

Abstract
In recent years the plasma proteomes of several different myelodysplastic syndrome (MDS) subgroups have been investigated and compared with those of healthy donors. However, the resulting data do not facilitate a direct and statistical comparison of the changes among the different MDS subgroups that would be useful for the selection and proposal of diagnostic biomarker candidates. The aim of this work was to identify plasma protein biomarker candidates for different MDS subgroups by reanalyzing the proteomic data of four MDS subgroups: refractory cytopenia with multilineage dysplasia (RCMD), refractory anemia or refractory anemia with ringed sideroblasts (RA-RARS), refractory anemia with excess blasts subtype 1 (RAEB-1), and refractory anemia with excess blasts subtype 2 (RAEB-2). Reanalysis of a total of 47 MDS patients revealed biomarker candidates, with alpha-2-HS-glycoprotein and leucine-rich alpha-2-glycoprotein as the most promising candidates.
1. Introduction
Myelodysplastic syndrome (MDS) is a group of heterogeneous oncohematological bone marrow disorders characterized by peripheral blood cytopenias, ineffective hematopoiesis, bone marrow hypercellularity, and so forth [1]. MDS classification covers a range from low-risk subgroups with good patient outlook and survival, to high-risk subgroups characterized by a progression of the disease toward acute myeloid leukemia and a poor outcome [2, 3]. The molecular mechanisms that lead to the genesis of MDS and its development are not yet fully understood. Moreover, our knowledge of the changes occurring in MDS remains limited. Some findings at the DNA (chromosomal aberrations [4], up- or downregulation of genes [5], DNA methylation changes [6], single nucleotide polymorphisms [7], etc.) and RNA levels (altered expression of microRNAs in CD34+ cells [8, 9]) have been observed; however, there is a lack of detailed characterization of the changes at the protein level. Protein changes, whether in protein levels or posttranslational modifications, are expected to play a crucial role in the modern diagnostic toolkit. Considerable effort has been expended in the preparation of such tools in recent years (from the studies of plasma protein interactions with antifouling surfaces [10] to the preparation of low- or even nonfouling surfaces suitable for biochip construction [11, 12]); the topic of clinical applications in oncohematology has been reviewed by Fracchiolla et al. [13]; however, the first step has to be the identification of protein biomarker candidates. In our previous studies, we used a proteomic approach to investigate plasma proteome changes in the different MDS subgroups, covering the range from low- to high-risk subgroups: refractory cytopenia with multilineage dysplasia [14], refractory anemia with excess blasts subtype 1 [15], refractory anemia and refractory anemia with ringed sideroblasts [16], and refractory anemia with excess blasts subtype 2 [17]. Several proteins were proposed as potential biomarkers of different MDS subgroups in comparison with control groups of healthy donors. Although the control group study designs were kept similar to maintain consistency in the interpretation of the results and to facilitate comparison of the changes among the different MDS subgroups, only rough estimation may be obtained on this basis. Moreover, some criteria (statistical significance) cannot be estimated by this method at all. Therefore, the goal of this work was to reanalyze the data from our four proteomic studies of different MDS subgroups in order to evaluate the protein biomarker candidates of these different MDS subgroups.
2. Methods
In this work, the data from four previous proteomic studies of different MDS subgroups has been reanalyzed: refractory cytopenia with multilineage dysplasia [14], refractory anemia with excess blasts subtype 1 [15], refractory anemia and refractory anemia with ringed sideroblasts [16], and refractory anemia with excess blasts subtype 2 [17]; only patient data (no healthy control donors) were used. There were 47 myelodysplastic syndrome patients: 22 patients with refractory cytopenia with multilineage dysplasia (RCMD), 10 patients with refractory anemia or refractory anemia with ringed sideroblasts (RA-RARS), 7 patients with refractory anemia with excess blasts subtype 1 (RAEB-1), and 8 patients with refractory anemia with excess blasts subtype 2 (RAEB-2). The median of age was 57, 71.5, 68, and 63.5 years, and the patient make-up was 50%, 40%, 57%, and 38% male in RCMD, RA-RARS, RAEB-1, and RAEB-2, respectively. Patients' characteristics are summarized in Table 1. Diagnoses were established according to the WHO classification criteria [2]. All individuals tested agreed to participate in the study on the basis of an informed consent. All samples were obtained and analyzed in accordance with the Ethical Committee regulations of the Institute of Hematology and Blood Transfusion in Prague.
Table 1.
Patients' characteristics.
| Patient | Sex | Age | Diagnosis | Karyotype | WBC [109/L] | PLT [109/L] | Blasts in PB [%] | NS [%] | IPSS | IPSS-R |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | f | 21 | RCMD | 46, XX | 3.51 | 31 | 0 | 22.9 | Good | Very good |
| 2 | f | 24 | RCMD | 46, XX | 4.79 | 238 | 0 | 52 | Good | Very good |
| 3 | f | 29 | RCMD | 46, XX | 3.93 | 19 | 0 | 27 | Good | Very good |
| 4 | m | 29 | RCMD | 46, XY-polyploidy | 3.81 | 50 | 0 | 73 | Good | Very good |
| 5 | f | 30 | RCMD | 46, XX | 2.53 | 134 | 0 | 36 | Good | Very good |
| 6 | m | 30 | RCMD | 46, XY | 7.96 | 107 | 0 | 69 | Good | Very good |
| 7 | m | 49 | RAEB2 | 46, XY | 4.34 | 15 | 3 | 23 | Good | Very good |
| 8 | f | 50 | RA | 46, XX | 3.64 | 184 | 0 | 59 | Good | Very good |
| 9 | f | 50 | RCMD | 46, XX, inv(9) | 1.81 | 108 | 0 | 48 | Intermediate | Intermediate |
| 10 | f | 51 | RCMD | 46, XX, 9qh+ | 3.80 | 20 | 0 | 58 | Intermediate | Intermediate |
| 11 | m | 55 | RCMD | 46, XY | 2.19 | 236 | 0 | 25 | Good | Very good |
| 12 | f | 56 | RCMD | 46, XX | 3.90 | 129 | 0 | 39 | Good | Very good |
| 13 | f | 56 | RCMD | 46, XX | 4.15 | 211 | 0 | 71 | Good | Very good |
| 14 | m | 58 | RAEB1 | 45, XY, −18, multiple aberrations | 1.26 | 28 | 0 | 30 | Poor | Very poor |
| 15 | f | 58 | RAEB2 | 42~47, XX, del(5)(q?), −7, +8, der(12)t(7; 12)(?; p?13)ins(12; 7)(q?12; ?)ins(12; 7)(q?13; ?), der(17)t(17; 20)(p11.2; ?) | 1.57 | 310 | 5.2 | 43.2 | Poor | Very poor |
| 16 | m | 58 | RCMD | 46, XY −46, XY, del(20)(q12) | 2.00 | 211 | 0 | 56 | Good | Good |
| 17 | m | 58 | RCMD | 46, XY | 2.48 | 153 | 0 | 55 | Good | Very good |
| 18 | m | 59 | RAEB2 | 46, XY −43~44, XY, multiple changes | 2.30 | 110 | 14 | 18 | Poor | Very poor |
| 19 | m | 59 | RCMD | 46, XY | 2.81 | 103 | 0 | 50 | Good | Very good |
| 20 | f | 60 | RA | 46, XX | 7.41 | 149 | 0 | 60 | Good | Very good |
| 21 | m | 60 | RAEB1 | 46, XY | 0.65 | 88 | 0 | 34 | Good | Very good |
| 22 | f | 60 | RAEB2 | 46, XX | 5.36 | 39 | 11 | 46 | Good | Very good |
| 23 | m | 61 | RARS | 46, XY | 5.84 | 218 | 0 | 67 | Good | Very good |
| 24 | m | 62 | RCMD | 46, XY −45, X, −Y | 6.82 | 89 | 0 | 65 | Good | Very good |
| 25 | f | 62 | RCMD | 46, XX | 2.58 | 28 | 0 | 51 | Good | Very good |
| 26 | m | 62 | RCMD | 46, XY | 4.49 | 56 | 0 | 61 | Good | Very good |
| 27 | f | 63 | RA | 46, XX −46, XX, del(5)(q13q13) | 3.54 | 146 | 0 | 52 | Good | Good |
| 28 | f | 64 | RAEB1 | 46, XX, t(2; 12)(p22; q13) | 5.91 | 121 | 1 | 44 | Intermediate | Intermediate |
| 29 | f | 65 | RA | 46, XX −46, XX, del(5)(q15q33) | 3.20 | 192 | 1 | 29 | Good | Good |
| 30 | m | 65 | RCMD | 46, XY −43~46, XY, der(2)t(2; 12)(q37; ?), del(11)(q13) | 2.93 | 297 | 0 | 43 | Poor | Very poor |
| 31 | m | 66 | RCMD | 46, XY, 21ps+ | 6.74 | 81 | 0 | 52 | Intermediate | Intermediate |
| 32 | f | 66 | RCMD | 46, XX | 2.79 | 16 | 0 | 84.3 | Good | Very good |
| 33 | f | 67 | RAEB2 | — | 3.93 | 22 | 14 | 28 | — | — |
| 34 | f | 68 | RAEB1 | 46, XX −46, XX, del(5)(q22q33) | 3.80 | 412 | 0 | 50 | Good | Good |
| 35 | m | 68 | RCMD | 46, XY −45, X, −Y | 19.94 | 399 | 3 | 51 | Good | Very good |
| 36 | f | 70 | RAEB1 | 46, XX | 1.13 | 150 | 0 | 46 | Good | Very good |
| 37 | m | 70 | RAEB2 | 5q31 deletion (8 of 11 tests) | 3.70 | 156 | 8 | 73 | Good | Good |
| 38 | m | 71 | RAEB1 | 46, XY | 4.25 | 85 | 3 | 17 | Good | Very good |
| 39 | m | 71 | RAEB1 | 46, XY | 1.39 | 21 | 0 | 20 | Good | Very good |
| 40 | f | 72 | RCMD | 46, XX, del(5)(q13.3q33.3) | 4.18 | 119 | 0 | 47 | Good | Good |
| 41 | f | 76 | RAEB2 | 47–51, XX-multiple changes | 1.52 | 8 | 2 | 42 | Poor | Very poor |
| 42 | m | 78 | RA | 46, XY | 8.65 | 162 | 0 | 75 | Good | Very good |
| 43 | f | 78 | RA | 46, XX, del(5)(q13q33) | 3.77 | 288 | 0 | 67 | Good | Good |
| 44 | m | 78 | RA | 46, XY | 5.36 | 243 | 1 | 68 | Good | Very good |
| 45 | f | 79 | RAEB2 | 46, XX -46, XX, del(5)(q13q33) | 0.43 | 6 | 38 | 8 | Good | Good |
| 46 | f | 86 | RARS | 46, XX | 8.86 | 375 | 0 | 55 | Good | Very good |
| 47 | m | 89 | RARS | 46, XY | 6.44 | 155 | 0 | 40 | Good | Very good |
WBC: white blood cells; PLT: platelets; PB: peripheral blood; NS: neutrophil segments.
Scanned gel images obtained from our previous four proteomic studies were used in this study; blood collection, sample preparation, high-abundance plasma protein depletion, 2D SDS-PAGE protein separation, protein visualization, and gel digitization have been described in detail [14, 18]. Digitized gel images were processed with Progenesis SameSpots software (Nonlinear Dynamics, Newcastle upon Tyne, UK); images were divided into four groups according to MDS diagnoses, and the fold and P values of all spots were computed by the software using one-way ANOVA analysis. Protein identification was performed for spots (proteins within the spots) that were not submitted for protein identification in the previous studies. An HCT ultra ion-trap mass spectrometer with nanoelectrospray ionization (Bruker Daltonics, Bremen, Germany) coupled to a nanoLC system UltiMate 3000 (Dionex, Sunnydale, CA, USA) was used to perform MS analysis. Mascot (Matrix Science, London, UK) was used for database searching (Swiss-Prot). Two unique peptides (with a higher Mascot score than the minimum for identification, P < 0.05) were necessary to identify a protein. The procedure was described in detail previously [18]. Western blot analysis was performed as previously described in detail [19]. Briefly, 6 samples (3 males and 3 females) were used for each MDS subgroup as a pooled sample. Proteins of pooled samples were precipitated with acetone, protein pellets were diluted in a sample buffer, and SDS-PAGE was performed, followed by protein transfer to a PVDF membrane. The following primary antibodies were used: monoclonal mouse anti-leucine-rich alpha-2-glycoprotein (ab57992), 1 : 400 (Abcam, Cambridge, UK); monoclonal mouse antialbumin (A6684), 1 : 2000 (Sigma-Aldrich, Prague, Czech Republic); polyclonal rabbit anti-alpha-2-HS-glycoprotein (ab112528), 1 : 1000 (Abcam); and polyclonal mouse antiapolipoprotein A-I (H00000335-B01P), 1 : 1000 (Abnova, Taipei, Taiwan). The following secondary antibodies were utilized: rabbit anti-mouse IgG antibody conjugated with peroxidase (A9044), 1 : 80000 (Sigma-Aldrich) and goat anti-rabbit IgG antibody conjugated with peroxidase (A0545), 1 : 80000 (Sigma-Aldrich). Protein bands were visualized using a 1-Step Ultra TMB-Blotting Solution (Thermo Scientific, Waltham, MA, USA).
3. Results and Discussion
The aim of this work was to evaluate plasma protein biomarker candidates of myelodysplastic syndrome subgroups by reanalyzing previously published proteomic data to allow direct and statistical comparisons. In order to select the most promising protein candidates, a two-step selection process was applied. In the first step, all four MDS subgroups were compared together and spots that were found to significantly differ (ANOVA P < 0.05) among the groups were selected; 42 different spots were found. In the second step, all the groups were compared mutually (each to each other), and the only spots selected in the first step were considered. In order to maintain the same level of significance, a Bonferroni correction was applied in the second step selection [20]. Therefore, the P value threshold for the second step comparison was lowered to P < 0.00833. As an additional criterion, only spots with at least a 50% change of their normalized spot volumes were accepted. There were then 23 different spots found which satisfied these criteria; proteins in 20 spots (Figure 1) were identified by mass spectrometry. The numbers of spots that were found to differ between the compared pairs of MDS subgroups are summarized in Table 2. A brief characterization of these spots (P value and the relative change between the groups) with regard to the compared pairs of MDS subgroups is shown in Table 3. Identification of the proteins, together with the number of unique identified peptides, accession numbers, and protein sequence coverage, is summarized in Table 4. From the results in Table 2 there is no direct correlation between the number of differences and the severity of the subgroups. Although the patient cohort is relatively small, the results support the notion proposed in our previous studies that “a degree of change” is the principal factor affecting proteome alterations observed for different MDS subgroups [16, 17]. Therefore, when protein posttranslational modifications are taken into consideration, it is not surprising that several proteins found to differ in this work were also identified to differentiate between MDS subgroups and the healthy control groups: alpha-2-HS-glycoprotein, leucine-rich alpha-2-glycoprotein, retinol-binding protein 4, hemopexin, apolipoprotein A-I, and so forth. These observations suggest that it is unlikely to find a single protein as a diagnostic MDS biomarker when only considering its plasma level change. However, finding a single protein biomarker with respect to its plasma level change can be possible for potential prognostic MDS biomarkers, as previously indicated for alpha-2-HS-glycoprotein [16, 17]. This protein seemed to decrease its plasma level relative to the severity of the MDS subgroups studied; and moreover, it was also shown that its plasma level decrease reflected the degree of malignancy found in other different tumor types [21].
Figure 1.
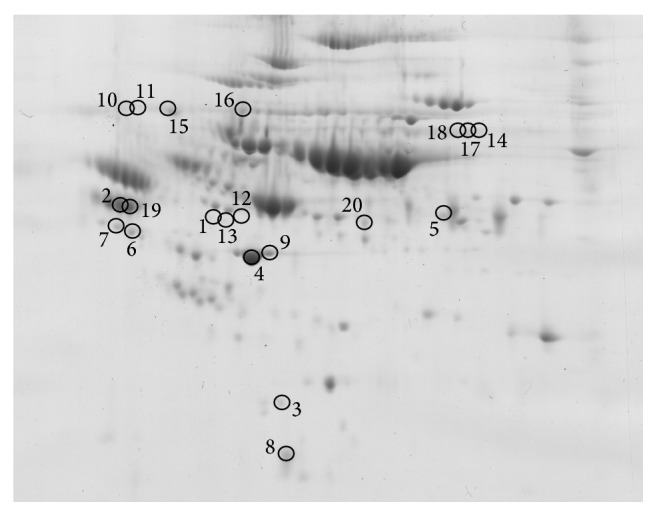
Positions of the spots. Positions of the spots with identified proteins were displayed on an illustrative 2D gel of a patient sample. For better clarity the gel image is shown as highlighted by brightness and contrast image adjustment.
Table 2.
The numbers of spots found to differ between the compared pairs of MDS subgroups.
| RCMD | RA-RARS | RAEB-1 | RAEB-2 | |
|---|---|---|---|---|
| RCMD | — | — | — | — |
| RA-RARS | 6 | — | — | — |
| RAEB-1 | 11 | 0 | — | — |
| RAEB-2 | 6 | 5 | 0 | — |
Table 3.
Brief characterization of the identified spots.
| RCMD versus RA-RARS | |||||
|---|---|---|---|---|---|
| Increase in RCMD | Increase in RA-RARS | ||||
| Spot | P | r | Spot | P | r |
| 8 | 0.00117 | 1.6 | 3 | 0.00002 | 2.3 |
| 15 | 0.00061 | 1.7 | 14 | 0.00534 | 1.6 |
| — | — | — | 17 | 0.00107 | 1.6 |
| — | — | — | 18 | 0.00093 | 1.6 |
|
| |||||
| RCMD versus RAEB-1 | |||||
| Increase in RCMD | Increase in RAEB-1 | ||||
| Spot | P | r | Spot | P | r |
|
| |||||
| 2 | 0.0032 | 1.8 | 1 | 0.00074 | 6.4 |
| 5 | 0.00382 | 1.6 | 12 | 0.00177 | 4.7 |
| 8 | 0.00011 | 2.0 | 13 | 0.00232 | 5.2 |
| 19 | 0.00124 | 1.6 | 14 | 0.00093 | 1.7 |
| 20 | 0.0035 | 1.5 | 16 | 0.00274 | 1.7 |
| — | — | — | 18 | 0.00519 | 1.6 |
|
| |||||
| RCMD versus RAEB-2 | |||||
| Increase in RCMD | Increase in RAEB-2 | ||||
| Spot | P | r | Spot | P | r |
|
| |||||
| 8 | 0.00392 | 1.6 | 6 | 0.00787 | 1.6 |
| 9 | 0.00335 | 1.6 | 7 | 0.00525 | 1.7 |
| — | — | — | 10 | 0.00659 | 1.6 |
| — | — | — | 18 | 0.00701 | 1.6 |
|
| |||||
| RA-RARS versus RAEB-2 | |||||
| Increase in RA-RARS | Increase in RAEB-2 | ||||
| Spot | P | r | Spot | P | r |
|
| |||||
| 3 | 0.00538 | 2.1 | 10 | 0.00435 | 1.8 |
| 4 | 0.00348 | 1.5 | 11 | 0.00104 | 1.6 |
| 9 | 0.00008 | 1.8 | — | — | — |
P: t-test P value, r: fold change value.
Table 4.
Protein identification.
| Spot | Protein identification | Peptides | AN | SC (%) |
|---|---|---|---|---|
| 1 | Alpha-1-antitrypsin | 12 | P01009 | 38 |
|
| ||||
| 2 | Alpha-2-HS-glycoprotein | 2 | P02765 | 13 |
|
| ||||
| 3 | Apolipoprotein A-I | 6 | P02647 | 25 |
|
| ||||
| 4 | Apolipoprotein A-IV | 10 | P06727 | 44 |
|
| ||||
| 5 | Hemopexin | 3 | P02790 | 11 |
|
| ||||
| 6 | Leucine-rich alpha-2-glycoprotein | 4 | P02750 | 23 |
|
| ||||
| 7 | Leucine-rich alpha-2-glycoprotein | 3 | P02750 | 18 |
|
| ||||
| 8 | Retinol-binding protein 4 | 2 | P02753 | 16 |
|
| ||||
| 9 | Actin, cytoplasmic 1; 2 | 4; 4 | P60709; P63261 | 22; 22 |
| Apolipoprotein A-IV | 5 | P06727 | 21 | |
|
| ||||
| 10 | Alpha-1-antichymotrypsin | 6 | P01011 | 23 |
| Plasma protease C1 inhibitor | 4 | P05155 | 13 | |
|
| ||||
| 11 | Alpha-1-antichymotrypsin | 4 | P01011 | 14 |
| Plasma protease C1 inhibitor | 4 | P05155 | 18 | |
|
| ||||
| 12 | Alpha-1-antitrypsin | 10 | P01009 | 29 |
| Antithrombin-III | 2 | P01008 | 8 | |
|
| ||||
| 13 | Alpha-1-antitrypsin | 9 | P01009 | 28 |
| Antithrombin-III | 2 | P01008 | 8 | |
|
| ||||
| 14 | Ig mu chain C region | 2 | P01871 | 17 |
| Prothrombin | 3 | P00734 | 20 | |
|
| ||||
| 15 | Plasma protease C1 inhibitor | 3 | P05155 | 13 |
| Alpha-1-antichymotrypsin | 2 | P01011 | 10 | |
|
| ||||
| 16 | Alpha-1-antitrypsin | 3 | P01009 | 24 |
| Prothrombin | 4 | P00734 | 24 | |
| Complement C4-A; B | 4 | P0C0L4; P0C0L5 | 4 | |
|
| ||||
| 17 | Prothrombin | 3 | P00734 | 25 |
| Serum albumin | 3 | P02768 | 11 | |
| Ig mu chain C region | 2 | P01871 | 13 | |
|
| ||||
| 18 | Serum albumin | 3 | P02768 | 8 |
| Ig mu chain C region | 2 | P01871 | 19 | |
| Prothrombin | 3 | P00734 | 27 | |
|
| ||||
| 19 | Alpha-1-antichymotrypsin | 3 | P01011 | 11 |
| Alpha-2-HS-glycoprotein | 2 | P02765 | 9 | |
| Kininogen-1 | 3 | P01042 | 8 | |
| Corticosteroid-binding globulin | 2 | P08185 | 14 | |
|
| ||||
| 20 | Pigment epithelium-derived factor | 4 | P36955 | 16 |
| Complement factor I | 4 | P05156 | 10 | |
| Beta-2-glycoprotein 1 | 3 | P02749 | 26 | |
| Alpha-1-antichymotrypsin | 2 | P01011 | 14 | |
AN: protein accession number (UniProt), SC: sequence coverage in %.
In order to estimate whether the changes determined by 2D SDS-PAGE reflect the plasma level changes or posttranslational modifications of proteins, we performed western blot analysis for the selected proteins. Alpha-2-HS-glycoprotein and leucine-rich alpha-2-glycoprotein were selected as previously proposed MDS biomarker candidates (differentiating MDS subgroups from healthy controls), as well as due to their possible role in MDS pathophysiology as previously described in detail in [16] and [15], respectively. Apolipoprotein A1 was selected as it was identified in all four of the MDS subgroups studied and because it has been observed to form posttranslationally modified isoforms in cardiovascular disease patients [19, 22]. Serum albumin was selected as a control protein of the acute phase reaction to reflect a possible inflammation influence. Our findings are illustrated in Figure 2.
Figure 2.

Western blot analysis. Western blot analysis was performed for alpha-2-HS-glycoprotein (a), leucine-rich alpha-2-glycoprotein (b), apolipoprotein A1 (c), and serum albumin (d) using pooled plasma samples of patients with four different MDS subgroups: refractory cytopenia with multilineage dysplasia (RCMD), refractory anemia or refractory anemia with ringed sideroblasts (RA-RARS), refractory anemia with excess blasts subtype 1 (RAEB-1), and refractory anemia with excess blasts subtype 2 (RAEB-2). For better clarity the western blot analysis results are shown as highlighted by brightness and contrast image adjustment.
Western blotting of alpha-2-HS-glycoprotein showed several bands of approximately 50 kDa with a trend of decreasing intensity in advanced MDS subgroups. This result is in agreement with our electrophoretic data; however, it is apparent from the western blot that the representation of individual bands differs more substantially relative to total protein levels. The most obvious change can be observed for the bottom band, whose intensity increases substantially in the RAEB-1 subgroup. This supports the need for precise characterization of A2HSG posttranslational modifications and their quantification. Western blot analysis of leucine-rich alpha-2-glycoprotein revealed two bands of approximately 48 and 60 kDa. The lower 48 kDa band corresponds to that identified in 2D electrophoresis and shows the trend of increasing its intensity in advanced MDS subgroups. This is in agreement with the data obtained by 2D electrophoresis. As in the case of alpha-2-HS-glycoprotein, the modifications need to be characterized. Apolipoprotein A1 was shown to increase the spot volume in RA-RARS compared to RCMD and RAEB-2. It is clear from the western blot that the highest intensities were observed for both the RA-RARS and RAEB-1 subgroups. The fact that the changes were found for RA-RARS by 2D electrophoresis and not for RAEB-1 was most probably caused by the low number of RAEB-1 samples. No obvious changes were observed for albumin; therefore, we assume a minimal influence of the acute phase reaction on the results. Nevertheless, it has been recently shown that there are many other factors (genetic, clinical, or lifestyle factors) that can strongly affect protein plasma levels [23]. Moreover, the alterations in plasma protein levels may be affected by defects in cells' functions. For example, Blalock et al. [24] reported that phosphorylated form (on Thr451) of the dsRNA-dependent kinase accumulates in the cell nucleus of high-risk MDS patients and thus probably alters nuclear signaling. The implications of this finding on the disease or plasma protein changes are not known. A study by Aivado et al. [25] showed that CXC chemokine ligands 4 and 7 decreased their serum levels in advanced MDS patients compared to non-MDS cytopenia patients. The authors also showed that this serum decrease was related to platelets and, therefore, both the chemokines should be considered as platelet-derived markers. That platelet function impaired in MDS patients was recently confirmed in the study by Fröbel et al. [26]. In our study, the CXCL4 and CXCL7 were not identified; this is, however, not surprising when considering their low plasma (serum) levels. Aivado et al. used mass spectrometry-based detection which is capable of detecting proteins of lower concentrations compared to electrophoresis-based studies; the advantages and limitations of this approach have been reviewed in literature [27]. Unfortunately, there is no all-purpose proteomic method and a subset of proteins can be observed at once using a specific proteomic method. In the study by Chen et al. [28] ProteinChip array technology and mass spectrometry were used to investigate disease-associated and therapy-associated differences in sera of del(5q) MDS patients. Platelet factor 4 (also known as CXCL4) was found to be a potential therapy-associated marker; therefore, the results supported the observation by Aivado et al. [25]. Several proteins were proposed to be potential disease-associated markers (e.g., ITIH4, transferrin, transthyretin); these proteins were not identified in our reanalysis; however, it is not surprising as there were no non-MDS control samples used in our reanalysis contrary to the work by Chen et al. [28]. Nevertheless, when the results obtained by Chen et al. are compared to our previous protemic studies (which were used for this reanalysis) investigating different MDS subgroup patients with the healthy control samples, there were the same proteins identified. Since the proteins are altered between MDS and control samples but seem to be unaltered among MDS subgroups as shown in our reanalysis, these results indicate that the protein changes may be related to other (patho)physiological processes and not to be specific to MDS.
Identifying new biomarkers could have a significant impact on the clinical practice. While for diagnostics it is important to investigate the differences between the healthy (or nondisease) and patient cohorts, it is essential to identify alterations in the disease progression for prognosis and therapy monitoring. Considering MDS, it is important to describe the changes among different subgroups (as was the aim of our study), alterations related to the therapy (e.g., the study by Chen et al.), or specific changes during the disease progression. The changes related to MDS progression toward acute myeloid leukemia were investigated in work by Braoudaki et al. [29] in plasma, bone marrow, and cell lysate samples of pediatric patients. The most promising protein candidates observed in our reanalysis were also identified by Braoudaki et al.: leucine-rich alpha-2-glycoprotein and alpha-2-HS-glycoprotein. This observation further highlights the potential and importance of those protein candidates. Moreover, another protein coidentified in two spots (antithrombin-III) in our reanalysis was also observed in the study by Braoudaki et al.; antithrombin-III was found to be differentially expressed in both bone marrow and peripheral blood plasma samples and it was shown to be altered after 3 months of treatment. Thus, antithrombin-III could be another promising target of future proteomic investigations.
4. Conclusions
In conclusion, plasma protein biomarker candidates have been presented in this work with respect to different MDS subgroups. Alpha-2-HS-glycoprotein and leucine-rich alpha-2-glycoprotein appear to be the most promising candidates with regard to western blot observations, as well as our previous results detailing the differences in plasma proteome patterns between MDS subgroups and healthy donors. The presented results should be a catalyst for further MDS biomarker validation requiring precise protein posttranslational modification characterization, profiling of the changes in MDS subgroups, and extended statistical validation with large patient cohorts.
Acknowledgments
This study was supported by the Czech Science Foundation P205/12/G118 and by the Ministry of Health of the Czech Republic project for the conceptual development of the research organization (Institute of Hematology and Blood Transfusion).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Malcovati L., Hellström-Lindberg E., Bowen D., et al. Diagnosis and treatment of primary myelodysplastic syndromes in adults: recommendations from the European LeukemiaNet. Blood. 2013;122(17):2943–2964. doi: 10.1182/blood-2013-03-492884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vardiman J. W., Harris N. L., Brunning R. D. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100(7):2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- 3.Vardiman J. W., Thiele J., Arber D. A., et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 4.Zemanova Z., Michalova K., Buryova H., et al. Involvement of deleted chromosome 5 in complex chromosomal aberrations in newly diagnosed myelodysplastic syndromes (MDS) is correlated with extremely adverse prognosis. Leukemia Research. 2014;38(5):537–544. doi: 10.1016/j.leukres.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Vasikova A., Belickova M., Budinska E., Cermak J. A distinct expression of various gene subsets in CD34+ cells from patients with early and advanced myelodysplastic syndrome. Leukemia Research. 2010;34(12):1566–1572. doi: 10.1016/j.leukres.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 6.Cechova H., Lassuthova P., Novakova L., et al. Monitoring of methylation changes in 9p21 region in patients with myelodysplastic syndromes and acute myeloid leukemia. Neoplasma. 2012;59(2):168–174. doi: 10.4149/neo_2012_022. [DOI] [PubMed] [Google Scholar]
- 7.Belickova M., Merkerova M. D., Stara E., et al. DNA repair gene variants are associated with an increased risk of myelodysplastic syndromes in a Czech population. Journal of Hematology and Oncology. 2013;6(1, article 9) doi: 10.1186/1756-8722-6-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Merkerova M. D., Krejcik Z., Votavova H., Belickova M., Vasikova A., Cermak J. Distinctive microRNA expression profiles in CD34+ bone marrow cells from patients with myelodysplastic syndrome. European Journal of Human Genetics. 2011;19(3):313–319. doi: 10.1038/ejhg.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Votavova H., Grmanova M., Dostalova Merkerova M., et al. Differential expression of MicroRNAs in CD34+ cells of 5q- syndrome. Journal of Hematology and Oncology. 2011;4, article 1 doi: 10.1186/1756-8722-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emmenegger C. R., Brynda E., Riedel T., Sedlakova Z., Houska M., Alles A. B. Interaction of blood plasma with antifouling surfaces. Langmuir. 2009;25(11):6328–6333. doi: 10.1021/la900083s. [DOI] [PubMed] [Google Scholar]
- 11.Riedel T., Brynda E., Dyr J. E., Houska M. Controlled preparation of thin fibrin films immobilized at solid surfaces. Journal of Biomedical Materials Research A. 2009;88(2):437–447. doi: 10.1002/jbm.a.31755. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez-Emmenegger C., Brynda E., Riedel T., et al. Polymer brushes showing non-fouling in blood plasma challenge the currently accepted design of protein resistant surfaces. Macromolecular Rapid Communications. 2011;32(13):952–957. doi: 10.1002/marc.201100189. [DOI] [PubMed] [Google Scholar]
- 13.Fracchiolla N. S., Artuso S., Cortelezzi A. Biosensors in clinical practice: focus on oncohematology. Sensors. 2013;13(5):6423–6447. doi: 10.3390/s130506423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Májek P., Reicheltová Z., Suttnar J., Čermák J., Dyr J. E. Plasma proteome changes associated with refractory cytopenia with multilineage dysplasia. Proteome Science. 2011;9, article 64 doi: 10.1186/1477-5956-9-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Májek P., Reicheltová Z., Suttnar J., Čermák J., Dyr J. E. Plasma protein alterations in the refractory anemia with excess blasts subtype 1 subgroup of myelodysplastic syndrome. Proteome Science. 2012;10(1, article 31) doi: 10.1186/1477-5956-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Májek P., Riedelová-Reicheltová Z., Suttnar J., Pečánková K., Čermák J., Dyr J. E. Plasma proteome changes associated with refractory anemia and refractory anemia with ringed sideroblasts in patients with myelodysplastic syndrome. Proteome Science. 2013;11(1, article 14) doi: 10.1186/1477-5956-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majek P., Riedelova-Reicheltova Z., Suttnar J., Pecankova K., Cermak J., Dyr J. E. Proteome changes in the plasma of myelodysplastic syndrome patients with refractory anemia with excess blasts subtype 2. Disease Markers. 2014;2014:8. doi: 10.1155/2014/178709.178709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Májek P., Reicheltová Z., Štikarová J., Suttnar J., Sobotková A., Dyr J. E. Proteome changes in platelets activated by arachidonic acid, collagen, and thrombin. Proteome Science. 2010;8, article 56 doi: 10.1186/1477-5956-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Májek P., Reicheltová Z., Suttnar J., et al. Plasma proteome changes in cardiovascular disease patients: novel isoforms of apolipoprotein A1. Journal of Translational Medicine. 2011;9, article 84 doi: 10.1186/1479-5876-9-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holm S. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics. 1979;6(2):65–70. [Google Scholar]
- 21.Petrik V., Saadoun S., Loosemore A., et al. Serum α2-HS glycoprotein predicts survival in patients with glioblastoma. Clinical Chemistry. 2008;54(4):713–722. doi: 10.1373/clinchem.2007.096792. [DOI] [PubMed] [Google Scholar]
- 22.Cubedo J., Padró T., Badimon L. Glycoproteome of human apolipoprotein A-I: N- and O-glycosylated forms are increased in patients with acute myocardial infarction. Translational Research. 2014;164(3):209–222. doi: 10.1016/j.trsl.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Enroth S., Johansson Å., Enroth S. B., Gyllensten U. Strong effects of genetic and lifestyle factors on biomarker variation and use of personalized cutoffs. Nature Communications. 2014;5, article 4684 doi: 10.1038/ncomms5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blalock W. L., Bavelloni A., Piazzi M., et al. Multiple forms of PKR present in the nuclei of acute leukemia cells represent an active kinase that is responsive to stress. Leukemia. 2011;25(2):236–245. doi: 10.1038/leu.2010.264. [DOI] [PubMed] [Google Scholar]
- 25.Aivado M., Spentzos D., Germing U., et al. Serum proteome profiling detects myelodysplastic syndromes and identifies CXC chemokine ligands 4 and 7 as markers for advanced disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(4):1307–1312. doi: 10.1073/pnas.0610330104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fröbel J., Cadeddu R.-P., Hartwig S., et al. Platelet proteome analysis reveals integrin-dependent aggregation defects in patients with myelodysplastic syndromes. Molecular and Cellular Proteomics. 2013;12(5):1272–1280. doi: 10.1074/mcp.M112.023168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beranova-Giorgianni S. Proteome analysis by two-dimensional gel electrophoresis and mass spectrometry: strengths and limitations. TrAC—Trends in Analytical Chemistry. 2003;22(5):273–281. doi: 10.1016/s0165-9936(03)00508-9. [DOI] [Google Scholar]
- 28.Chen C., Bowen D. T., Giagounidis A. A. N., Schlegelberger B., Haase S., Wright E. G. Identification of disease- and therapy-associated proteome changes in the sera of patients with myelodysplastic syndromes and del(5q) Leukemia. 2010;24(11):1875–1884. doi: 10.1038/leu.2010.182. [DOI] [PubMed] [Google Scholar]
- 29.Braoudaki M., Tzortzatou-Stathopoulou F., Anagnostopoulos A. K., et al. Proteomic analysis of childhood de novo acute myeloid leukemia and myelodysplastic syndrome/AML: correlation to molecular and cytogenetic analyses. Amino Acids. 2011;40(3):943–951. doi: 10.1007/s00726-010-0718-9. [DOI] [PubMed] [Google Scholar]