. This could lead to recruitment of additional T cells into the CNS in general or into the parenchyma.
In a clinical study, 10 of 16 of our PND patients treated with FK506 showed improvement of their symptoms,7 which when combined with observed decreases in CSF WBC count and CXCL10 levels reported here, suggests that this treatment strategy warrants further study, particularly if used early in the disease. In addition, the specific mechanism by which FK506 decreases CXCL10 in PND patient CSF is of interest. Although the inhibition of calcineurin activity in T cells is a known effect of FK506, additional intracellular targets such as p38 and c‐Jun N‐terminal kinase have been suggested.33 Interestingly, our laboratory has recently shown that FK506 preferentially accumulates in DCs, thereby targeting T cells engaged in antigen‐specific T cell–APC interactions.34 It is possible that FK506 may similarly act upon APCs in the brain. Inhibition of the EAE animal model of MS has been observed with FK506 treatment.35, 36 It would be interesting to determine whether treatment of EAE or MS patients with FK506 decreases CXCL10 levels in a fashion similar to that shown in our PND patients. A limitation of our study is that patients were treated with both FK506 and a tapered prednisone dose. Thus, it is possible that some of the effect of treatment on CSF CXCL10 may be due to prednisone. However, based on studies by others,37, 38 it is reasonable to expect FK506 to be responsible for most or all of the effect.
In conclusion, we have shown a significant effect of FK506 immunosuppressive therapy on the important inflammatory chemokine CXCL10 in patients with PND. As elevated levels of CXCL10 are also found in other autoimmune neurological disorders, the presence of this chemokine in inflammatory CSF is significant and may be directly involved in the autoimmune response. It is not known whether the arrival of T cells or peripheral APCs in the CNS or another process altogether is the initiating event for diseases such as PND. Interestingly, Bailey and colleagues have shown that CNS myeloid DCs present endogenous peptide to T cells in the brain to drive EAE relapses in mouse models.39 The mechanism of immunosuppression and CXCL10 reduction in PND patients treated with FK506 may include decreased recruitment of T cells and/or APC from the periphery to the CNS, or interruption of the interaction of these two cell types or their intracellular signaling. Inhibition of T cell access to the brain might consequently slow the progression of disease, and may argue for combination therapy with agents that prevent T cell access to the CNS.
Authorship
W.K.R. designed and performed the experiments, analyzed the data, and wrote the manuscript. N.E.B. helped with designing and performing mouse experiments, data analysis, and writing the manuscript. M.O.F. recruited, enrolled, and followed patients in the study, obtained patient samples, and helped with patient data analysis and writing the manuscript. A.D. recruited, enrolled, and followed patients in the study, obtained patient samples, and edited the manuscript. R.M.R. provided advice about chemokine analysis and experiments and edited the manuscript. R.B.D. supervised the project, helped with the design of experiments and data analysis, followed patients in the study, and wrote the manuscript.
Potential Conflicts of Interest
R.M.R.: employment, Biogen.
Acknowledgment
This study was supported by grants UL1RR024143 8 and KL2TR000151 from the NIH National Center for Research Resources and National Center for Advancing Sciences to the Rockefeller University Hospital. R.B.D. is an Investigator of the Howard Hughes Medical Institute.
We thank the patients with paraneoplastic neurologic disorders who participated in this study and their families; the Rockefeller University Hospital staff for facilitating patient studies; Dr J. Posner for referring many of the patients; Dr A. Luster for kindly providing CXCL10−/− mice; Dr C. Tu for statistics advice; and members of our laboratory for many helpful discussions of this work.
References
- 1. Darnell RB, Posner JB. Paraneoplastic syndromes affecting the nervous system. Semin Oncol 2006;33:270–298. [DOI] [PubMed] [Google Scholar]
- 2. Darnell RB. Onconeural antigens and the paraneoplastic neurologic disorders: at the intersection of cancer, immunity, and the brain. Proc Natl Acad Sci U S A 1996;93:4529–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roberts WK, Darnell RB. Neuroimmunology of the paraneoplastic neurological degenerations. Curr Opin Immunol 2004;16:616–622. [DOI] [PubMed] [Google Scholar]
- 4. Albert ML, Austin LM, Darnell RB. Detection and treatment of activated T cells in the cerebrospinal fluid of patients with paraneoplastic cerebellar degeneration. Ann Neurol 2000;47:9–17. [PubMed] [Google Scholar]
- 5. Albert ML, Darnell JC, Bender A, et al. Tumor‐specific killer cells in paraneoplastic cerebellar degeneration. Nat Med 1998;4:1321–1324. [DOI] [PubMed] [Google Scholar]
- 6. Roberts WK, Deluca IJ, Thomas A, et al. Patients with lung cancer and paraneoplastic Hu syndrome harbor HuD‐specific type 2 CD8+ T cells. J Clin Invest 2009;119:2042–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Orange D, Frank M, Tian S, Dousmanis A, et al. Cellular immune suppression in paraneoplastic neurologic syndromes targeting intracellular antigens. Arch Neurol 2012;69:1132–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schreiber S, Crabtree G. The mechanism of action of cyclosporin A and FK506. Immunol Today 1992;13:136–142. [DOI] [PubMed] [Google Scholar]
- 9. Sawada S, Suzuki G, Kawase Y, Takaku F. Novel immunosuppressive agent, FK506. In vitro effects on the cloned T cell activation. J Immunol 1987;139:1797–1803. [PubMed] [Google Scholar]
- 10. Snyder SH, Lai MM, Burnett PE. Immunophilins in the nervous system. Neuron 1998;21:283–294. [DOI] [PubMed] [Google Scholar]
- 11. Klein RS, Lin E, Zhang B, et al. Neuronal CXCL10 directs CD8+ T cell recruitment and control of West Nile virus encephalitis. J Virol 2005;79:11457–11466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Engelhardt B, Ransohoff RM. The ins and outs of T‐lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol 2005;26:485–495. [DOI] [PubMed] [Google Scholar]
- 13. Sørensen T, Trebst C, Kivisakk P, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co‐localization in the inflamed central nervous system. J Neuroimmunol 2002;127:59–68. [DOI] [PubMed] [Google Scholar]
- 14. Blachere NE, Darnell RB, Albert ML. Apoptotic cells deliver processed antigen to dendritic cells for cross‐presentation. PLoS Biol 2005;3:e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sorensen TL, Tani M, Jensen J, et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest 1999;103:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang CC, Omarjee S, Lim A, et al. Chemokine levels and chemokine receptor expression in the blood and the cerebrospinal fluid of HIV‐infected patients with cryptococcal meningitis and cryptococcosis‐associated immune reconstitution inflammatory syndrome. J Infect Dis 2013;208:1604–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kamat A, Lyons JL, Misra V, et al. Monocyte activation markers in cerebrospinal fluid associated with impaired neurocognitive testing in advanced HIV infection. J Acquir Immune Defic Syndr 2012;60:234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Henningsson AJ, Tjernberg I, Malmvall BE, et al. Indications of Th1 and Th17 responses in cerebrospinal fluid from patients with Lyme neuroborreliosis: a large retrospective study. J Neuroinflammation 2011;8:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dufour JH, Dziejman M, Liu MT, et al. IFN‐gamma‐inducible protein 10 (IP‐10; CXCL10)‐deficient mice reveal a role for IP‐10 in effector T cell generation and trafficking. J Immunol 2002;168:3195–3204. [DOI] [PubMed] [Google Scholar]
- 20. Luster AD, Ravetch JV. Biochemical characterization of a gamma interferon‐inducible cytokine (IP‐10). J Exp Med 1987;166:1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang J, Campbell IL. Innate STAT1‐dependent genomic response of neurons to the antiviral cytokine alpha interferon. J Virol 2005;79:8295–8302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balashov KE, Rottman JB, Weiner HL, Hancock WW. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP‐1alpha and IP‐10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A 1999;96:6873–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Santomasso BD, Roberts WK, Thomas A, et al. A T cell receptor associated with naturally occurring human tumor immunity. Proc Natl Acad Sci U S A 2007;104:19073–19078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Groom JR, Richmond J, Murooka TT, et al. CXCR3 chemokine receptor‐ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 2012;37:1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flynn G, Maru S, Loughlin J, et al. Regulation of chemokine receptor expression in human microglia and astrocytes. J Neuroimmunol 2003;136:84–93. [DOI] [PubMed] [Google Scholar]
- 26. Millward JM, Caruso M, Campbell IL, et al. IFN‐gamma‐induced chemokines synergize with pertussis toxin to promote T cell entry to the central nervous system. J Immunol 2007;178:8175–8182. [DOI] [PubMed] [Google Scholar]
- 27. Gangur V, Simons FE, Hayglass KT. Human IP‐10 selectively promotes dominance of polyclonally activated and environmental antigen‐driven IFN‐gamma over IL‐4 responses. FASEB J 1998;12:705–713. [DOI] [PubMed] [Google Scholar]
- 28. Liu L, Huang D, Matsui M, et al. Severe disease, unaltered leukocyte migration, and reduced IFN‐gamma production in CXCR3‐/‐ mice with experimental autoimmune encephalomyelitis. J Immunol 2006;176:4399–4409. [DOI] [PubMed] [Google Scholar]
- 29. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res 2011;317:620–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klein RS, Izikson L, Means T, et al. IFN‐inducible protein 10/CXC chemokine ligand 10‐independent induction of experimental autoimmune encephalomyelitis. J Immunol 2004;172:550–559. [DOI] [PubMed] [Google Scholar]
- 31. Park C, Lee S, Cho IH, et al. TLR3‐mediated signal induces proinflammatory cytokine and chemokine gene expression in astrocytes: differential signaling mechanisms of TLR3‐induced IP‐10 and IL‐8 gene expression. Glia 2006;53:248–256. [DOI] [PubMed] [Google Scholar]
- 32. Glabinski AR, Tani M, Strieter RM, et al. Synchronous synthesis of alpha‐ and beta‐chemokines by cells of diverse lineage in the central nervous system of mice with relapses of chronic experimental autoimmune encephalomyelitis. Am J Pathol 1997;150:617–630. [PMC free article] [PubMed] [Google Scholar]
- 33. Matsuda S, Shibasaki F, Takehana K, et al. Two distinct action mechanisms of immunophilin‐ligand complexes for the blockade of T cell activation. EMBO Rep 2000;1:428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Orange DE, Blachere NE, Fak J, et al. Dendritic cells loaded with FK506 kill T cells in an antigen‐specific manner and prevent autoimmunity in vivo. Elife 2013;2:e00105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gold BG, Voda J, Yu X, et al. FK506 and a nonimmunosuppressant derivative reduce axonal and myelin damage in experimental autoimmune encephalomyelitis: neuroimmunophilin ligand‐mediated neuroprotection in a model of multiple sclerosis. J Neurosci Res 2004;77:367–377. [DOI] [PubMed] [Google Scholar]
- 36. Inamura N, Hashimoto M, Nakahara K, et al. Immunosuppressive effect of FK506 on experimental allergic encephalomyelitis in rats. Int J Immunopharmacol 1988;10:991–995. [DOI] [PubMed] [Google Scholar]
- 37. Chang KT, Lin HY, Kuo CH, Hung CH. Tacrolimus suppresses atopic dermatitis‐associated cytokines and chemokines in monocytes. J Microbiol Immunol Infect. http://dx.doi.org/10.1016/j.jmii.2014.07.006 (in press). [DOI] [PubMed] [Google Scholar]
- 38. Cristillo AD, Macri MJ, Bierer BE. Differential chemokine expression profiles in human peripheral blood T lymphocytes: dependence on T cell coreceptor and calcineurin signaling. Blood 2003;101:216–225. [DOI] [PubMed] [Google Scholar]
- 39. Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides 'preferentially' polarize CD4+ T(H)−17 cells in relapsing EAE. Nat Immunol 2007;8:172–180. [DOI] [PubMed] [Google Scholar]