

Abstract
Objective: To investigate the roles of macrophage stimulating protein (MSP) and its tyrosine kinase receptor RON in smoke-induced airway inflammation of rats. Methods: Inhalation of combustion smoke was administered in rats to induce airway inflammation. Alveolar macrophages (AM) of healthy and smoking rats were isolated at different time points, cultured and then treated with different concentrations of MSP for 24 h. Results: When compared with healthy rats, MSP increased in the serum and bronchoalveolar lavage fluid (BALF) of smoking rats in a time dependent manner. In smoking rats, the RON expression in the lung and AM was higher than in healthy rats, and these increases were time dependent. MSP stimulated the production of malondialdehyde (MDA) and reduced superoxide dismutase (SOD) activity in rat AM cells in a dose dependent manner. MSP also stimulated the release of inflammatory factors TNF-α, IL-8, IL-1β and IL-10 in rat AM in a dose-dependent manner. Moreover, at the same MSP concentration, the contents of MDA, TNF-α, IL-8 and IL-1β in the AM of smoking rates were higher than in healthy rats, while the IL-10 content and SOD activity were lower than in healthy rats. Conclusion: MSP and its receptor RON are involved in the smoke-induced airway inflammation in rats via promoting AM to release inflammatory cytokines and inducing the increase of oxygen free radical.
Keywords: Macrophage stimulating protein, alveolar macrophage, inflammatory factor
Introduction
Chronic obstructive pulmonary disease (COPD) is a disease characterized by chronic persistent airflow limitation that is usually progressive and is associated with chronic inflammation of the lungs [1,2]. The pathogenesis of COPD has involvement of a variety of inflammatory cells. Alveolar macrophages (AM) may secrete a lot of inflammatory cytokines to initiate and regulate the local immune inflammatory response [3,4]. Studies have confirmed that AM plays important roles in the pathogenesis of COPD [5]. Macrophage stimulating protein (MSP), also known as hepatocyte growth factor-like protein [6], is a kind of glycoprotein synthesized in the liver and has immunoregulatory activity. MSP may bind to its specific receptor tyrosine kinase RON to stimulate the macrophages to regulate macrophage motility and its phagocytic activity [7] and induce the production of oxygen free radicals and cytokines, inducing the activation of NF-κB and regulating cell differentiation, migration and matrix invasion [8,9]. Studies have found that MSP at high concentrations are detectable in the bronchial alveolar lavage fluid (BALF) and induced sputum of patients with bronchiectasis [10,11]. Brunelleschi et al. [12] also found that tyrosine receptor kinase RON was expressed on the AM of patients with lung cancer, pulmonary fibrosis and pulmonary sarcoidosis. However, no study has been conducted to investigate the roles of MSP/RON in the airway inflammatory diseases. This study was undertaken to detect the MSP concentration in the serum and BALF of rats with smoke inhalation, the expressions of tyrosine kinase receptor RON in the lung, and the contents of IL-1β, TNF-α, IL-8, IL-10, malondialdehyde (MDA) as well as superoxide dismutase (SOD) activity in the supernatant of AM following MSP treatment. This study aimed to evaluate the roles of MSP/RON in the pathogenesis of smoke-induced airway inflammation.
Materials and methods
Materials and reagents
Male Wistar rats (Animal Center of North Sichuan Medical College); Hong Mei cigarette (Yuxi cigarette factory, tar: 12 mg, nicotine: 1.1 mg, carbon monoxide [CO] content in smudge: 13 mg); fetal bovine serum (America Hyclone company); Wright-Giemsa dye (Nanjing Jiancheng limited company of science and Technology); low glucose DMEM medium (American Hyclone company), recombinant human MSP (American R&D company); MSP and IL-8 detection kits (Wuhan Xinqidi company packaged); IL-1β, TNF-α and IL-10 detection kits (Neobioscience); superoxide dismutase (SOD) and malondialdehyde (MDA) detection kits (Nanjing Jiancheng Technology Co. Ltd.); primers (Shanghai SANGON Biotech Corp); reverse transcription kit (Chengdu B-ray grams Biotechnology Co. Ltd.); PCR reaction system (Tiangen Biotech Co. Ltd.); mouse anti human RON β (E-3) monoclonal antibody (American SANTA company); RON immunohistochemistry kit (Neobioscience).
Establishment of airway inflammation rat model and grouping
A total of 32 male Wistar rats weighing 180-200 g were randomly divided into (1) normal control group, (2) 1-month smoking group, (3) 2-month smoking group and (4) 3-month smoking group. Rats were given ad libitum access to food and water. Rats receiving smoke inhalation were place into self-made plexiglass boxes sized 60 cm × 60 cm × 35 cm and inhalation of combustion smoke was administered. Ten cigarettes were burned within 1 h at each smoke inhalation which was performed once daily.
Preparation of serum and BALF
Rats were intraperitoneally injected with 1% pentobarbital sodium at 30 mg/kg for anesthesia, 5 ml of blood was collected from the inferior vena cava and centrifuged (3000 r/min, 20 min), and the serum was collected and stored -80°C. The trachea and lungs were exposed. Then, the right main bronchus were clamped, and the left lung was lavaged with 15 ml of normal saline at 37°C 4 times. The recovery rate of each lavage fluid was 90%-95%. The lavage was filtrated through a 200-mesh sterile and monolayer cell strainer, and the lavage fluid was harvested and centrifuged at 1500 r/min for 5 min at 4°C. The supernatant was collected and stored at -80°C for use.
Preparation of lung samples
The left lungs were put into liquid nitrogen for RNA extraction. The right lungs were fixed in 4% paraformaldehydefor 24 h and embedded in paraffin, and sections were prepared for HE staining and immunohistochemistry.
Isolation, culture and processing of AM
After centrifugation of the BALF, the precipitated cells were washed with PBS and then maintained in phenol red free DMEM medium (low glucose) containing 10% FBS, 50 μg/ml streptomycin and 5 U/ml penicillin. Then, single cell suspension was prepared. After Wright-Giemsa staining, cells were counted (at least 200 cells) and cell viability was determined after trypan blue staining. The purity of AM was > 95%, and the survival rate was > 98%. The cell density was adjusted to 1 × 109/L and then seeded into 6-wellplates containing coverslips. Two hours later, cells were adherent to the bottom. AMs were randomly divided into blank control group (PBS) and MSP groups (high [500 ng], median [50 ng] and low [5 ng] doses). Two hours later, 200 μL of cell supernatant was collected and centrifuged at 1000 r/min for 10 min. The supernatant was stored at -80°C for further detections of SOD activity and MDA content. Then, AMs were further cultured up to 24 h. The remaining cell supernatant was collected and centrifuged at 1000 r/min for 10 min, and the supernatant was collected and stored at -80°C for further detections of cytokines. After cell supernatant was collected, the plate was washed with PBS, and cells in the plate were fixed in 4% paraformaldehyde at room temperature. The coverslips were harvested, washed in PBS twice, and stored at -20°C after air-drying.
Detection of MSP concentration
MSP detection kit was used to determine the concentration of MSP in the BALF and serum, and corresponding detection kits were used to detect the concentrations of IL-1β, TNF-α, IL-8 and IL-10 in the supernatant.
Detection of SOD activity and MDA content in the supernatant of cells
The SOD activity was determined with xanthine oxidase colorimetric method; MDA content was measured by thiobarbituric acid (TBA) assay. Determination and calculation were conducted according to the manufacturer’s instructions.
Detection of RON mRNA expression
RT-PCR was used to detect the mRNA expression of RON in the lung, and the internal control was glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Primers used for PCR were as follows: RON forward: 5-GAGAGCCTTCAGACCTACAGA-3’, reverse: 5’-TTGACGTGCTCCTGTGATGCA-3’, length: 400 bp; GAPDH: forward: 5’-ACCACAGTCCATGCCATCAC-3’, reverse: 5-TCCACCACC CTGTTGCTGTA-3’, length: 450 bp.
Immunohistochemical staining
Streptavidin peroxidase (SP) conjugated method was used for immunohistochemistry of adherent AMs and lung sections according to the manufacturer’s instructions. Mouse anti human RON β (E-3) monoclonal antibody (1:20) was used. For the immunohistochemistry of lung sections, antigen retrieval was performed with a microwave oven. Visualization was done with DAB and counterstaining with hematoxylin. Positive cells had brown granules.
Five fields were randomly selected from each section at a high magnification and captured (OLYMPUS DP70). The optical density was analyzed with the IMAGE-PRO plus 6 image analysis software, and an average was calculated.
Statistical analysis
Data are expressed as means ± standard deviation (SD) when normal distribution was identified. One-way analysis of variance (ANOVA) was employed for intergroup comparisons among groups, and the SNK method was used for post hoc multiple comparisons. Factorial analysis was used to analyze the interaction of two factors. Linear correlation analysis was applied to evaluate the relationship between different parameters. A value of P < 0.05 was considered statistically significant.
Results
Pathological changes in rat lungs
Light microscopy showed that, in normal control group, the mucosal columnar epithelial cells of the trachea and bronchi were abundant, ciliated cells were arranged regularly, scattered goblet cells were seen between ciliated cells, and there were a small amount of lymphocytes along the inner wall of the pipe (Figure 1A and 1B). In 1-month smoking rats, there were hyperemia, edema and infiltration of inflammatory cells at different levels of the trachea and bronchi, and degeneration, necrosis and shedding were also observed in some epithelial cells, the increase in goblet cells was not obvious, and there were no obvious hyperplasia and hypertrophy of vascular smooth muscle (Figure 1C and 1D). While in 3-month smoking rats, there were infiltration of inflammatory cells (lymphocytes, plasma cells, neutrophils and monocytes) to different extents in the trachea and bronchi at different levels. The degeneration, necrosis and shedding of bronchial ciliated epithelial cells were also observed. Goblet cells increased significantly, the cell body enlarged and there was abundant mucin. Bronchial smooth muscle was thickening. There were many secretions, macrophages and neutrophils in the lumen, and widespread emphysema was observed in the tissues surrounding the lungs, accompanied by disordered alveolar structure, alveolar wall thinning or fracture and expansion of the alveolar cavity. Some alveoli merged and form pulmonary bulla (Figure 2). Compared with 3-month smoking rats, pathological changes in 2-month smoking group were relatively mild (Figure 1E and 1F).
Figure 1.
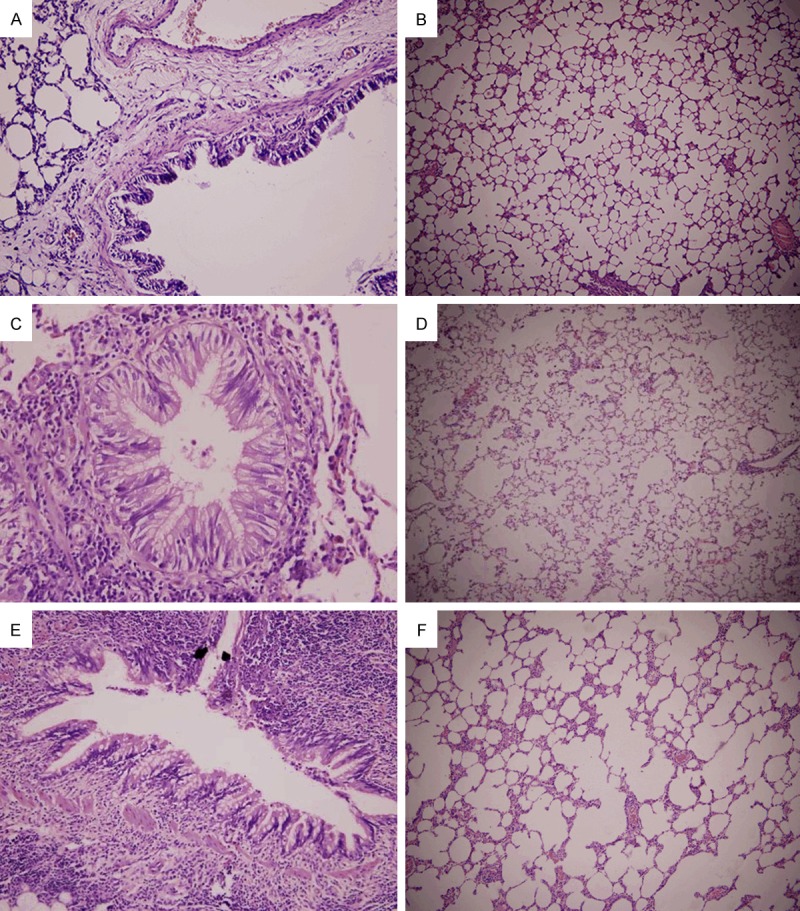
The lungs of normal control group. A. The bronchial columnar epithelial cells were intact, cilia were arranged regularly, scattered goblet cells were observed among ciliated columnar epithelial cells, and a small amount of lymphocytes was noted in the bronchial wall (HE staining; × 200). B. Normal alveoli (HE staining; × 100). Rats received smoke inhalation for 1 month. C. The bronchial ciliated columnar epithelial cells were nearly normal, the cilia were lodging, the shedding was observed in a small amount of epithelial cells, goblet cells slightly increased, and obvious lymphocyte infiltration was noted on the wall (HE staining; × 400). D. The alveolar structure was normal, and infiltration of a few macrophages was observed in the alveolar cavity (HE staining; × 100). Rats received smoke inhalation for 2 months. E. The bronchial epithelium goblet cells increased when compared with 1-month smoke inhalation group, the goblet cell body enlarged and there was abundant mucin, and the bronchial smooth muscle was thickening. (HE staining; × 400). F. The alveolar structure was destructed, the alveolar cavity expanded, the alveolar wall was thinning and fractured, infiltration of inflammatory cells was found in part of alveolar wall, and the alveolar number significantly reduced when compared with 1-month smoke inhalation group. (HE staining; × 100).
Figure 2.
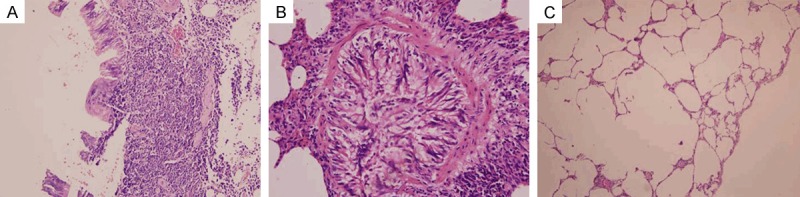
Rats received smoke inhalation for 3 months. A. The bronchial epithelial breakage, epithelial cell exfoliation, and infiltration of numerous inflammatory cells into the mucosa, submucosa and muscular layer were observed (HE staining × 200). B. The bronchial epithelial cilia were lodging, adherent and shedding, goblet cell body enlarged and there was abundant mucin. Bronchial smooth muscle was thickening and there were abundant secretions in the lumen (HE staining, × 400). C. Emphysema, disordered alveolar structure, alveolar wall thinning or fracture, expansion of the alveolar cavity, alveoli merging to form pulmonary bulla were also observed (HE staining, × 100).
Total cells and AM in the BALF
As shown in Table 1, the total cells and AM in the BALF increased gradually with the prolongation of smoke inhalation. Total cells and AM in the BALF of rats receiving smoke inhalation were significantly different from those in normal control group (P < 0.01); Post hoc analysis also showed significant differences among groups (P < 0.05).
Table 1.
Total cells and AM in the BALF of different groups (n = 8, mean ± s)
| Groups | Total cells (× 108/L) | AM (× 108/L) |
|---|---|---|
| Control | 1.46±0.07 | 1.39±0.11 |
| 1-month smoking | 3.05±0.26** | 2.36±0.24** |
| 2-month smoking | 5.41±0.35**,ΔΔ | 4.04±0.52**,ΔΔ |
| 3-month smoking | 6.03±0.42**,ΔΔ,# | 5.75±0.57**,ΔΔ,## |
P < 0.01 vs. normal control group;
P < 0.01 vs. 1-month smoking group;
P < 0.05 vs. 2-month smoking group;
P < 0.01 vs. 2-month smoking group.
Concentrations of MSP in the BALF and serum
The longer the smoke inhalation, the higher the MSP concentration of the BALF and serum was. Statistical analysis showed that MSP concentrations of the serum and BALF in rats receiving smoke inhalation (except for 1-month smoke inhalation) were significantly higher than in rats of normal control group (P < 0.05). Post hoc analysis also showed significant differences (P < 0.05) (Table 2).
Table 2.
Concentrations of MSP in the BALF and serum (pg/ml) (n = 8, mean ± s)
| Groups | BALF (pg/ml) | Serum (pg/ml) |
|---|---|---|
| Control | 37.80±2.14 | 35.51±1.65 |
| 1-month smoking | 42.43±2.70* | 38.52±3.33 |
| 2-month smoking | 51.55±5.04**,ΔΔ | 47.45±1.99**,ΔΔ |
| 3-month smoking | 65.82±7.00**,ΔΔ,## | 57.97±6.17**,ΔΔ,## |
P < 0.05 vs. normal control group;
P < 0.01 vs. normal control group;
P < 0.01 vs. 1-month smoking group;
P < 0.01 vs. 2-month smoking group.
MDA content and SOD activity of AM following MSP treatment
MSP at different concentrations was used to treat AM from healthy rats and those after smoke inhalation (Figure 3). With the increase in MSP concentration, the MDA content of the AM supernatant increased while the SOD activity decreased (P < 0.05). MSP could promote the MDA production in AM of healthy rats and smoking rats, but inhibited the SOD activity in a dose-dependent manner. Moreover, the MDA content increased and SOD activity decreased in a time-dependent manner. These results indicated that MSP promotes the MDA production by rat AM, and decrease the SOD activity in a time-dependent manner.
Figure 3.
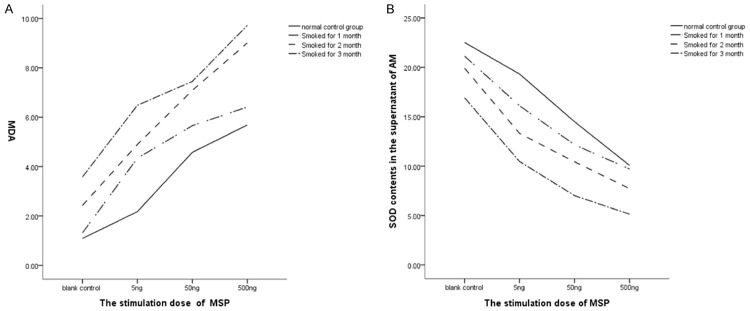
A. MDA content of the supernatant of AM. B. SOD activity of the supernatant of AM.
Factorial analysis showed that the interaction between MSP and smoke inhalation (P < 0.01). The higher the MSP concentration and the longer the smoke inhalation, the higher of MDA content was and the lower the SOD activity was.
Contents of TNF-α, IL-8, IL-1β and IL-10 of rat AM after MSP treatment
With the increase in MSP concentration, the contents of IL-8, TNF-α, IL-1β and IL-10 in the AM supernatant increased gradually in each group (Figure 4). When compared with control group, the contents of TNF-α, IL-8 and IL-1β in the supernatant of smoking rats remained unchanged after treatment with 5-ng MSP, while increased dramatically after treatment with 50 ng of MSP (P<0.05). The highest increase was observed in AM after treated with 500 ng of MSP (P < 0.01). Significant difference was also found in the IL-10 content between two groups (P < 0.05). These results indicate that MSP induces AM to secrete TNF-α, IL-8, IL-1β and IL-10.
Figure 4.

A. TNF-α content of the AM supernatant. B. IL-8 content of the AM supernatant. C. IL-1β content of the AM supernatant. D. IL-10 content of the AM supernatant.
With the prolongation of smoke inhalation, contents of TNF-α, IL-8, and IL-1β in the AM supernatant increased significantly (P < 0.05), but the IL-10 content decreased. Statistical analysis showed there was no marked difference in the IL-10 content between two smoke inhalation groups (P < 0.05).
Factorial analysis showed that MSP and smoke stimulation exerted synergistic effects on the secretion of TNF-α, IL-8 and IL-1β in AM (P < 0.05). The higher the MSP concentration and the longer the smoke inhalation, the higher the contents of TNF-α, IL-8 and IL-1β were in the AM. Moreover, MSP and smoke inhalation also conferred synergistic effects on the secretion of IL-10 in AM of normal rats (P < 0.05), but these synergistic effects were not observed in AM from smoke inhalation rats (P > 0.05).
RON expression in the lung
The mRNA expression of RON in the lung of 1-month smoking rats was similar to that of healthy rats (P > 0.05), but the mRNA expression of RON in the lung of 2-month smoking rats was significantly higher than in healthy rats and 1-month smoking rats (P < 0.01); the mRNA expression of RON in the lung of 3-month smoking rats was significantly higher than in other groups (P < 0.05). These results indicated that, as the prolongation of smoke inhalation, mRNA expression of RON increased in the rat lung (Table 3 and Figure 5).
Table 3.
mRNA and protein expression of RON in the lung and RON protein expression in the AM (n = 8, mean ± SD)
| Groups | RON mRNA in lung | RON protein in lung | RON protein in AM |
|---|---|---|---|
| Control | 0.35±0.08 | 0.0725±0.0093 | 0.1097±0.0062 |
| 1-month smoking | 0.45±0.10 | 0.0972±0.0437 | 0.1465±0.3260ΔΔ |
| 2-month smoking | 0.64±0.08ΔΔ,* | 0.1342±0.0283ΔΔ,* | 0.2140±0.0432ΔΔ,** |
| 3-month smoking | 0.89±0.09ΔΔ,**,# | 0.1706±0.0270ΔΔ,**,## | 0.3103±0.1005ΔΔ,**,## |
P < 0.01 vs. normal control group;
P < 0.05 vs. 1-month smoking group;
P < 0.01 vs. 1-month smoking group;
P < 0.05 vs. 2-month smoking group;
P < 0.01 vs. 2-month smoking group.
Figure 5.
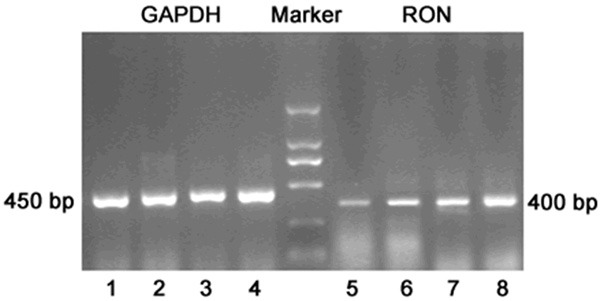
RON mRNA expression in the lung: 1, 2, 3, 4: GAPDH of normal control group, 1 month smoking group, 2 month smoking group and 3 month smoking group, respectively; 5, 6, 7, 8: RON of normal control group, 1 month smoking group, 2 month smoking group and 3 month smoking group, respectively.
RON protein expression of in the lung and AM
Immunohistochemistry showed RON protein expression in the airway ciliated epithelium and AM of rats in normal control group and smoke inhalation group. RON protein was expressed only on top surface of cilia in normal control group and 1-month smoking group, RON protein was expressed in the ciliary epithelial cytoplasm and on the top surface of airway cilia in 2-month and 3-month smoking groups. In AM of healthy rats, RON protein was mainly expressed on the cell membrane, the RON protein expression of 1-month smoking group enhanced, little RON protein was expressed in the cytoplasm, RON was still mainly expressed on rat AM cell membrane; after 2-month and 3-month smoke inhalation, the RON expression in the cytoplasm significantly increased. When compared with normal control group, RON protein expression in the airway epithelium and AM increased significantly in rats after smoke inhalation for 2 months and 3 months (P < 0.05), while the RON protein expressions in the airway epithelium in 1-month smoking group was similar to that in normal group. Post hoc comparisons of different smoked groups also had significant differences (P < 0.05) (Table 3; Figures 6 and 7).
Figure 6.
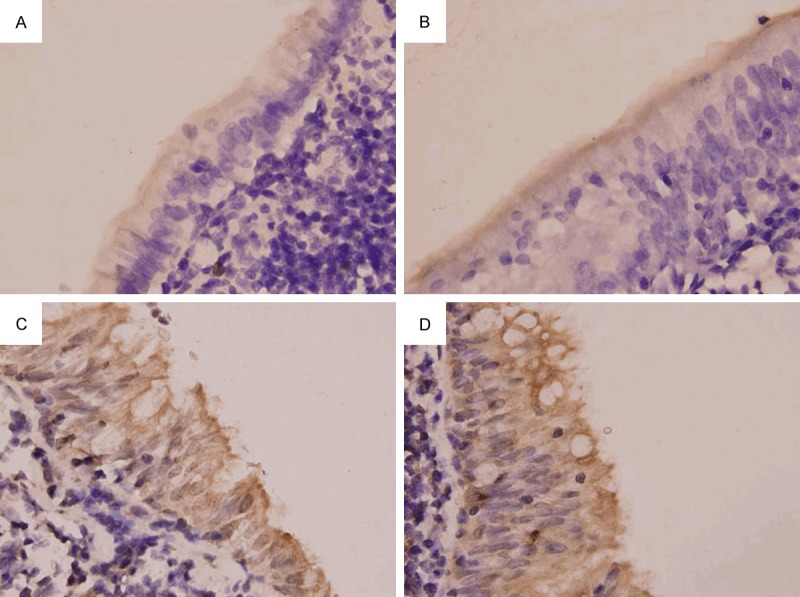
RON protein expression in the bronchial epithelium (SP method; × 1000). A. Normal control group. B. 1-month smoking group. C. 2-month smoking group. D. 3-month smoking group.
Figure 7.
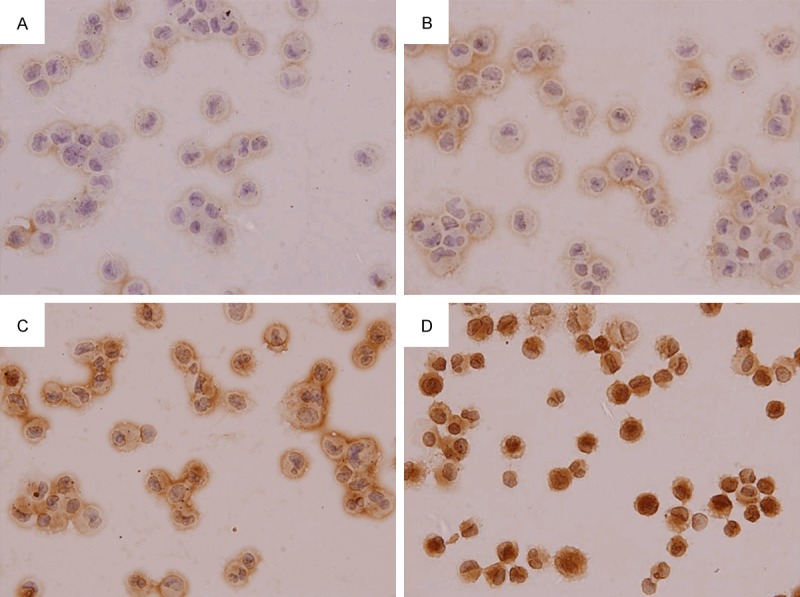
RON protein expression in AM (SP method; × 1000). A. Normal control group. B. 1-month smoking group. C. 2-month smoking group. D. 3-month smoking group.
Discussion
It has been widely accepted that smoking is the main risk factor of respiratory system disease including chronic bronchitis and COPD [13-15], and about 15% of long-term smokers will develop COPD. However, it has been established that airway inflammation plays an important role in the pathogenesis of COPD, and many inflammatory cells (neutrophils, macrophages, mast cells, etc) and cytokines (TNF-α, IL-8, IL-1β and leukotrienes) are found to be involved in it [16]. Airway inflammation will cause airway remodeling and lead to airflow limitation. Therefore, airway inflammation is a basic pathological feature of respiratory system diseases including COPD and bronchial asthma.
Our findings demonstrated the airway inflammation in a rat model during the smoke inhalation, and a large number of inflammatory cells infiltrated into the airway with the passive smoking. Further investigations on BALF showed that the total cells and AM increased with the prolongation of smoke inhalation, and the numbers of total cells and AM in the BALF of 3-month smoking group were significantly higher than those in normal control group. Studies have shown that the inflammatory cells in the lower respiratory tract markedly increase in smokers [17], and AM in the BALF of smokers and COPD patients increase several times [18]. In addition, with the infiltration of inflammatory cells, the bronchial epithelial cells became shedding gradually, goblet cells increased, bronchial smooth muscle was also progressively thickening, the alveolar cavity enlarged and alveolar wall became thinning and fractured, which were characteristics of emphysema. Therefore, AM is closely related to the airway inflammation, which may subsequently lead to the airway remodeling.
Our results indicated that the MSP concentration of the serum and BALF of smoke inhalation rats was significantly higher than that in control group, and the increase in MSP concentration was dependent on the duration of smoke inhalation: the longer the smoke inhalation, the higher the MSP concentration of the serum and BALF, indicating that smoke inhalation may increase the synthesis and release of MSP, which is consistent with findings in the study of Takano et al. in which the MSP expression increased significantly in some inflammatory diseases of the respiratory system [11].
Gunella et al. [19] found that the MSP treated AM of smokers and patients with interstitial lung disease such as sarcoidosis would lead to a significant increase in oxygen free radicals (OFR) generation, a significant increase in cytokines and an enhancement of NF-κB activation, suggesting that MSP plays an important role in the pathogenesis of some respiratory diseases through activating AM. Our findings revealed that the MSP treated AM from healthy rats and smoking rats showed significant increases in the TNF-α, IL-8 and IL-1β and MDA and marked reduction in SOD activity, which were dependent on the concentration of MSP. These findings were consistent with previously reported [12,19]. Moreover, with the prolongation of smoke inhalation, the contents of TNF-α, IL-8, IL-1β and MDA in supernatants of AM of rats increased while the SOD activity decreased. These suggest that smoke inhalation alone may cause the increase in MDA and proinflammatory cytokines TNF-α, IL-8 and IL-1β and reduction in the SOD activity. Furthermore, in the presence of MSP, above effects became more obvious in a MSP dose dependent manner.
TNF-α has a wide range of biological activities and can activate neutrophils and meditate the inflammatory response. IL-1β is also a pro-inflammatory cytokine, and can stimulate the degranulation of monocytes and eosinophils [20,21]. Both cytokines are the key pro-inflammatory cytokines in humans, may be secreted by a variety of inflammatory cells following LPS treatment, and then induce the secretion of IL-6, IL-8 and other cytokines, leading to a cascade of inflammation. AM may secrete a large amount of cytokines, and regulate and initiate local immune response, playing an important role in the pathogenesis of inflammation and other airway inflammatory diseases including COPD. In this study, results showed that there was an interaction between MSP and smoke inhalation. MSP may further aggravate the oxidative stress caused by smoke inhalation and increase OFR and release of inflammatory cytokines. Therefore, we hypothesize that the initiation and regulation of inflammation may be associated with MSP.
Interestingly, in the supernatant of AM from both healthy rats and smoking rats, IL-10 increased after MSP treatment; but the increase was higher in normal control group than in other groups. This may be ascribed to the anti-inflammatory capability of IL-10 and the pro-inflammatory activities of TNF-α, IL-8 and IL-1β. Thus, MSP may not only have pro-inflammatory activity, but possess the anti-inflammatory activity to a certain extent. In healthy rats, both effects of MSP are in a state of equilibrium. However, after smoke inhalation, this balance is broken, resulting in the enhancement of pro-inflammatory effect and weakening of anti-inflammatory effect.
RON is a MSP specific receptor and mainly expressed in the human epithelial cells, granulocytes, monocytes, macrophages and tonsil germinal layer, small intestine, colon, kidney, lung and bone marrow. RON expression is also detectable in human ciliated epithelial cells [10,22]. There is evidence showing that both tracheobronchial epithelium and AM of rats have RON expression. In the present study, results showed that the RON mRNA expression in the lung of smoking rats and the RON protein expression in the ciliated epithelial cells and AM increased with the prolongation of smoke inhalation.RON mRNA expression in 3-month smoking rats was significantly higher than that in other groups. Although no marked differences were observed in the mRNA and protein expressions of RON of bronchial epithelial cells between 1-month smoking rats and control group, the RON expression in bronchial epithelial cells and AM were significantly higher in remaining smoking rats than in normal group. This suggests that smoke inhalation promotes the synthesis and release of MSP and increases the mRNA and protein expressions of RON in the AM and ciliated epithelial cells.
In summary, our present study demonstrates that smoke inhalation increases the synthesis and release of MSP, and elevates the protein and mRNA expressions of RON in the lung. The MSP and RON may synergistic effect to activate AM, leading to increases in TNF-α, IL-8, IL-1β, IL-10 and MDA, and a decrease in SOD activity. These findings suggest that MSP and its receptor RON are involved in the initiation and regulation of airway inflammation in rats receiving smoke inhalation through activating AM to release OFR and inflammatory mediators.
Acknowledgements
This study was supported by Sichuan provincial department of public health.
Disclosure of conflict of interest
None.
References
- 1.Sevenoaks MJ, Stockley RA. Chronic Obstructive Pulmonary Disease, inflammation and co-morbidity--a common inflammatory phenotype? Respir Res. 2006;7:70. doi: 10.1186/1465-9921-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomoda K, Kubo K, Dairiki K, Yamaji T, Yamamoto Y, Nishii Y, Nakamura A, Yoshikawa M, Hamada K, Kimura H. Whey peptide-based enteral diet attenuated elastase-induced emphysema with increase in short chain fatty acids in mice. BMC Pulm Med. 2015;15:64. doi: 10.1186/s12890-015-0059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Seimetz M, Parajuli N, Pichl A, Bednorz M, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, Weissmann N. Cigarette Smoke-Induced Emphysema and Pulmonary Hypertension Can Be Prevented by Phospho-diesterase 4 and 5 Inhibition in Mice. PLoS One. 2015;10:e0129327. doi: 10.1371/journal.pone.0129327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tetley TD. Macrophages and the pathogenesis of COPD. Chest. 2002;121:156s–159s. doi: 10.1378/chest.121.5_suppl.156s. [DOI] [PubMed] [Google Scholar]
- 5.Vlahos R, Bozinovski S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front Immunol. 2014;5:435. doi: 10.3389/fimmu.2014.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Han S, Stuart LA, Degen SJ. Characterization of the DNF15S2 locus on human chromosome 3: identification of a gene coding for four kringle domains with homology to hepatocyte growth factor. Biochemistry. 1991;30:9768–9780. doi: 10.1021/bi00104a029. [DOI] [PubMed] [Google Scholar]
- 7.Chaudhuri A. Regulation of Macrophage Polarization by RON Receptor Tyrosine Kinase Signaling. Front Immunol. 2014;5:546. doi: 10.3389/fimmu.2014.00546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camp ER, Liu W, Fan F, Yang A, Somcio R, Ellis LM. RON, a tyrosine kinase receptor involved in tumor progression and metastasis. Ann Surg Oncol. 2005;12:273–281. doi: 10.1245/ASO.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 9.Nikolaidis NM, Gray JK, Gurusamy D, Fox W, Stuart WD, Huber N, Waltz SE. Ron receptor tyrosine kinase negatively regulates TNF-alpha production in alveolar macrophages by inhibiting NF-kappaB activity and Adam17 production. Shock. 2010;33:197–204. doi: 10.1097/SHK.0b013e3181ae8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakamoto O, Iwama A, Amitani R, Takehara T, Yamaguchi N, Yamamoto T, Masuyama K, Yamanaka T, Ando M, Suda T. Role of macrophage-stimulating protein and its receptor, RON tyrosine kinase, in ciliary motility. J Clin Invest. 1997;99:701–709. doi: 10.1172/JCI119214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takano Y, Sakamoto O, Suga M, Suda T, Ando M. Elevated levels of macrophage-stimulating protein in induced sputum of patients with bronchiectasis. Respir Med. 2000;94:784–790. doi: 10.1053/rmed.2000.0822. [DOI] [PubMed] [Google Scholar]
- 12.Brunelleschi S, Penengo L, Lavagno L, Santoro C, Colangelo D, Viano I, Gaudino G. Macrophage stimulating protein (MSP) evokes superoxide anion production by human macrophages of different origin. Br J Pharmacol. 2001;134:1285–1295. doi: 10.1038/sj.bjp.0704356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1:1645–1648. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherrill DL, Holberg CJ, Enright PL, Lebowitz MD, Burrows B. Longitudinal analysis of the effects of smoking onset and cessation on pulmonary function. Am J Respir Crit Care Med. 1994;149:591–597. doi: 10.1164/ajrccm.149.3.8118623. [DOI] [PubMed] [Google Scholar]
- 15.Sherrill DL, Lebowitz MD, Knudson RJ, Burrows B. Smoking and symptom effects on the curves of lung function growth and decline. Am Rev Respir Dis. 1991;144:17–22. doi: 10.1164/ajrccm/144.1.17. [DOI] [PubMed] [Google Scholar]
- 16.Barnes PJ, Shapiro SD, Pauwels RA. Chronic obstructive pulmonary disease: molecular and cellular mechanisms. Eur Respir J. 2003;22:672–688. doi: 10.1183/09031936.03.00040703. [DOI] [PubMed] [Google Scholar]
- 17.Taylor JD. COPD and the response of the lung to tobacco smoke exposure. Pulm Pharmacol Ther. 2010;23:376–383. doi: 10.1016/j.pupt.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Shapiro SD. The macrophage in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160:S29–32. doi: 10.1164/ajrccm.160.supplement_1.9. [DOI] [PubMed] [Google Scholar]
- 19.Gunella G, Bardelli C, Amoruso A, Viano I, Balbo P, Brunelleschi S. Macrophage-stimulating protein differently affects human alveolar macrophages from smoker and non-smoker patients: evaluation of respiratory burst, cytokine release and NF-kappaB pathway. Br J Pharmacol. 2006;148:478–489. doi: 10.1038/sj.bjp.0706751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murugan V, Peck MJ. Signal transduction pathways linking the activation of alveolar macrophages with the recruitment of neutrophils to lungs in chronic obstructive pulmonary disease. Exp Lung Res. 2009;35:439–485. doi: 10.1080/01902140902759290. [DOI] [PubMed] [Google Scholar]
- 21.Chung KF. Cytokines in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2001;34:50s–59s. [PubMed] [Google Scholar]
- 22.Wang MH, Padhye SS, Guin S, Ma Q, Zhou YQ. Potential therapeutics specific to c-MET/RON receptor tyrosine kinases for molecular targeting in cancer therapy. Acta Pharmacol Sin. 2010;31:1181–1188. doi: 10.1038/aps.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]