

Abstract
High mobility group box 1 (HMGB1) has been widely reported to mediate damage caused by inflammatory responses. The aim of our study is to investigate the role of HMGB1 in endotoxin tolerance (ET) alleviating inflammation of acute liver failure (ALF) rats and its possible signaling mechanism. To mimic ET, male Sprague-Dawley rats were pretreated with low dose of lipopolysaccharide (LPS) (0.1 mg/kg once a day intraperitoneally for consecutive five days) before subsequent ALF induction. ALF was induced by intraperitoneal administration of D-GalN/LPS. ET induced by LPS pretreatment significantly improved the survival rate of ALF rats. Moreover, after ALF induction, ET+ALF rats exhibited lower serum enzyme (ALT, AST and TBiL) levels, lower production of inflammatory cytokines (IL-6, TNF-a and HMGB1) and more minor liver histopathological damage than ALF rats. ET+ALF rats showed enhanced expression levels of HMGB1, decreased levels of STAT1 and p-STAT1, augmented expression of SOCS1 in liver tissues than ALF rats. These results indicated that ET induced by low-dose LPS pretreatment may alleviate inflammation and liver injury in experimental acute liver failure rats mainly through inhibition of hepatic HMGB1 translocation and release.
Keywords: Endotoxin tolerance, high mobility group box 1, acute liver failure, JAK/STAT1 signaling, lipopolysaccharide (LPS)
Introduction
Acute liver failure (ALF) is a clinical syndrome in which acute loss of metabolic and synthetic liver function leads to coagulopathy, hepatic encephalopathy and multiorgan failure within a short time in patients. Pathology of ALF is characterized by massive hepatocellular necrosis and large scales of inflammation in liver [1]. Despite considerable advances have been made in the understanding of its pathophysiology, ALF is still a serious clinical condition with an unacceptably high mortality rate. Currently, liver transplantation is the most effective treatment for ALF, although shortage of donor organs limits this option. In addition, bioartificial and artificial liver support systems are safe, beneficial but need to be evaluated in more detail [2]. Therefore, further investigation is required so as to dissect the exact pathogenesis of ALF. Disorder of immune balance is considered to contribute to formation and development of ALF, with massive release of various inflammatory cytokines from necrotic and apoptosis hepatocytes [3].
Endotoxin tolerance (ET) is defined as a reduced inflammatory response of the host (in vivo) or of cultured macrophage/monocyte (in vitro) to LPS following a previous exposure to this stimulus. Although the phenomenon of ET was first described by Beeson [4] more than 60 years ago, its mechanism has still not been fully clarified, and thus is far from being applied in clinical practice.
Recently, illumination of the relationship between ET and some inflammatory mediators suggests a new direction for its promising clinical application in diseases, such as sepsis [5]. ET has been demonstrated to ameliorate experimental ALF [6], however, molecular mechanism of protective effect of ET on ALF remains unclear.
HMGB1, a protein generally found in the nucleus that promotes DNA bending by binding to nucleosomes, has been reported to mediate damage caused by inflammatory responses [7]. Aneja et al. demonstrated that preconditioning with low-dose recombinant HMGB1 induced endotoxin tolerance in mice [8]. Moreover, endogenous HMGB1 has been confirmed as a major mediator in ET alleviating sepsis induced by lethal-dose LPS [7]. We hypothesized that the protective effect of ET on ALF might rely on regulation of serum HMGB1. SOCS1 [9,10], the negative regulator of JAK/STAT1 signaling pathway, is involved in the regulation of ET and JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation [11]. In present study, we further confirmed the molecular mechanism of ET regulating translocation and release of HMGB1.
Materials and methods
Animals
Male Sprague-Dawley rats weighing 180-200 g were purchased from the Shanghai Laboratory Animal Center (Shanghai, China). All rats were housed at the Experimental Animal Laboratory of Wenzhou Medical University (Wenzhou, Zhejiang, China) under standard laboratory conditions (laminar-flow, temperature-controlled by 21 ± 2°C, 12 h light/dark cycle). The rats were fed with a standard chow diet and free water. They were conducted under the standard procedure set by the Committee for the Purpose of Control and Supervision of Experiments on Animals and the National Institutes of Health for the specification use of the experimental animals. The research protocol was approved by the Animal Ethics Committee of Wenzhou Medical University.
Regents
D-GalN (G0500-1 g) and LPS (L-2880-10 mg) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Enzyme-linked immunosorbent assay (ELISA) kits for tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6), BCA protein assay kits were acquired from the Beyotime institute of biotechnology (Nanjing, China). Enzyme-linked immunosorbent assay (ELISA) kits for High Mobility Group Box 1 (HMGB1) were from Shanghai Westang Bio-Tech Co, Ltd. (Shanghai, China). Antibodies against HMGB1 were purchased from Cell Signaling Technology (Danvers, MA, USA) and Antibodies against p-STAT1, STAT1, SOCS1 and GAPDH were obtained from Santa Cruz Biotechnology (CA, USA). Inhibitors of protease cocktail were acquired from Roche (Summerville, NJ, USA). Automatic biochemistry analyzer was purchased from Abbott Laboratories, USA.
Study design and grouping
D-GalN and LPS were dissolved in sterile 0.9% sodium chloride according to the product description. The rats were randomly divided into three groups: control group (n=6), ALF group (n=40), ET+ALF group (n=20). For induction of ET, rats in ET+ALF model were given 0.1 mg/kg LPS intraperitoneally once every day for consecutive five days. Rats in the ALF group and control group were injected with the same volume of sterile 0.9% sodium chloride instead of LPS for the same treatment. For induction of ALF, rats in ET+ALF group and ALF group were given one intra- peritoneal injection of 500 µL saline containing 800 mg/kg D-GalN and 8 µg LPS on the sixth day. Meanwhile, rats in control group were injected with the same volume of sterile 0.9% sodium chloride. In a separate experiment, survival rate was monitored for 3 days after injection of D-GalN/LPS in both ALF group (n=20) and ET+ALF group (n=20). Except rats for the survival rate, all rats were finally sacrificed with chloral hydrate (1.0 g/kg, intraperitoneally (i.p.)) at 2, 6, 12, 24, 48 hours.
Serum cytokine and biochemical markers assays
After the experimental animals were sacrificed, blood of 2 ml was collected from the portal vein, 30 minutes’ standing, centrifuged at 3000 r/min for 10 min, the upper serum was kept and stored in -80°C deep-freezer. Serum concentrations of tumor necrosis factor-α (TNF-a), interleukin-6 (IL-6) and HMGB1 were determined by using antibody enzyme-linked immunosorbent assay (ELISA) kits. All procedures were performed in accordance with manufacturer instructions. The levels of AST, ALT and total bilirubin (TBiL) in the serum were tested by an automatic biochemistry analyzer.
Liver histological examination
After the blood collection, liver samples were dissected and fixed in 10% neutral formalin solution and embedded in paraffin. The embedded liver tissues were cut into 4 μm sections, stained in hematoxylin and eosin (H&E) and viewed under a video microscope for histolopathological changes.
Western blot analysis
After the blood collection and livers dissection for histological examination, livers were removed immediately, cut into pieces and stored at -80°C for further research. The isolation of total protein was used lyses buffer (1 ml Radiation immune precipitation (RIPA) paralysis liquid, 10 µl PMSF, 10 µl sodium orthovanadate activating agent) accompanied by a inhibitor of protease cocktail. Samples were placed on ice bath for 30 minutes and centrifugated at 13000 r/min for 10 min, the supernatant was collected and stored at -80°C for preservation, and the protein concentration was measured with BCA protein assay kit. The samples (15 μg protein each) were suffered from polyacrylamide gel electrophoresis (SDS-PAGE) after heat denaturation at 95°C for 5 min. Subsequently, the target proteins in gel were shifted onto polyvinylidene fluoride membrane (PVDF) membranes. Then, the membranes were blocked by 5% bovine albumin (5% BSA) for 1.5 h at room temperature. The primary antibodies of HMGB1, p-STAT1, STAT1, SOCS1 and GAPDH were used to incubate the membranes at 4°C overnight, respectively. The membranes were washed with TBST for 7 minutes four times, incubated with the secondary antibody at room temperature for 1 h, washed again with TBST for 7 minutes four times. Then the film exposure was performed after covered with chemiluminescent reagents.
Statistical analysis
All data were analyzed with SPSS version 19.0 (IBM, Chicago, USA) and expressed as means ± standard deviation (SD). Statistical significance was evaluated by one-way analysis of variance (ANOVA) or the least significant difference (LSD) test. P values less than 0.05 were considered statistically significant (P<0.05).
Results
ET induced by LPS pretreatment improved the survival rate of ALF rats
We randomly adopted twenty rats for each group to exclusively investigate the effect of endotoxin tolerance on the survival rate of rats. The mortalities in 72 hours after injection of D-GalN/LPS or saline for each group were monitored.
The mortality rate of rats in ALF group was 60% in 72 hours. Twelve in twenty rats of the ALF group died totally and majority of them died in the first 12 h. However, the mortality rate in ET+ALF group was 10%. Only two in twenty rats of the ET+ALF group died, at 5 h and 10 h, respectively. None of the twenty rats in control group died in 72 hours (Figure 1).
Figure 1.
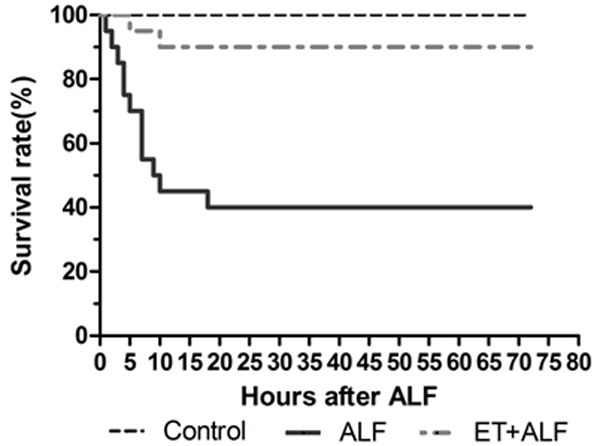
Significant difference in survival rates was found between ALF group and ET+ALF group by Kaplan-Meier survival analysis (P<0.01 by Log-rank Test).
Comparison of the mortality rates of 72 hours after injection of D-GalN/LPS or saline in different groups clearly revealed that pre-existed endotoxin tolerance condition induced by LPS pretreatment significantly improved the survival of ALF rats.
Effect of ET induced by LPS pretreatment on histopathology characteristics of liver tissues
Liver sections from each group were stained with hematoxylin and eosin (HE). In ALF group, liver tissues represented serious injuries with damaged hepatic lobules, ruined hepatic cords, and large numbers of infiltrated inflammatory cells, including mainly monocytes and lymphocytes. Necrosis and apoptosis can also be found. However, liver injuries in ET+ALF group were less severe with mild necrosis of hepatocytes and less infiltrated inflammatory cells. Normal hepatic cells, central veins and lobules were shown in control group (Figure 2).
Figure 2.
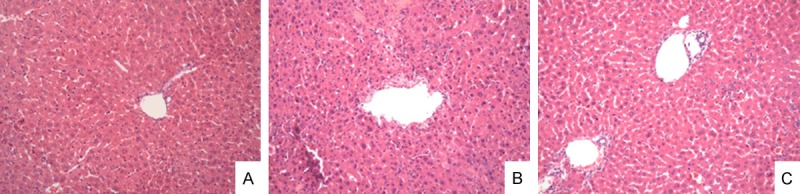
Representative photographs of liver sections using H&E staining (original magnification: × 200). A. Liver sections from the control group; B. Liver sections from the ALF group; C. Liver sections from the ET+ALF group.
We concluded pre-existed endotoxin tolerance condition induced by LPS pretreatment ameliorated inflammation in liver of experimental ALF rats.
ET induced by low-dose LPS pretreatment alleviated liver injuries in ALF
To study the effect of ET on liver function of experimental ALT rats, the levels of serum ALT, AST and TBiL in 48 h after ALF induction for each group were examined. Levels of ALT, AST and TBiL were elevated in both ALF group and ET+ALF group than that in control group. However, the increases in ALF group were significantly higher than that in ET+ALF group at 6 h, 12 h, 24 h, 48 h (P<0.05). Moreover, levels of ALT and AST in both groups reached the peak at 12 h and levels of TBiL continuously rise in 48 hours (Figure 3A-C). No obvious variation occurred in control group.
Figure 3.
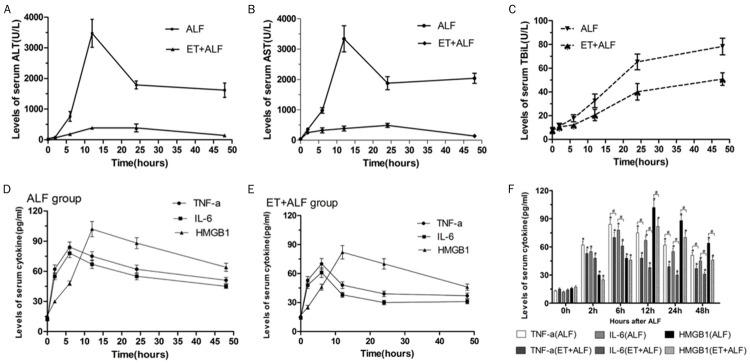
Effect of ET induced by LPS pretreatment on liver function and levels of serum cytokine in ALF rats. The serum levels of alanine aminotransferase (ALT) (A), aspartate aminotransferase (AST) (B) and total bilirubin (TBiL) (C) were assayed using an automated blood chemistry analyzer. The serum concentrations of TNF-a, IL-6 and HMGB1 in ALF group (D) and ET+ALF group (E) were detected by ELISA assay and compared with that in control group (F).
After comparing levels of serum markers for liver function in 48 hours, we concluded that pre-existed endotoxin tolerance condition induced by low-dose LPS pretreatment significantly alleviated liver injuries in ALF rats.
ET induced by low-dose LPS pretreatment decreased serum levels of HMGB1 and pro-inflammatory cytokines in ALF
Previous studies have shown the release of cytoplasmic HMGB1 in activated immune cells and hepatocytes can activate macrophages to produce pro-inflammatory cytokines [12-14]. To investigate the role of ET on alleviating inflammation of ALF rats, we detected serum levels of TNF-α, IL-6 and HMGB1 in 48 hours after ALF induction. Data indicated that TNF-α, IL-6 levels started to increase immediately after injection of D-GalN/LPS and peaked at 6 hour in both ALF group and ET+ALF group when compared to the control group (P<0.05). In addition, levels of both TNF-a and IL-6 in ET+ALF group were lower than that in ALF group at each time point (P<0.05). However, HMGB1 levels increased slowly during the first 6 hours and peaked at 12 hour in both groups. Also, levels of HMGB1 in ET+ALF group were lower than their counterparts in ALF group at each time point (P<0.05) (Figure 3D-F).
ET induced by low-dose LPS pretreatment enhanced liver tissues levels of HMGB1 in ALF
To clarify whether alleviated inflammation and liver damage in ET+ALF rats were related to levels of HMGB1 in liver tissues, the protein levels of hepatic HMGB1 were measured by western blotting. As shown in Figure 4E, protein levels of hepatic HMGB1 gradually decreased after ALF induction and reached the nadir at 12 hour postinduction in both ALF group and ET+ALF group. Also, the levels of hepatic HMGB1 in both groups were decreased at every time point after ALF induction, as compared to the control group (P<0.05). However, relatively higher protein levels of HMGB1 in ET+ALF group indicated that ET induced by low-dose LPS pretreatment could partly reverse the decline of hepatic HMGB1 in ALF group (P<0.05).
Figure 4.

Effect of ET induced by low-dose LPS pretreatment on the changes of SOCS1 (B), STAT1 (C), p-STAT1 (D) and HMGB1 (E) protein expression in liver of ALF rats. (A) displays typical pictures of protein abundance in the liver. (B-E) shows the quantitative levels of SOCS1, STAT1, p-STAT1, and HMGB1 protein in the liver measured by Western blot respectively. Lane 1 represents rat liver of the control group; Lane 2-6 represent rat liver of ALF group at 2 h, 6 h, 12 h, 24 h, 48 h after ALF induction; Lane 7-11 represent rat liver of ET+ALF group at 2 h, 6 h, 12 h, 24 h, 48 h after ALF induction. Levels of SOCS1 (B), STAT1 (C), p-STAT1 (D) and P65 (E) were standardized to GAPDH content. All data were expressed as mean ± SD of six rats at every point of time. *Represents P<0.05 versus control group; #indicates P<0.05 versus ALF group.
ET induced by low-dose LPS pretreatment reduced expression of p-STAT1 and STAT1 in ALF
Researchers previously reported that activation of JAK/STAT1 signaling promotes hyperacetylation, mobilization from the nucleus to the cytoplasm and secretion of HMGB1 [11]. After phosphorylation, STAT1 proteins are activated. Phosphorylated STAT1 proteins dimerize via Src-homology 2 (SH2)-domain-phospho-tyrosine interactions and translocate to the nucleus, where they function as transcriptional or translocational activators [15]. To investigate the effect of ET on STAT1 phosphorylation, the protein levels of p-STAT1 in liver tissues were initially tested by western blotting. As shown in Figure 4D, the expression of p-STAT1 in ALF group reached the peak at 24 hour after ALF induction and the expression of p-STAT1 in ALF+ET group continuously increased in 48 hours. Moreover, the expression of p-STAT1 were significantly increased in ALF group and slightly augmented in ET+ALF group, as compared to the control group (P<0.05). Results showed the protein levels of p-STAT1 in ET+ALF group were lower at all five time points than that in ALF group (P<0.05).
To confirm the possible effect of ET on the expression of STAT1, the protein levels of STAT1 were confirmed by western blotting. Although the proteins levels of STAT1 in both ET+ALF group and ALF group were increased (P<0.05) as shown in Figure 4C, levels in ET+ALF group were much lower than that in ALF group (P<0.05). The proteins levels of STAT1 persistently increased after ALF induction and reached the peak at 12 h and 24 h postinduction in ALF group and ET+ALF group, respectively.
ET induced by low-dose LPS pretreatment augmented expression of SOCS1 in ALF
To clarify the effect of ET on expression of SOCS1 in ALF, western blotting was used to measure the protein levels of SOCS1 in liver tissues. The protein levels of SOCS1 in ALF group gradually increased and reached the top at 12 h after ALF induction while the protein levels of SOCS1 in ALF+ET group continuously increased in 48 hours (Figure 4B). Moreover, the protein levels of SOCS1 in both ET+ALF group and ALF group were significantly increased at each of the five time points when compared to the control group (P<0.05). However, the expression of SOCS1 in ET+ALF group were significantly higher at all five time points than those in ALF group (P<0.05).
Discussion
Prior exposure of organisms or innate immune cells like monocytes/macrophages to minute amounts of endotoxin cause them to become refractory to subsequent endotoxin challenge, a phenomenon called “endotoxin tolerance” [16]. We have previously reported that endotoxin tolerance (ET) induced by low-dose LPS pretreatment resulted in a marked improvement of acute liver failure (ALF) as evidenced by decreased serum transaminase, reduced hepatocellular damage along with suppressed immune cell infiltration, inhibited pro-inflammatory cytokine production and mortality [6,10]. However, the underlying molecular mechanisms implicated in protective effect of endotoxin tolerance on ALF remain elusive.
Previous studies consistently demonstrated that high levels of and various pro-inflammatory cytokine production participate in the pathogenesis of ALF and majority of ALF are characterized by systemic inflammatory response syndrome(SIRS) [2]. Both high mortality and severe liver damages existed in ALF were closely related with high levels of inflammation. Researchers have reported that high levels of HMGB1 were detected in rat ALF model, administration of exogenous recombinant HMGB1 enhanced hepatic injury while blockade of extracellular HMGB1 by a neutralizing antibody inhibited pro-inflammatory cytokine production [17]. These results indicated that HMGB1 might play an important role in ALF progression. Considering the significant role of HMGB1 in ALF, we speculated that the protective effect of ET on ALF might be set on regulation of serum HMGB1. In this study, we finally discovered ET ameliorated hepatic injury and inflammation in ALF through reducing levels of serum HMGB1 and further investigated the signaling mechanism of ET suppressing translocation and secretion of HMGB1.
D-GalN/LPS-induced ALF in mice has been demonstrated as one of the most widely investigated and the most representative animal models for mimicking human ALF and illuminating its exact pathogenesis mechanism [18]. D-galactosamine is a toxin that causes injury specific to the liver by selective depletion of uridine nucleotides in hepatocytes. LPS, a main part of gram-negative bacteria cell wall, causes release of a wide variety of inflammatory mediators via activation of hepatic sinus gap endothelial cells and Kupffer cells. Taken together, D-galactosamine companied by LPS can extend liver injury. Consistent of clinical observation of patients with ALF, high levels of serum ALT, AST, TBiL and severe liver pathology were discovered in our present research, indicating the ALF model was successfully established. The ET model was established as previously described [5].
Extracellular HMGB1 has been found related with the pathogenesis of ALF [5,17,19] and other pro-inflammatory diseases, such as sepsis [20], T-cell mediated hepatitis [12], rheumatoid arthritis [21], acute allograft rejection [22], acute lung injury [23]. HMGB1 functions through forming specific complexes with other signaling molecules such as inflammatory cytokines, single-stranded DNA, lipopolysaccharide and the complexes exert their biological actions via multiple receptors including the receptor for advanced glycationend products (RAGE) [24], TLR4 and TLR2 [25]. Production of HMGB1 box-A protein in the liver appeared to have a therapeutic effect in a rat model of ALF [26]. HMGB1 is instrumental in mediating a response to liver damage. In our study, the plasma concentrations of HMGB1 in the ALF group, determined by ELISA, were significantly increased and reached a peak at 24 h after ALF induction. Although serum levels of HMGB1 in the ET+ALF group also peaked at 24 h, the levels at each of the five time points were much lower than that in ALF group. Also, we detected protein levels of HMGB1 in liver tissues by western blot analysis. Much higher levels of HMGB1 in the ET+ALF group than the ALF group were found, although levels of HMGB1 in both groups were lower than that in the control group.
NF-κB, binding to its inhibitory IκB proteins in resting cells, is a pivotal transcription factor required for the synthesis of inflammatory mediators, such as TNF-a and IL-6. Upon activation, NF-kB is phosphorylated, separated from Iκ-Bα, translocated from cytoplasm into the nucleus and binds to NF-kappa B promoter sites on DNA, activating gene transcription of cytokines such as TNF-α, and accelerating liver injury [27]. In our research, reduction of serum HMGB1 in ET group inhibited NF-κB DNA-binding activity, which was associated with suppressed Iκ-Bα phosphorylation, thereby, blocking the production of TNF-α and IL-6, and as a consequence, ameliorating hepatic inflammation and injury. We concluded that ET induced by LPS pretreatment might ameliorate inflammation in ALF rats by downregulation of serum HMGB1, further reduce hepatocellular damage and improve the survival.
HMGB1 can be either released passively from injured or necrotic cells or secreted by activated immune cells such as monocytes, macrophages, dendritic cells (DCs) or some parenchymal cells, including hepatocytes, intestinal epithelial cells and cardiomyocytes [14,28,29]. The intrahepatic innate immune cells (Kuffers and dentritic cells) are activated by extracellular HMGB1 to produce large amounts of inflammatory mediators such as TNF-a and IL-6 that exacerbate hepatocytes injury, therefore triggers a vicious cycle of hepatic injury. As reported, high plasma concentrations of HMGB1 in ALF model were confirmed to correlate with low serum levels of HMGB1 since severe liver damages cause passively leak of HMGB1 from cytoplasm and nucleus of hepatocytes [17]. Immunochemistry staining indicated that HMGB1 in normal livers was localized predominantly in the nucleus of hepatocytes. HMGB1 released from injured or necrotic hepatocytes play a pivotal role in development of hepatic injury [12]. In our research, both inflammatory cells and necrotic hepatocytes were found by histopathological examination in liver tissues of the ALF group. In this study, we explored the possibility in active secretion of HMGB1 from normal hepatocytes and immune cells in liver, including Kuffer cells, DC, lymphocytes, NK cells.
High-mobility group box 1 (HMGB1) is a highly conserved nuclear protein that is involved in transcriptional activation and DNA folding in the eukaryotic cells [30]. The first step for the release of HMGB1 is mobilization from the nucleus to the cytoplasm and the second step is release of cytoplasmic HMGB1. Researchers have identified a critical role of the JAK/STAT1 pathway in promoting HMGB1 nuclear translocation to cytoplasm for subsequent release [11]. To dissect the underlying mechanism for ET-mediated downregulation of serum HMGB1 in ALF, we explored the expression of STAT1 in liver tissues since STAT1 has been suggested to be an activator for translocation of HMGB1. Our results showed the protein expression of STAT1 were obviously lower in liver tissues of the ET group rats when compared to the ALF groups, indicating that ET induced by LPS pretreatment might inhibit the expression of STAT1. Given this effect, levels of HMGB1 nuclear translocation from the nucleus to the cytoplasm were reduced, leading to lower levels of release of cytoplasmic HMGB1.
Suppressors of cytokine signaling (SOCS) proteins function as feedback inhibitors of the JAK/STAT signaling pathway, terminating innate and adaptive immune responses [31]. Researchers confirmed the regulation of endotoxin tolerance is multifaceted, involving receptors, signaling molecules, and negative regulators such as IRAK-M, SOCS1, short-vision MyD88, and SHIP [9,32]. In our present study, we observed higher expression of hepatic SOCS1 in ET+ALF group than that in ALF group, which indicated SOCS1 may involve in the mechanism of the protective effect of ET on ALF. We concluded that ET might inhibit the expression of STAT1 by enhancing the expression of SOCS1, finally leading to lower plasma concentrations of HMGB1.
For the first time, we demonstrated ET might protect rats from D-GalN/LPS induced ALF by decreasing serum HMGB1, which means decline in both passive release from necrotic hepatocytes and active secretion from normal hepatocytes and inflammatory cells. Inhibition of JAK/STAT1 signaling plays a pivotal role in decline of active secretion of HMGB1. Further studies are required to elucidate this issue. However, our conclusions prompt that reduction or blockage of serum HMGB1 may represent a novel therapeutic strategy to prevent acute liver failure, at least.
Acknowledgements
This work was supported by science and technology planned projects of Wenzhou city (Y20140669).
Disclosure of conflict of interest
None.
References
- 1.Willars C. Update in intensive care medicine: acute liver failure. Initial management, supportive treatment and who to transplant. Curr Opin Crit Care. 2014;20:202–209. doi: 10.1097/MCC.0000000000000073. [DOI] [PubMed] [Google Scholar]
- 2.Stadlbauer V, Jalan R. Acute liver failure: liver support therapies. Curr Opin Crit Care. 2007;13:215–221. doi: 10.1097/MCC.0b013e328052c4cc. [DOI] [PubMed] [Google Scholar]
- 3.Nakama T, Hirono S, Moriuchi A, Hasuike S, Nagata K, Hori T, Ido A, Hayashi K, Tsubouchi H. Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology. 2001;33:1441–1450. doi: 10.1053/jhep.2001.24561. [DOI] [PubMed] [Google Scholar]
- 4.Beeson PB. Development of tolerance to typhoid bacterial pyrogen and its abolition by reticulo-endothelial blockade. Proc Soc Exp Biol Med. 1946;61:248–250. doi: 10.3181/00379727-61-15291p. [DOI] [PubMed] [Google Scholar]
- 5.Shi DW, Zhang J, Jiang HN, Tong CY, Gu GR, Ji Y, Summah H, Qu JM. LPS pretreatment ameliorates multiple organ injuries and improves survival in a murine model of polymicrobial sepsis. Inflamm Res. 2011;60:841–849. doi: 10.1007/s00011-011-0342-5. [DOI] [PubMed] [Google Scholar]
- 6.Zhang S, Yang N, Ni S, Li W, Xu L, Dong P, Lu M. Pretreatment of lipopolysaccharide (LPS) ameliorates D-GalN/LPS induced acute liver failure through TLR4 signaling pathway. Int J Clin Exp Pathol. 2014;7:6626–6634. [PMC free article] [PubMed] [Google Scholar]
- 7.Li S, Luo C, Yin C, Peng C, Han R, Zhou J, He Q, Zhou J. Endogenous HMGB1 is required in endotoxin tolerance. J Surg Res. 2013;185:319–328. doi: 10.1016/j.jss.2013.05.062. [DOI] [PubMed] [Google Scholar]
- 8.Aneja RK, Tsung A, Sjodin H, Gefter JV, Delude RL, Billiar TR, Fink MP. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J Leukoc Biol. 2008;84:1326–1334. doi: 10.1189/jlb.0108030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Piao W, Song C, Chen H, Diaz MA, Wahl LM, Fitzgerald KA, Li L, Medvedev AE. Endotoxin tolerance dysregulates MyD88- and Toll/IL-1R domain-containing adapter inducing IFN-beta-dependent pathways and increases expression of negative regulators of TLR signaling. J Leukoc Biol. 2009;86:863–875. doi: 10.1189/jlb.0309189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dong JZ, Wang LP, Zhang SN, Shi KQ, Chen SL, Yang NB, Ni SL, Zhu JH, Lu MQ. LPS pretreatment ameliorates D-galactosamine/lipopolysaccharide-induced acute liver failure in rat. Int J Clin Exp Pathol. 2014;7:7399–7408. [PMC free article] [PubMed] [Google Scholar]
- 11.Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, Robinson M, Van Zoelen MA, Yang H, Li J, Erlandsson-Harris H, Chavan SS, Wang H, Andersson U, Tracey KJ. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A. 2014;111:3068–3073. doi: 10.1073/pnas.1316925111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong Q, Zhang H, Li JH, Duan LH, Zhong S, Kong XL, Zheng F, Tan Z, Xiong P, Chen G, Fang M, Gong FL. High-mobility group box 1 exacerbates concanavalin A-induced hepatic injury in mice. J Mol Med (Berl) 2010;88:1289–1298. doi: 10.1007/s00109-010-0681-7. [DOI] [PubMed] [Google Scholar]
- 13.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR. HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med. 2007;204:2913–2923. doi: 10.1084/jem.20070247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A. High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem. 2010;285:39888–39897. doi: 10.1074/jbc.M110.128348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li WX. Canonical and non-canonical JAK-STAT signaling. Trends Cell Biol. 2008;18:545–551. doi: 10.1016/j.tcb.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–487. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Takano K, Shinoda M, Tanabe M, Miyasho T, Yamada S, Ono S, Masugi Y, Suda K, Fukunaga K, Hayashida T, Hibi T, Obara H, Takeuchi H, Kawachi S, Kawasako K, Okamoto M, Yokota H, Maruyama I, Kitagawa Y. Protective effect of high-mobility group box 1 blockade on acute liver failure in rats. Shock. 2010;34:573–579. doi: 10.1097/SHK.0b013e3181df0433. [DOI] [PubMed] [Google Scholar]
- 18.Park JH, Kim KH, Lee WR, Han SM, Park KK. Protective effect of melittin on inflammation and apoptosis in acute liver failure. Apoptosis. 2012;17:61–69. doi: 10.1007/s10495-011-0659-0. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka M, Shinoda M, Takayanagi A, Oshima G, Nishiyama R, Fukuda K, Yagi H, Hayashida T, Masugi Y, Suda K, Yamada S, Miyasho T, Hibi T, Abe Y, Kitago M, Obara H, Itano O, Takeuchi H, Sakamoto M, Tanabe M, Maruyama I, Kitagawa Y. Gene transfer of high-mobility group box 1 box-A domain in a rat acute liver failure model. J Surg Res. 2015;194:571–580. doi: 10.1016/j.jss.2014.11.022. [DOI] [PubMed] [Google Scholar]
- 20.Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andersson U, Tracey KJ. HMGB1 as a mediator of necrosis-induced inflammation and a therapeutic target in arthritis. Rheum Dis Clin North Am. 2004;30:627–637. xi. doi: 10.1016/j.rdc.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, Yin H, Han J, Huang B, Xu J, Zheng F, Tan Z, Fang M, Rui L, Chen D, Wang S, Zheng X, Wang CY, Gong F. Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant. 2007;7:799–808. doi: 10.1111/j.1600-6143.2007.01734.x. [DOI] [PubMed] [Google Scholar]
- 23.Abraham E, Arcaroli J, Carmody A, Wang H, Tracey KJ. HMG-1 as a mediator of acute lung inflammation. J Immunol. 2000;165:2950–2954. doi: 10.4049/jimmunol.165.6.2950. [DOI] [PubMed] [Google Scholar]
- 24.Musumeci D, Roviello GN, Montesarchio D. An overview on HMGB1 inhibitors as potential therapeutic agents in HMGB1-related pathologies. Pharmacol Ther. 2014;141:347–357. doi: 10.1016/j.pharmthera.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 25.Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107:11942–11947. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka M, Shinoda M, Takayanagi A, Oshima G, Nishiyama R, Fukuda K, Yagi H, Hayashida T, Masugi Y, Suda K, Yamada S, Miyasho T, Hibi T, Abe Y, Kitago M, Obara H, Itano O, Takeuchi H, Sakamoto M, Tanabe M, Maruyama I, Kitagawa Y. Gene transfer of high-mobility group box 1 box-A domain in a rat acute liver failure model. J Surg Res. 2015;194:571–80. doi: 10.1016/j.jss.2014.11.022. [DOI] [PubMed] [Google Scholar]
- 27.Gong X, Luo FL, Zhang L, Li HZ, Wu MJ, Li XH, Wang B, Hu N, Wang CD, Yang JQ, Wan JY. Tetrandrine attenuates lipopolysaccharide-induced fulminant hepatic failure in D-galactosamine-sensitized mice. Int Immunopharmacol. 2010;10:357–363. doi: 10.1016/j.intimp.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 28.Fujii K, Luo Y, Sasahira T, Denda A, Ohmori H, Kuniyasu H. Co-treatment with deoxycholic acid and azoxymethane accelerates secretion of HMGB1 in IEC6 intestinal epithelial cells. Cell Prolif. 2009;42:701–709. doi: 10.1111/j.1365-2184.2009.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu H, Su Z, Wu J, Yang M, Penninger JM, Martin CM, Kvietys PR, Rui T. The alarmin cytokine, high mobility group box 1, is produced by viable cardiomyocytes and mediates the lipopolysaccharide-induced myocardial dysfunction via a TLR4/phosphatidylinositol 3-kinase gamma pathway. J Immunol. 2010;184:1492–1498. doi: 10.4049/jimmunol.0902660. [DOI] [PubMed] [Google Scholar]
- 30.Muller S, Ronfani L, Bianchi ME. Regulated expression and subcellular localization of HMGB1, a chromatin protein with a cytokine function. J Intern Med. 2004;255:332–343. doi: 10.1111/j.1365-2796.2003.01296.x. [DOI] [PubMed] [Google Scholar]
- 31.Linossi EM, Babon JJ, Hilton DJ, Nicholson SE. Suppression of cytokine signaling: the SOCS perspective. Cytokine Growth Factor Rev. 2013;24:241–248. doi: 10.1016/j.cytogfr.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong Y, Medvedev AE. Induction of endotoxin tolerance in vivo inhibits activation of IRAK4 and increases negative regulators IRAK-M, SHIP-1, and A20. J Leukoc Biol. 2011;90:1141–1148. doi: 10.1189/jlb.0611273. [DOI] [PMC free article] [PubMed] [Google Scholar]