

Abstract
The cyclic AMP-dependent protein kinase (PKA), which activates prosurvival signaling proteins, has been implicated in the expression of long-term potentiation and hippocampal long-term memory. It has come to light that H89 commonly known as the PKA inhibitor have diverse roles in the nervous system that are unrelated to its role as a PKA inhibitor. We have investigated the role of H89 in ischemic and reperfusion injury. First, we examined the expression of postsynaptic density protein 95 (PSD95), microtubule-associated protein 2 (MAP2), and synaptophysin in mouse brain after middle cerebral artery occlusion injury. Next, we examined the role of H89 pretreatment on the expression of brain-derived neurotrophic factor (BDNF), PSD95, MAP2, and the apoptosis regulators Bcl2 and cleaved caspase-3 in cultured neuroblastoma cells exposed to hypoxia and reperfusion injury. In addition, we investigated the alteration of AKT activation in H89 pretreated neuroblastoma cells under hypoxia and reperfusion injury. The data suggest that H89 may contribute to brain recovery after ischemic stroke by regulating neuronal death and proteins related to synaptic plasticity.
1. Introduction
Protein kinase A (PKA) [1] acts to phosphorylate other proteins, regulating them in a reversible manner. When cyclic adenosine monophosphate (cAMP) binds to the subunits of PKA, they undergo a conformational change that promotes phosphorylation [2]. PKA is implicated also in neural health. It stimulates neurite outgrowth in neurons and neuronal cell lines [3, 4] and promotes axon regeneration in vivo [5, 6]. cAMP/PKA signaling affects long-term synaptic plasticity and long-term memory [7].
Many studies that evaluate the role of PKA, which include smooth muscle cells [8, 9], neuronal tissue [10, 11], and epithelial cells [12, 13], have relied on the isoquinoline derivative N-[2-p-bromocinnamylamino-ethyl]-5-isoquinolinesulfonamide (H89), an inhibitor of PKA. H89 has an inhibition constant (K i) of 0.05 mM in its inhibition of PKA [14, 15]. However, effects of H89 that are unrelated to its inhibition have been observed. In a kinase study, at a concentration of 10 μM, H89 inhibited the activity of the protein kinases Rho-associated kinase- (ROCK-) II, MSK1 and the ribosomal protein S6 kinase β-1 (S6K1) far more potently than it inhibited PKA itself [16]. In addition, H89 10 μM maintains the neurite outgrowth of neuroblastoma cells [17]. There are several reports that H89 reduced Ca2+ uptake into the sarcoplasmic reticulum by attenuating the Ca2+-ATPase's [18] affinity for calcium [19]. At 20 μM, H89 prevented the glucose-induced increase in cytosolic calcium in pancreatic islets and attenuated the release of calcium in a differentiated β-cell line. In a study of expression of myelin basic protein in oligodendrocytes, H89 is involved in the phosphorylation of extracellular-signal–regulated kinase 1 and 2 (ERK 1 and 2) phosphorylation in response to insulin-like growth factor-1 [20] and it lowered potassium current through voltage-gated channels in rat myocytes [21].
Of particular interest is the H89 inhibition of S6K1, noted above. S6K1 is a downstream target of the mammalian target of rapamycin (mTOR) protein, which regulates the autophagy pathway [22] and is a mechanism target for regulation of cell size [23]. Several researchers have questioned the role of PKA in autophagy, since the studies rely at least in part on the selectivity of H89, which they consider uncertain [24, 25]. The second issue involves the action of H89 itself. Clearly, it has physiological effects unrelated to PKA. We have elected to examine those effects and chose to focus on H89's role in neural health, especially ischemic stroke.
Cerebral ischemia leads to neuronal death and synaptic dysfunction, resulting in cognitive decline [26–29]. Understanding the pathogenesis after ischemic stroke should inform medical care and maximize recovery. In the present study, we investigated the role of H89 in many aspects of nervous system function. Specifically, we examined its role in the expression of brain-derived neurotrophic factor (BDNF) in the development of neurites to axons [30–32], learning and memory [33], synaptic plasticity [34], the expression of B-cell lymphoma 2 (Bcl2) [35, 36] as it relates to neuronal death, the expression of synaptophysin [37], postsynaptic density protein 95 (PSD-95) [38, 39] as it relates to synaptic plasticity, and the expression of microtubule-associated protein 2 (MAP2). The latter interacts with actin filaments, shown to be necessary for neurite outgrowth [40–43] in a middle cerebral artery occlusion (MCAO) animal model and in an in vitro study. In present study, we suggest that H89 may confer protection from brain damage following cerebral ischemia.
2. Materials and Methods
2.1. Animal Model
Male C57BL/6 mice (Orient, GyeongGi-Do, Korea) that were eight-to-twelve weeks old were used in this study. Hypoxia followed by reperfusion (H/R) was imposed by subjecting mice to transient focal cerebral ischemia by intraluminal middle cerebral artery blockade with a nylon suture, as previously described [44]. After 60 min of MCAO, blood flow was restored by withdrawing the suture and regional cerebral blood flow was monitored with a laser Doppler flow meter (Transonic Systems, Inc., Ithaca, NY, USA). All animal procedures and experiments were performed in accordance with the Guide to the Care and Use of Laboratory Animals and were approved by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures were done at room temperature unless indicated otherwise. We used 5 rats in each group for study. Each measurement included 3 repeats per animal.
2.2. Immunohistochemistry
Frozen brain sections were cut into 5 μm sections and mounted on clean glass slides (Thermo Scientific, Waltham, MA, USA), air-dried, and fixed in cold acetone for 10 min at −20°C. The slides were washed in Tris-buffered saline (TBS; 20 nM Tris (pH 7.2), 150 mM NaCl), incubated with 0.3% H2O2 in methanol to quench endogenous peroxidase activity, and washed three times with distilled water, and the sections were blocked with 10% normal rabbit serum. Additional frozen brain sections (20 μm) were fixed in ice-cold acetone for 20 min. To block nonspecific labeling, sections were incubated in 5% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, USA) in 0.1% phosphate-buffered saline (PBS) for 30 min before addition of primary and secondary antibodies. Primary antibodies for PSD-95 (1 : 100, Millipore, Massachusetts, MA, USA), synaptophysin (1 : 100, Millipore, Massachusetts, MA, USA), and MAP2 (1 : 100, Abcam, Cambridge, MA, USA) were applied to the samples for 24 h at 4°C; then the samples were incubated with the appropriate florescence secondary antibody (1 : 100, Invitrogen, Carlsbad, CA, USA) for 90 min, washed three times for 10 min in PBS with Tween-20 (PBST), and incubated with rhodamine-conjugated sheep anti-rabbit or fluorescein isothiocyanate- (FITC-) conjugated sheep anti-mouse secondary antibody (both diluted to 1 : 200 with 5% BSA fraction V in 0.1% PBST) for 2 h in the dark. This was followed by three washes in PBS and incubation in 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich, St. Louis, MO, USA) for counterstaining. Tissues were then visualized under a confocal microscope (Zeiss LSM 700, Carl Zeiss, Thornwood, NY, USA).
2.3. Cell Culture
Neuro2A (N2A) cells purchased from ATCC biotechnology (ATCC, Manassas, VA, USA) were derived from mouse neuroblastoma. The cells exhibited properties of neuronal stem cells and were capable of differentiating into neuron-like cells in the presence of retinoic acid (RA). Undifferentiated N2A cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) and 100 μg/mL penicillin-streptomycin (Gibco, Grand Island, NY, USA). N2A cells were passaged at least twice and then plated at 5 × 104 cells/mL in DMEM supplemented with 10% FBS for 24 h, after which the medium was changed to DMEM supplemented with 2% FBS and 20 μM RA for differentiation. Cultures were maintained in a humidified atmosphere of 5% CO2 at 37°C. The medium was changed every two days [45].
2.4. Hypoxia and Reperfusion (H/R) and H89 Treatment
Confluent cells were transferred to an anaerobic chamber (Forma Scientific, OH, USA, O2 tension = 0.1%). They were washed three times with PBS and the culture medium was replaced with deoxygenated, glucose-free balanced salt solution and incubated for 4 h. Following H/R injury, cells were incubated for 18 h under normal growth conditions [46]. H89 (10 μM, Sigma-Aldrich, St. Louis, MO, USA) was treated in the N2A cells at 2 h before H/R injury. In present study, we used the 10 μM concentration of H89, considering previous researches regarding other functions except from PKA inhibitor[17–19, 47, 48].
2.5. Neurite Length Measurement
To determine the length of their neurites, the cells were fixed for 20 min in 3.7% formaldehyde. Neurite formation was defined as an outgrowth from the cell body that was longer than the diameter of the cell body. N2A cells in three randomly selected fields (30–100 cells per field) were measured using ImageJ software (ImageJ, Madison, WI, USA) [49]. At least 30 cells per treatment were scored [50].
2.6. Reverse Transcription PCR (RT-PCR)
To examine the expression of BDNF, Bcl2, and MAP2 in N2A cells after H/R injury, RT-PCR was performed. Briefly, samples were lysed with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and total RNA was extracted according to the manufacturer's protocol. Complementary DNA synthesis from mRNA and sample normalization was performed. PCR was performed using the following thermal cycling conditions: 10 min at 95°C, 35 cycles of denaturing at 95°C for 15 sec, annealing for 30 sec at 70°C, elongation at 72°C for 30 sec, final extension for 10 min at 72°C, and maintenance at 4°C. PCR was performed using the following primers (5′ to 3′); BDNF (F): AGT GAT GAC CAT CCT TTT CCT TAC, (R): CCT CAA ATG TGT CAT CCA AGG A, Bcl2 (F): AAG CTG TCA CAG AGG GGC TA, (R): CAG GCT GGA AGG AGA AGA TG, MAP2 (F): TGA AGA ATG GCA GAT GAA C, (R): AGA AGG AGG CAG ATT AGC, GAPDH (F): GGCATGGACTGTGGTCATGAG, (R): TGCACCACCAACTGCTTAGC. PCR products were electrophoresed in 1.5% agarose gels and stained with ethidium bromide.
2.7. Western Blot Analysis
After H/R injury, cells were washed rapidly with ice-cold PBS, scraped, and collected. Cell pellets were lysed with ice-cold RIPA buffer (Sigma-Aldrich, St. Louis, MO, USA). The lysates were centrifuged at 13,200 rpm for 1 h at 4°C to produce whole-cell extracts. Protein was quantified with the bicinchoninic acid (BCA) method (Pierce biotechnology, Rockford, IL, USA). Protein (20 μg) was separated on a 10% SDS–polyacrylamide (PAGE) gel and transferred onto a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% BSA (in TBS/Tween [TBS-T]) for 1 h, immunoblots were incubated overnight at 4°C with primary antibodies specific for Bcl2 (1 : 2000, Millipore, Massachusetts, MA, USA), cleaved caspase-3 (1 : 2000, Santa Cruz, Santa Cruz, CA, USA), PSD-95 (1 : 2000, Millipore, Massachusetts, MA, USA), AKT (1 : 2000, Cell signaling, Danvers, MA, USA), p-AKT (1 : 2000, Cell signaling, Danvers, MA, USA), or β-actin (1 : 2000, Santa Cruz, Santa Cruz, CA, USA). Next, blots were incubated with horseradish peroxidase- (HRP-) linked anti-mouse and anti-rabbit IgG antibodies purchased from Abcam (Abcam, Cambridge, MA, USA) for 1 h. Enhanced chemiluminescence was performed by electrochemiluminescence (ECL: Pierce Biotechnology, Rockford, IL, USA) [51].
2.8. Immunocytochemistry
The expression of BDNF, cleaved caspase-3, Bcl2, and PSD-95 in N2A cells was confirmed by immunocytochemistry. Cells in all experimental groups were washed three times with PBS, fixed with 4% paraformaldehyde for 3 h, and then washed with PBS. N2A cells were permeabilized with 0.025% Triton X-100 and blocked for 1 h with dilution buffer (Invitrogen, Carlsbad, CA, USA). The following primary antibodies: anti-rabbit BDNF (1 : 500, Abcam, Cambridge, MA, USA), anti-rabbit cleaved caspase-3 (1 : 500, Santa Cruz, Santa Cruz, CA, USA), anti-rabbit PSD-95 (1 : 500, Millipore, Massachusetts, MA, USA), anti-mouse Bcl2 (1 : 500, Millipore, Massachusetts, MA, USA) were prepared in dilution buffer, added to samples, and incubated for 3 h. Primary antibody was then removed and cells were washed three times for 3 min each with PBS. Later, samples were incubated with FITC-conjugated goat, anti-rabbit (1 : 200, Jackson Immunoresearch, PA, USA), or rhodamine-conjugated donkey, anti-mouse secondary antibodies (1 : 500, Millipore, Massachusetts, MA, USA) for 2 h. Cells were washed again three times for 3 min each with PBS and stained with 1 μg/mL DAPI (1 : 100, Sigma-Aldrich, St. Louis, MO, USA) for 10 min at room temperature. Fixed samples were imaged using a Zeiss LSM 700 confocal microscope (Carl Zeiss, Thornwood, NY, USA).
2.9. Statistical Analysis
Statistical analyses were carried out using SPSS 18.0 software (IBM Corp., Armonk, NY, USA). Data are expressed as mean ± S.E.M. Significant intergroup differences were determined by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc multiple-comparison test. Each experiment included four replicates per treatment. Differences were considered significant at P < 0.05 (∗) or P < 0.001 (∗∗).
3. Results
3.1. MCAO Mouse Brain Exhibited Neuronal Death and Synaptic Plasticity Damage
We performed immunohistochemistry of the brain of H/R injured and control mice, using antibodies to synaptophysin (Figure 1), PSD-95 (Figure 2), and MAP2 (Figures 1 and 2). The former two were used as markers of synaptic plasticity; the latter is considered to be a neuronal microtubule protein marker. The immunoreactivity of all three proteins was less in the H/R injured group than in the control group. These results indicate that cerebral ischemia suppresses the expression of synaptophysin, PSD-95, and MAP2 in ischemic brain and that synaptic neuronal microtubule proteins were damaged by ischemic injury.
Figure 1.
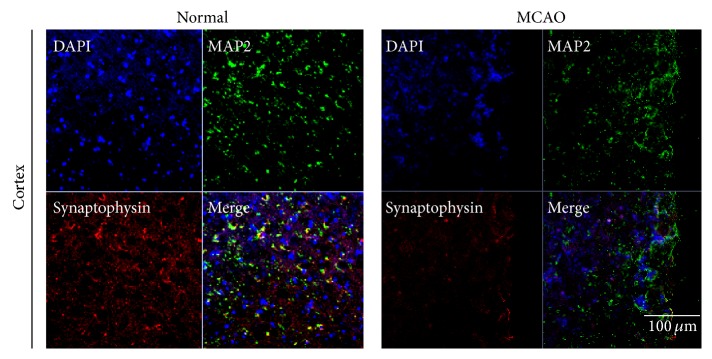
Immunochemical image for confirmation reduced synaptophysin expression in MCAO mouse brain. Immunochemical images showed that synaptophysin-positive cells (red) were decreased as expressed in MCAO mouse cortex. In addition, immunochemical images showed that MAP2- (considered as the neuron specific microtubule protein) positive cells (green) were strongly decreased in MCAO mouse cortex compared to the normal group. We used 5 rats in each groups for study. Each measurement included 3 repeats per animal. Scale bar = 100 μm, synaptophysin: red, MAP2: green, 4′, 6-diamidino-2-phenylindole (DAPI): blue, normal: normal control group, and MCAO: reperfusion 24 hr after MCAO injury.
Figure 2.
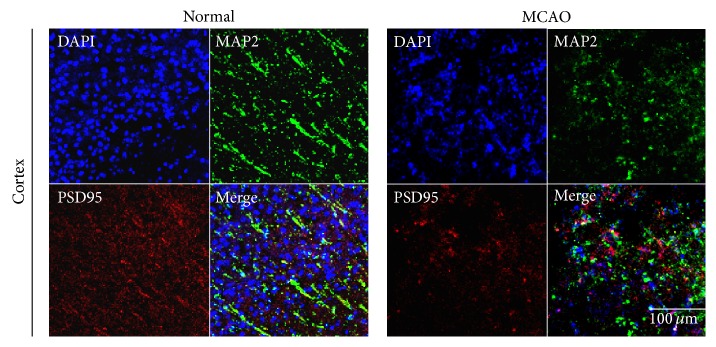
Immunochemical image for confirmation reduced PSD-95 expression in MCAO mouse brain. Immunochemical images showed that PSD-95- (as the post synaptic density protein) positive cells (red) were decreased as expressed in MCAO mouse cortex. Postsynaptic proteins were hardly observed in MCAO mouse brain cortex, whereas the normal cortex was observed evidently. We used 5 rats in each groups for study. Each measurement included 3 repeats per animal. Scale bar = 100 μm, PSD-95: red, MAP2: green, 4′,6-diamidino-2-phenylindole (DAPI): blue, normal: normal control group, and MCAO: reperfusion 24 hr after MCAO injury.
3.2. H/R Injury in Neuro2A Cells Inhibited, and H89 Pretreatment Restored, Neurite Outgrowth
Neurite outgrowth of Neuro2A cells was assessed by measuring neurite length with ImageJ software (Figure 3). The average length of normal N2A cells was approximately 65 μm, whereas neurites of cells subjected to H/R injury were approximately 26 μm long (Figure 3(a)). Neurites from cells that had been pretreated with H89 before H/R injury were, on average, approximately 45 μm, or almost twice that of the injured cells that were not pretreated (Figure 3(a)). Bright-field images showed the neurite length in all groups (Figures 3(b), 3(c), and 3(d)). The yellow line in all images permits easy comparison of neurite lengths.
Figure 3.

The measurement of neurite outgrowth in Neuro2A cells. (a) The graph of neurite outgrowth length (μm) in all groups. The neurite length significantly decreases in N2A cells against hypoxia reperfusion injury. Data are expressed as mean ± S.E.M. Significant intergroup differences were determined by one-way analysis of variance (ANOVA) followed by Bonferroni post hoc multiple-comparison test. Differences were considered significant at ∗ P < 0.05. (b) The image using bright field microscope in the normal group shows well developed neurite of N2A cells. (c) The image using bright field microscope in hypoxia reperfusion group shows shorter neurite outgrowth of N2A cells than the normal group. (d) The image using bright field microscope in H89 group shows well developed neurite of N2A cells compared to the hypoxia reperfusion group. Each experiment included 3 repeats per condition. H89 protected N2A cells against the neurite damage under H/R injury. Normal: the normal control group, H/R: 4 hr hypoxia and 18 hr reperfusion injury group, and H89: 2 hr PKA inhibitor H89 treatment group before 4 hr hypoxia and 18 hr reperfusion injury.
We also performed RT-PCR (Figure 4) to assess MAP2, a protein essential to neurite growth [41, 42]. The mRNA level of MAP2 in H/R injured N2A cells was reduced considerably compared to the control group (Figure 4). We conclude that H/R injury leads to reduction of neurite outgrowth, which can be alleviated by H89 pretreatment. Thus, H89 may ameliorate the effects of H/R injury.
Figure 4.
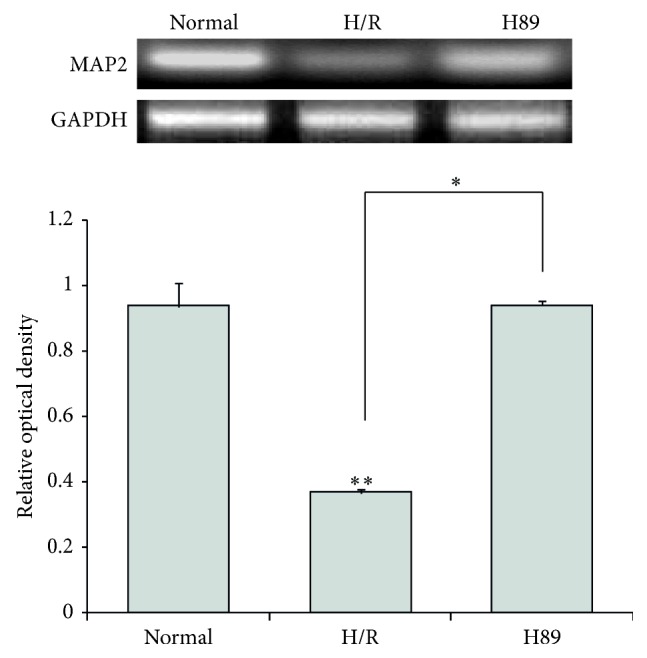
The measurement of MAP2 mRNA level in Neuro2A cells after PKA inhibitor treatment. MAP2 mRNA levels were measured by using RT-PCR. The H89 group showed higher mRNA levels of MAP2 compared to the hypoxia reperfusion injury group. Data were expressed as mean ± S.E.M, and each experiment included 3 repeats per condition. GAPDH was used as a control. Differences were considered significant at ∗ P < 0.05 and ∗∗ P < 0.001. Normal: the normal control group, H/R: 4 hr hypoxia and 18 hr reperfusion injury group, and H89: 2 hr PKA inhibitor H89 treatment group before 4 hr hypoxia and 18 hr reperfusion injury.
3.3. Cell Survival Was Increased in H89 Pretreated Neuro2A Cells after H/R Injury
To confirm whether or not H89 is involved in the neuronal cell death during H/R injury, we conducted the immunocytochemistry (Figures 5(a) and 5(b)), western blot analysis (Figures 5(c) and 5(d)), and RT-PCR (Figure 7(b)) using cleaved caspase-3 (as a marker of mitochondrial cell death) and Bcl2 (as a marker of anti-apoptosis) antibodies. H/R injured N2A cells were observed: the reduced Bcl2 immunoreactivity (Figure 5(b)), the decreased Bcl2 mRNA level (Figure 7(b)), the attenuated Bcl2 protein level (Figure 5(d)), the increased cleaved caspase-3 immunoreactivity (Figure 5(a)), and the increased cleaved caspase-3 protein level (Figure 5(c)). H89 pretreatment before H/R injury group showed the increased Bcl2 expression (Figures 5(b), 5(d), and 7(b)) and the reduced cleaved caspase-3 expression (Figures 5(a) and 5(c)) compared with the H/R group. These results indicated that the cell death in N2A cells was attenuated by H89 pretreatment in spite of hypoxia and reperfusion injury. Thus, we suggest that H89 may contribute to the neuronal cell survival pathway under hypoxia and reperfusion injury.
Figure 5.

The measurement of cleaved caspase-3 and Bcl2 expression in Neuro2A cells after H/R-induced injury. (a) The level of cleaved caspase-3 was evaluated by immunocytochemistry. This image shows that the expression of cleaved caspase-3 in the H/R group was strongly increased compared to the normal group. Cleaved caspase-3 expression was attenuated in H89 pretreatment treatment group under H/R-induced injury. (b) The level of Bcl2 was evaluated by immunocytochemistry. This image shows that the expression of Bcl2 in the H/R group was increased compared to the normal group. PKA inhibitor H89 pretreatment preserved the expression of Bcl2 in spite of hypoxia reperfusion injury. (c) Western blotting experiments showed that the relative protein expression of cleaved caspase-3 evidently attenuated in the H89 group compared to the hypoxia reperfusion group. (d) Western blotting experiments showed that the relative protein expression of Bcl2 slightly increased in the H89 group compared to the hypoxia reperfusion group. Data were expressed as mean ± S.E.M, and each experiment included 4 repeats per condition. Differences were considered significant ∗∗ P < 0.001. Scale bar: 200 μm, cleaved caspase-3: red, Bcl2: green, 4′,6-diamidino-2-phenylindole (DAPI): blue, normal: the normal control group, H/R: 4 hr hypoxia and 18 hr reperfusion injury group, and H89: 2 hr PKA inhibitor H89 treatment group before 4 hr hypoxia and 18 hr reperfusion injury.
Figure 7.

The measurement of BDNF and Bcl2 mRNA level in Neuro2A cells after H/R-induced injury. (a) BDNF and (b) Bcl2 mRNA levels were measured by using RT-PCR. The H89 pretreatment group showed higher mRNA levels of (a) BDNF and (b) Bcl2 compared to the hypoxia reperfusion injury group. Data were expressed as mean ± S.E.M, and each experiment included 3 repeats per condition. GAPDH was used as a control. Differences were considered significant at ∗ P < 0.05 and ∗∗ P < 0.001. Normal: the normal control group, H/R: 4 hr hypoxia and 18 hr reperfusion injury group, H89: 2 hr PKA inhibitor H89 treatment group before 4 hr hypoxia and 18 hr reperfusion injury.
3.4. The Increase of BDNF Expression in Neuro2A Cells Pretreated with H89 in Hypoxia Reperfusion Injury
We performed immunocytochemistry analysis (Figure 6) and RT-PCR (Figure 7(a)) using BDNF as the representative of neurotrophic factors in N2A cells to examine whether there was the alteration of neurotrophic factor expression in H89 pretreated N2A cells under hypoxia and reperfusion injury. We observed evidently lesser immunoreactivity of BDNF (Figure 6) in the H/R injured N2A cells compared to the normal control group. However, BDNF- (Figure 6) positive cells were obviously more expressed in H89 pretreated N2A cells than the H/R injury group. In addition, the BDNF mRNA level in N2A cells was higher in H89 pretreated N2A cells than the H/R injury group. These results showed that hypoxia and reperfusion stress suppresses the expression of BDNF in N2A cells, whereas H89 pretreatment H/R injured N2A cells considerably did not reduced the expression of BDNF against H/R injury. Based on these consequences, our results suggest that neurotrophic factor BDNF's expression was not reduced by H89 pretreatment despite ischemic injury. Thus, H89 may contribute to the expression of BDNF in N2A cells following hypoxia and reperfusion stress.
Figure 6.
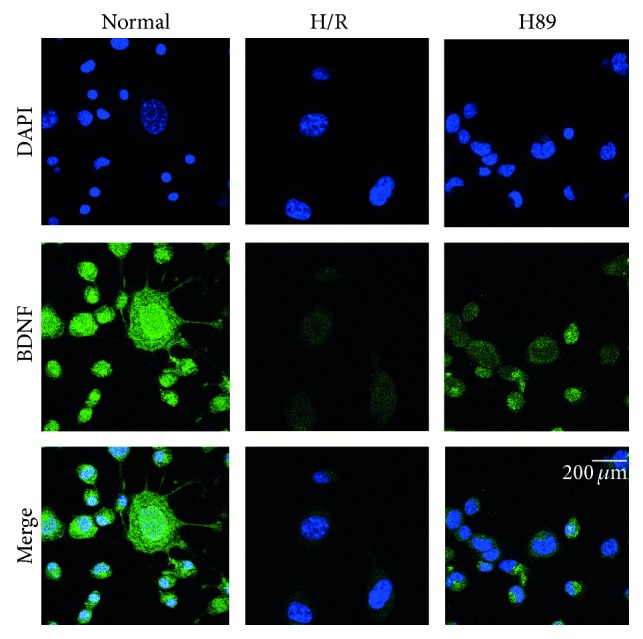
The measurement of BDNF expression in Neuro2A cells after H/R-induced injury. The level of BDNF was evaluated by immunocytochemistry. This image shows that the expression of BDNF in the H/R group was reduced compared to the normal group. H89 pretreatment increased the expression of BDNF in N2A cells in spite of hypoxia reperfusion injury. Scale bar: 200 μm, BDNF: green, 4′,6-diamidino-2-phenylindole (DAPI): blue, normal: the normal control group, H/R: 4 hr hypoxia and 18 hr reperfusion injury group, and H89: 2 hr PKA inhibitor H89 treatment group before 4 hr hypoxia and 18 hr reperfusion injury.
3.5. The Preservation of PSD-95 Expression in Neuro2A Cells Pretreated with H89 during Hypoxia Reperfusion Injury
We performed immunocytochemistry analysis (Figure 8(a)) and western blot analysis (Figure 8(b)) using PSD-95 antibody in N2A cells to investigate whether there was the alteration of synaptic plasticity related proteins in H89 pretreated N2A cells under hypoxia and reperfusion injury. In addition, we confirmed evidently decreased immunoreactivity of PSD-95 (Figure 8(a)) in the H/R injured N2A cells compared to the normal control group. On the other hand, the immunoreactivity of PSD-95 was more increased in H89 pretreated N2A cells than the H/R injury group (Figure 8(a)). Moreover, the protein level of PSD-95 (Figure 8(b)) in N2A cells was slightly higher in H89 pretreated N2A cells than the H/R injury group. These results indicated that hypoxia and reperfusion stress reduced the expression of PSD-95 in N2A cells, whereas H89 pretreatment H/R injured N2A cells considerably did not reduce expression of PSD-95 against H/R injury compared to H/R injured N2A cells. It is possible to extrapolate these results to suggest that the H/R injury reduced the expression of PSD-95. Data tend to support the conclusion that H89 may alleviate the synaptic plasticity damage of N2A cells against ischemic stress.
Figure 8.

The measurement of PSD-95 expression in Neuro2A cells after H/R-induced injury. The level of PSD-95 was evaluated by immunocytochemistry. This image shows that the expression of PSD-95 in the H/R group was decreased compared to the normal group. PKA inhibitor H89 pretreatment increased the expression of PSD-95 in spite of hypoxia reperfusion injury. (b) Western blotting experiments showed that the relative protein expression of PSD-95 slightly increased in the H89 group compared to the hypoxia reperfusion group. β-actin was used as an internal control. Data were expressed as mean ± S.E.M, and each experiment included 4 repeats per condition. Differences were considered significant at ∗ P < 0.05 and ∗∗ P < 0.001. Scale bar: 200 μm, PSD-95: green, 4′, 6-diamidino-2-phenylindole (DAPI): blue, normal: the normal control group, H/R: 4 hr hypoxia and 18 hr reperfusion injury group, and H89: 2 hr PKA inhibitor H89 treatment group before 4 hr hypoxia and 18 hr reperfusion injury.
3.6. The Measurement of Phosphorylation AKT Protein Level in H89 Pretreated Neuro2A Cells against Hypoxia Reperfusion Injury
We performed western blot analysis (Figure 9) using AKT and phosphorylation-AKT (p-AKT) antibody in N2A cells to investigate the change of AKT phosphorylation in H89 pretreated N2A cells under hypoxia and reperfusion injury. The protein level of phosphorylation-AKT (Figure 9) was evidently increased in H89 pretreated H/R injured N2A cells than the H/R injury group. This result shows that H89 considerably promotes the activation of AKT signaling in N2A cells against H/R injury. Our data supports the hypothesis that H89 may boost the phosphorylation of AKT in N2A cells to survive the cells against ischemic stress.
Figure 9.
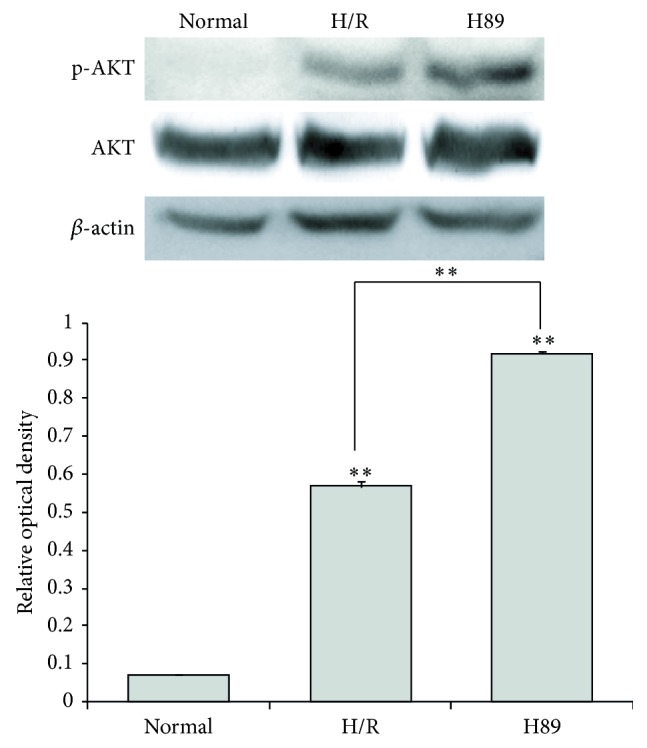
The measurement of AKT phosphorylation in Neuro2A cell against H/R injury. Western blotting experiments showed that the relative protein level of phosphorylation-AKT (p-AKT)/AKT significantly was increased in the H89 pretreatment group compared to the hypoxia and reperfusion group. β-actin was used as an internal control. Data were expressed as mean ± S.E.M, and each experiment included 4 repeats per condition. Differences were considered significant at ∗∗ P < 0.001. Normal: the normal control group, H/R: 4 hr hypoxia and 18 hr reperfusion injury group, H89: 2 hr PKA inhibitor H89 treatment group before 4 hr hypoxia and 18 hr reperfusion injury, and p-AKT: phosphorylation-AKT.
4. Discussion
In cerebral ischemia, the reduction of synaptic dysfunction and neuronal cell loss are important issues and are implicated in severe pathogenesis such as memory impairment following ischemic stroke [26–29, 52, 53]. In the search for a solution, many researchers study the molecules and the signal pathways that lead to reduced synaptic plasticity and cell death [54–56]; an example of one is PKA signaling [3–7]. H89, known as the molecule commonly used to inhibit PKA action, recently has been reported to have a variety of functions unrelated to its effect on PKA inhibition [16, 18, 21, 57]. H89 affects ROCK II and, through that effect, cell morphology [48] and neurite extension [58, 59]. The data presented here indicate that H89 promotes neurite outgrowth and protects it after hypoxia stress. MAP2 (known as the neuron specific cytoskeletal protein) is present during all stages of neuromorphogenesis [60] and is necessary for neurite initiation [60–62]. Our MAP2 expression data support the contention that H89 may also support neurite outgrowth through MAP2. We speculate that the maintenance of neurite outgrowth after ischemic stroke is central to the role of H89. Several studies have demonstrated that H89 induces autophagy in cells independent of PKA signaling [24, 25] and increases cell survival after inflammation [54, 63]. In the present study, we observed reduced expression of cleaved caspase-3 and increased expression of Bcl2 following pretreatment with H89, supporting the conclusion that H89 protects against hypoxia injury, specifically, that it increases neuronal cell survival rate after ischemic stroke. Neurotrophic molecules regulate synaptic plasticity of the nervous system [64–66]. Specifically, many researches demonstrated that BDNF accelerates the axogenesis [30–32], promotes poststroke plasticity in an in vivo study [32, 67–71], and contributes to healthy brain function, notably, neuronal survival and maintenance, neurogenesis, modulation of dendritic branching and dendritic spine morphology [72, 73], and development of neuronal connections required for learning and memory [74–76]. BDNF, through phosphorylation of its TrkB receptor, activates a neuron-specific protein, controls the actin cytoskeleton in dendritic spines [77] and their regression [78, 79], and promotes the actin polymerization [80]. Inhibition of BDNF synthesis results in smaller spine heads and impairs long-term potentiation of synaptic transmission [81, 82]. Moreover, BDNF signaling plays a crucial role in the development of synapses by controlling the transport of PSD-95, which is the major scaffolding protein at mature glutamate synapses [83, 84]. PSD-95 itself and its interaction with BDNF signaling have been implicated in diverse brain diseases [85–87]. When localized in postsynaptic terminals, PSD-95 has an important role in postsynaptic function and plasticity [88–90]. The loss of PSD-95 results in severe cognitive decline due to loss of neurons and synaptic disruption [91–93]. In addition, synaptophysin as a marker of the pre-synaptic nerve terminal density is essential for vesicle fusion and the release of neurotransmitter [94]. The reduction of synaptophysin has been reported to reduce synaptic plasticity in the brain [95, 96]. Our results suggest that H89 may enhance synaptic plasticity by promoting the BDNF expression in neuronal cells under ischemic brain injury. Also H89 may be involved in neurite outgrowth by regulating the preservation of synaptic proteins, such as PSD-95 and synaptophysin, following ischemic brain damage. AKT which is activated by phosphatidylinositol 3-kinase activity [97] has known to promote a cellular protection after ischemic injury in the brain [98]. Moreover, AKT has been reported that it mediates anti-apoptosis signalings in ischemic stroke studies [99, 100]. Some study indicated that H89 markedly enhances the phosphorylation of AKT [101]. Considering our results, we assume that H89 may contribute to the survival of neuronal cells against ischemic injury through the activation of AKT. In present study, although learning and memory were not assessed in the animal model used here and we has some limitations to identify the specific molecular mechanism by H89, we propose that H89 may ameliorate the pathophysiology following ischemic stroke by reducing neuronal cell death and involving synaptic plasticity.
Acknowledgment
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science, and Technology (NRF-2014R1A2A2A01006556).
Conflict of Interests
The authors declare that they have no conflict of interests regarding the publication of this paper.
References
- 1.Walsh D. A., Perkins J. P., Krebs E. G. An adenosine 3′,5′-monophosphate-dependant protein kinase from rabbit skeletal muscle. The Journal of Biological Chemistry. 1968;243(13):3763–3765. [PubMed] [Google Scholar]
- 2.Castellucci V. F., Kandel E. R., Schwartz J. H. Intracellular injection of the catalytic subunit of cyclic AMP-dependent protein kinase simulates facilitation of transmitter release underlying behavioral sensitization in Aplysia . Proceedings of the National Academy of Sciences of the United States of America. 1980;77(12):7492–7496. doi: 10.1073/pnas.77.12.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunelli M., Castellucci V., Kandel E. R. Synaptic facilitation and behavioral sensitization in Aplysia: possible role of serotonin and cyclic AMP. Science. 1976;194(4270):1178–1181. doi: 10.1126/science.186870. [DOI] [PubMed] [Google Scholar]
- 4.Kandel E. R. The molecular biology of memory storage: a dialog between genes and synapses. Bioscience Reports. 2001;21(5):565–611. doi: 10.1023/a:1014775008533. [DOI] [PubMed] [Google Scholar]
- 5.Rydel R. E., Greene L. A. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(4):1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu J., Cai D., Dai H., et al. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34(6):895–903. doi: 10.1016/S0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- 7.Burrell B. D., Sahley C. L. Learning in simple systems. Current Opinion in Neurobiology. 2001;11(6):757–764. doi: 10.1016/S0959-4388(01)00281-1. [DOI] [PubMed] [Google Scholar]
- 8.Ferro A., Coash M., Yamamoto T., Rob J., Ji Y., Queen L. Nitric oxide-dependent beta2-adrenergic dilatation of rat aorta is mediated through activation of both protein kinase A and Akt. British Journal of Pharmacology. 2004;143(3):397–403. doi: 10.1038/sj.bjp.0705933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sobolewski A., Jourdan K. B., Upton P. D., Long L., Morrell N. W. Mechanism of cicaprost-induced desensitization in rat pulmonary artery smooth muscle cells involves a PKA-mediated inhibition of adenylyl cyclase. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2004;287(2):L352–L359. doi: 10.1152/ajplung.00270.2003. [DOI] [PubMed] [Google Scholar]
- 10.Kaneishi K., Sakuma Y., Kobayashi H., Kato M. 3′,5′-cyclic adenosine monophosphate augments intracellular Ca2+ concentration and gonadotropin-releasing hormone (GnRH) release in immortalized GnRH neurons in an Na+-dependent manner. Endocrinology. 2002;143(11):4210–4217. doi: 10.1210/en.2002-220508. [DOI] [PubMed] [Google Scholar]
- 11.Kim S. H., Won S. J., Mao X. O., Jin K., Greenberg D. A. Involvement of protein kinase A in cannabinoid receptor-mediated protection from oxidative neuronal injury. Journal of Pharmacology and Experimental Therapeutics. 2005;313(1):88–94. doi: 10.1124/jpet.104.079509. [DOI] [PubMed] [Google Scholar]
- 12.Burvall K., Palmberg L., Larsson K. Expression of TNFalpha and its receptors R1 and R2 in human alveolar epithelial cells exposed to organic dust and the effects of 8-bromo-cAMP and protein kinase A modulation. Inflammation Research. 2005;54(7):281–288. doi: 10.1007/s00011-005-1356-7. [DOI] [PubMed] [Google Scholar]
- 13.Skinn A. C., MacNaughton W. K. Nitric oxide inhibits cAMP-dependent CFTR trafficking in intestinal epithelial cells. The American Journal of Physiology—Gastrointestinal and Liver Physiology. 2005;289(4):G739–G744. doi: 10.1152/ajpgi.00425.2004. [DOI] [PubMed] [Google Scholar]
- 14.Chijiwa T., Mishima A., Hagiwara M., et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-Bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. The Journal of Biological Chemistry. 1990;265(9):5267–5272. [PubMed] [Google Scholar]
- 15.Hidaka H., Inagaki M., Kawamoto S., Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23(21):5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- 16.Davies S. P., Reddy H., Caivano M., Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochemical Journal. 2000;351, part 1:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singleton D. W., Lu C. L., Colella R., Roisen F. J. Promotion of neurite outgrowth by protein kinase inhibitors and ganglioside GM1 in neuroblastoma cells involves MAP kinase ERK1/2. International Journal of Developmental Neuroscience. 2000;18(8):797–805. doi: 10.1016/s0736-5748(00)00047-2. [DOI] [PubMed] [Google Scholar]
- 18.Hussain M., Drago G. A., Bhogal M., Colyer J., Orchard C. H. Effects of the protein kinase A inhibitor H-89 on Ca2+ regulation in isolated ferret ventricular myocytes. Pflugers Archiv. 1999;437(4):529–537. doi: 10.1007/s004240050814. [DOI] [PubMed] [Google Scholar]
- 19.Lahouratate P., Guibert J., Camelin J.-C., Bertrand I. Specific inhibition of cardiac and skeletal muscle sarcoplasmic reticulum Ca2+ pumps by H-89. Biochemical Pharmacology. 1997;54(9):991–998. doi: 10.1016/s0006-2952(97)00320-1. [DOI] [PubMed] [Google Scholar]
- 20.Palacios N., Sánchez-Franco F., Fernández M., Sánchez I., Villuendas G., Cacicedo L. Opposite effects of two PKA inhibitors on cAMP inhibition of IGF-I-induced oligodendrocyte development: a problem of unspecificity? Brain Research. 2007;1178(1):1–11. doi: 10.1016/j.brainres.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 21.Pearman C., Kent W., Bracken N., Hussain M. H-89 inhibits transient outward and inward rectifier potassium currents in isolated rat ventricular myocytes. British Journal of Pharmacology. 2006;148(8):1091–1098. doi: 10.1038/sj.bjp.0706810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Um S. H., D'Alessio D., Thomas G. Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metabolism. 2006;3(6):393–402. doi: 10.1016/j.cmet.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 23.Ruvinsky I., Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends in Biochemical Sciences. 2006;31(6):342–348. doi: 10.1016/j.tibs.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Lochner A., Moolman J. A. The many faces of H89: a review. Cardiovascular Drug Reviews. 2006;24(3-4):261–274. doi: 10.1111/j.1527-3466.2006.00261.x. [DOI] [PubMed] [Google Scholar]
- 25.Murray A. J. Pharmacological PKA inhibition: all may not be what it seems. Science Signaling. 2008;1(22, article re4) doi: 10.1126/scisignal.122re4. [DOI] [PubMed] [Google Scholar]
- 26.Khatri N., Man H.-Y. Synaptic activity and bioenergy homeostasis: implications in brain trauma and neurodegenerative diseases. Frontiers in Neurology. 2013;4, article 199 doi: 10.3389/fneur.2013.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neumann J. T., Cohan C. H., Dave K. R., Wright C. B., Perez-Pinzon M. A. Global cerebral ischemia: synaptic and cognitive dysfunction. Current Drug Targets. 2013;14(1):20–35. doi: 10.2174/138945013804806514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofmeijer J., Mulder A. T. B., Farinha A. C., van Putten M. J. A. M., le Feber J. Mild hypoxia affects synaptic connectivity in cultured neuronal networks. Brain Research. 2014;1557:180–189. doi: 10.1016/j.brainres.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Li W., Huang R., Shetty R. A., et al. Transient focal cerebral ischemia induces long-term cognitive function deficit in an experimental ischemic stroke model. Neurobiology of Disease. 2013;59:18–25. doi: 10.1016/j.nbd.2013.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattson M. P., Partin J. Evidence for mitochondrial control of neuronal polarity. Journal of Neuroscience Research. 1999;56(1):8–20. doi: 10.1002/(sici)1097-4547(19990401)56:1x003C;8::aid-jnr2x0003e;3.3.co;2-7. [DOI] [PubMed] [Google Scholar]
- 31.Mattson M. P. Pathways towards and away from Alzheimer's disease. Nature. 2004;430(7000):631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lipsky R. H., Marini A. M. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Annals of the New York Academy of Sciences. 2007;1122:130–143. doi: 10.1196/annals.1403.009. [DOI] [PubMed] [Google Scholar]
- 33.Lu B., Wang K. H., Nose A. Molecular mechanisms underlying neural circuit formation. Current Opinion in Neurobiology. 2009;19(2):162–167. doi: 10.1016/j.conb.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burkhalter J., Fiumelli H., Allaman I., Chatton J.-Y., Martin J.-L. Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. The Journal of Neuroscience. 2003;23(23):8212–8220. doi: 10.1523/JNEUROSCI.23-23-08212.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kashyap M. P., Singh A. K., Yadav D. K., et al. 4-Hydroxy-trans-2-nonenal (4-HNE) induces neuronal SH-SY5Y cell death via hampering ATP binding at kinase domain of Akt1. Archives of Toxicology. 2014;89(1-2):243–258. doi: 10.1007/s00204-014-1260-4. [DOI] [PubMed] [Google Scholar]
- 36.Chiarotto G. B., Drummond L., Cavarretto G., Bombeiro A. L., de Oliveira A. L. R. Neuroprotective effect of tempol (4 hydroxy-tempo) on neuronal death induced by sciatic nerve transection in neonatal rats. Brain Research Bulletin. 2014;106:1–8. doi: 10.1016/j.brainresbull.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 37.Scheff S. W., Price D. A., Ansari M. A., et al. Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer's disease. Journal of Alzheimer's Disease. 2015;43(3):1073–1090. doi: 10.3233/JAD-141518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sultana R., Banks W. A., Butterfield D. A. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: insights into their potential roles for loss of synapses and memory, accumulation of Aβ, and neurodegeneration in a prodromal stage of Alzheimer's disease. Journal of Neuroscience Research. 2010;88(3):469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Q., Zhao H. F., Zhang Z. F., et al. Long-term green tea catechin administration prevents spatial learning and memory impairment in senescence-accelerated mouse prone-8 mice by decreasing Abeta1-42 oligomers and upregulating synaptic plasticity-related proteins in the hippocampus. Neuroscience. 2009;163(3):741–749. doi: 10.1016/j.neuroscience.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 40.Kim H., Binder L. I., Rosenbaum J. L. The periodic association of MAP2 with brain microtubules in vitro. The Journal of Cell Biology. 1979;80(2):266–276. doi: 10.1083/jcb.80.2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selden S. C., Pollard T. D. Phosphorylation of microtubule-associated proteins regulates their interaction with actin filaments. The Journal of Biological Chemistry. 1983;258(11):7064–7071. [PubMed] [Google Scholar]
- 42.Selden S. C., Pollard T. D. Interaction of actin filaments with microtubules is mediated by microtubule-associated proteins and regulated by phosphorylation. Annals of the New York Academy of Sciences. 1986;466:803–812. doi: 10.1111/j.1749-6632.1986.tb38464.x. [DOI] [PubMed] [Google Scholar]
- 43.Sattilaro R. F. Interaction of microtubule-associated protein 2 with actin filaments. Biochemistry. 1986;25(8):2003–2009. doi: 10.1021/bi00356a025. [DOI] [PubMed] [Google Scholar]
- 44.Unterberg A. W., Stover J., Kress B., Kiening K. L. Edema and brain trauma. Neuroscience. 2004;129(4):1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- 45.Nakamura Y., Nakamichi N., Takarada T., Ogita K., Yoneda Y. Transferrin receptor-1 suppresses neurite outgrowth in neuroblastoma Neuro2A cells. Neurochemistry International. 2012;60(5):448–457. doi: 10.1016/j.neuint.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 46.Mei Z. Y., Chin C. M., Yoon J. C., et al. Agmatine inhibits matrix metalloproteinase-9 via endothelial nitric oxide synthase in cerebral endothelial cells. Neurological Research. 2007;29(7):749–754. doi: 10.1179/016164107X208103. [DOI] [PubMed] [Google Scholar]
- 47.Wieprecht M., Wieder T., Geilen C. C. N-[2-bromocinnamyl(amino)ethyl]-5-isoquinolinesulphonamide (H-89) inhibits incorporation of choline into phosphatidylcholine via inhibition of choline kinase and has no effect on the phosphorylation of CTP:phosphocholine cytidylyltransferase. Biochemical Journal. 1994;297, part 1:241–247. doi: 10.1042/bj2970241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leemhuis J., Boutillier S., Schmidt G., Meyer D. K. The protein kinase A inhibitor H89 acts on cell morphology by inhibiting Rho kinase. The Journal of Pharmacology & Experimental Therapeutics. 2002;300(3):1000–1007. doi: 10.1124/jpet.300.3.1000. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe T., Yasutaka Y., Nishioku T., et al. Atorvastatin stimulates neuroblastoma cells to induce neurite outgrowth by increasing cellular prion protein expression. Neuroscience Letters. 2012;531(2):114–119. doi: 10.1016/j.neulet.2012.10.032. [DOI] [PubMed] [Google Scholar]
- 50.Yuasa K., Nagame T., Dohi M., et al. cGMP-dependent protein kinase I is involved in neurite outgrowth via a Rho effector, rhotekin, in Neuro2A neuroblastoma cells. Biochemical and Biophysical Research Communications. 2012;421(2):239–244. doi: 10.1016/j.bbrc.2012.03.143. [DOI] [PubMed] [Google Scholar]
- 51.Jung H.-J., Jeon Y.-H., Bokara K. K., et al. Agmatine promotes the migration of murine brain endothelial cells via multiple signaling pathways. Life Sciences. 2013;92(1):42–50. doi: 10.1016/j.lfs.2012.10.018. [DOI] [PubMed] [Google Scholar]
- 52.Tjepkema-Cloostermans M. C., Hindriks R., Hofmeijer J., van Putten M. J. A. M. Generalized periodic discharges after acute cerebral ischemia: reflection of selective synaptic failure? Clinical Neurophysiology. 2014;125(2):255–262. doi: 10.1016/j.clinph.2013.08.005. [DOI] [PubMed] [Google Scholar]
- 53.Zhao Y.-D., Cheng S.-Y., Ou S., Chen P.-H., Ruan H.-Z. Functional response of hippocampal CA1 pyramidal cells to neonatal hypoxic-ischemic brain damage. Neuroscience Letters. 2012;516(1):5–8. doi: 10.1016/j.neulet.2012.02.067. [DOI] [PubMed] [Google Scholar]
- 54.Park H., Licznerski P., Alavian K. N., Shanabrough M., Jonas E. A. Bcl-xL is necessary for neurite outgrowth in hippocampal neurons. Antioxidants & Redox Signaling. 2015;22(2):93–108. doi: 10.1089/ars.2013.5570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thundyil J., Manzanero S., Pavlovski D., et al. Evidence that the EphA2 receptor exacerbates ischemic brain injury. PLoS ONE. 2013;8(1) doi: 10.1371/journal.pone.0053528.e53528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bazan N. G., Marcheselli V. L., Cole-Edwards K. Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Annals of the New York Academy of Sciences. 2005;1053:137–147. doi: 10.1196/annals.1344.011. [DOI] [PubMed] [Google Scholar]
- 57.Eftekharzadeh B., Ramin M., Khodagholi F., et al. Inhibition of PKA attenuates memory deficits induced by β-amyloid (1–42), and decreases oxidative stress and NF-κB transcription factors. Behavioural Brain Research. 2012;226(1):301–308. doi: 10.1016/j.bbr.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 58.Bito H., Furuyashiki T., Ishihara H., et al. A critical role for a Rho-associated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron. 2000;26(2):431–441. doi: 10.1016/S0896-6273(00)81175-7. [DOI] [PubMed] [Google Scholar]
- 59.Tigyi G., Fischer D. J., Sebök Á., Marshall F., Dyer D. L., Miledi R. Lysophosphatidic acid-induced neurite retraction in PC12 cells: neurite-protective effects of cyclic AMP signaling. Journal of Neurochemistry. 1996;66(2):549–558. doi: 10.1046/j.1471-4159.1996.66020549.x. [DOI] [PubMed] [Google Scholar]
- 60.Caceres A., Banker G. A., Binder L. Immunocytochemical localization of tubulin and microtubule-associated protein 2 during the development of hippocampal neurons in culture. The Journal of Neuroscience. 1986;6(3):714–722. doi: 10.1523/JNEUROSCI.06-03-00714.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernhardt R., Matus A. Light and electron microscopic studies of the distribution of microtubule-associated protein 2 in rat brain: a difference between dendritic and axonal cytoskeletons. Journal of Comparative Neurology. 1984;226(2):203–221. doi: 10.1002/cne.902260205. [DOI] [PubMed] [Google Scholar]
- 62.Caceres A., Mautino J., Kosik K. S. Suppression of MAP2 in cultured cerebellar macroneurons inhibits minor neurite formation. Neuron. 1992;9(4):607–618. doi: 10.1016/0896-6273(92)90025-9. [DOI] [PubMed] [Google Scholar]
- 63.Sousa L. P., Carmo A. F., Rezende B. M., et al. Cyclic AMP enhances resolution of allergic pleurisy by promoting inflammatory cell apoptosis via inhibition of PI3K/Akt and NF-κB. Biochemical Pharmacology. 2009;78(4):396–405. doi: 10.1016/j.bcp.2009.04.030. [DOI] [PubMed] [Google Scholar]
- 64.Mattson M. P. Neurotransmitters in the regulation of neuronal cytoarchitecture. Brain Research. 1988;472(2):179–212. doi: 10.1016/0165-0173(88)90020-3. [DOI] [PubMed] [Google Scholar]
- 65.Loers G., Schachner M. Recognition molecules and neural repair. Journal of Neurochemistry. 2007;101(4):865–882. doi: 10.1111/j.1471-4159.2006.04409.x. [DOI] [PubMed] [Google Scholar]
- 66.Gottmann K., Mittmann T., Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Experimental Brain Research. 2009;199(3-4):203–234. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- 67.Binder D. K., Scharfman H. E. Brain-derived neurotrophic factor. Growth Factors. 2004;22(3):123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen J., Zhang C., Jiang H., et al. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. Journal of Cerebral Blood Flow and Metabolism. 2005;25(2):281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schäbitz W.-R., Berger C., Kollmar R., et al. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke. 2004;35(4):992–997. doi: 10.1161/01.str.0000119754.85848.0d. [DOI] [PubMed] [Google Scholar]
- 70.Kim M. W., Bang M. S., Han T. R., et al. Exercise increased BDNF and trkB in the contralateral hemisphere of the ischemic rat brain. Brain Research. 2005;1052(1):16–21. doi: 10.1016/j.brainres.2005.05.070. [DOI] [PubMed] [Google Scholar]
- 71.Schäbitz W.-R., Steigleder T., Cooper-Kuhn C. M., et al. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke. 2007;38(7):2165–2172. doi: 10.1161/strokeaha.106.477331. [DOI] [PubMed] [Google Scholar]
- 72.Horch H. W., Katz L. C. BDNF release from single cells elicits local dendritic growth in nearby neurons. Nature Neuroscience. 2002;5(11):1177–1184. doi: 10.1038/nn927. [DOI] [PubMed] [Google Scholar]
- 73.Tanaka J.-I., Horiike Y., Matsuzaki M., Miyazaki T., Ellis-Davies G. C. R., Kasai H. Protein synthesis and neurotrophin-dependent structural plasticity of single dendritic spines. Science. 2008;319(5870):1683–1687. doi: 10.1126/science.1152864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.McAllister A. K., Lo D. C., Katz L. C. Neurotrophins regulate dendritic growth in developing visual cortex. Neuron. 1995;15(4):791–803. doi: 10.1016/0896-6273(95)90171-X. [DOI] [PubMed] [Google Scholar]
- 75.Patterson S. L., Abel T., Deuel T. A. S., Martin K. C., Rose J. C., Kandel E. R. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16(6):1137–1145. doi: 10.1016/S0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 76.Horch H. W., Krüttgen A., Portbury S. D., Katz L. C. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23(2):353–364. doi: 10.1016/S0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- 77.Sala C., Piëch V., Wilson N. R., Passafaro M., Liu G., Sheng M. Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron. 2001;31(1):115–130. doi: 10.1016/S0896-6273(01)00339-7. [DOI] [PubMed] [Google Scholar]
- 78.Bennett M. Positive and negative symptoms in schizophrenia: the NMDA receptor hypofunction hypothesis, neuregulin/ErbB4 and synapse regression. Australian and New Zealand Journal of Psychiatry. 2009;43(8):711–721. doi: 10.1080/00048670903001943. [DOI] [PubMed] [Google Scholar]
- 79.Xu B., Zang K., Ruff N. L., et al. Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron. 2000;26(1):233–245. doi: 10.1016/s0896-6273(00)81153-8. [DOI] [PubMed] [Google Scholar]
- 80.Bramham C. R., Wells D. G. Dendritic mRNA: transport, translation and function. Nature Reviews Neuroscience. 2007;8(10):776–789. doi: 10.1038/nrn2150. [DOI] [PubMed] [Google Scholar]
- 81.An J. J., Gharami K., Liao G.-Y., et al. Distinct role of Long 3′ UTR BDNF mRNA in spine morphology and synaptic plasticity in hippocampal neurons. Cell. 2008;134(1):175–187. doi: 10.1016/j.cell.2008.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Waterhouse E. G., Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Molecular and Cellular Neuroscience. 2009;42(2):81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yoshii A., Constantine-Paton M. BDNF induces transport of PSD-95 to dendrites through PI3K-AKT signaling after NMDA receptor activation. Nature Neuroscience. 2007;10(6):702–711. doi: 10.1038/nn1903. [DOI] [PubMed] [Google Scholar]
- 84.Yoshii A., Murata Y., Kim J., Zhang C., Shokat K. M., Constantine-Paton M. TrkB and protein kinase M ζ regulate synaptic localization of PSD-95 in developing cortex. The Journal of Neuroscience. 2011;31(33):11894–11904. doi: 10.1523/jneurosci.2190-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tsai N.-P., Wilkerson J. R., Guo W., et al. Multiple autism-linked genes mediate synapse elimination via proteasomal degradation of a synaptic scaffold PSD-95. Cell. 2012;151(7):1581–1594. doi: 10.1016/j.cell.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cao C., Rioult-Pedotti M. S., Migani P., et al. Impairment of TrkB-PSD-95 signaling in Angelman syndrome. PLoS Biology. 2013;11(2) doi: 10.1371/journal.pbio.1001478.e1001478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mukai J., Dhilla A., Drew L. J., et al. Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nature Neuroscience. 2008;11(11):1302–1310. doi: 10.1038/nn.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.El-Husseini A. E.-D., Schnell E., Chetkovich D. M., Nicoll R. A., Bredt D. S. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290(5495):1364–1368. [PubMed] [Google Scholar]
- 89.Vickers C. A., Stephens B., Bowen J., Arbuthnott G. W., Grant S. G. N., Ingham C. A. Neurone specific regulation of dendritic spines in vivo by post synaptic density 95 protein (PSD-95) Brain Research. 2006;1090(1):89–98. doi: 10.1016/j.brainres.2006.03.075. [DOI] [PubMed] [Google Scholar]
- 90.Radwanska K., Medvedev N. I., Pereira G. S., et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(45):18471–18475. doi: 10.1073/pnas.1109680108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Han K., Kim E. Synaptic adhesion molecules and PSD-95. Progress in Neurobiology. 2008;84(3):263–283. doi: 10.1016/j.pneurobio.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 92.Terry R. D., Masliah E., Salmon D. P., et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Annals of Neurology. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 93.Scheff S. W., Price D. A., Schmitt F. A., Scheff M. A., Mufson E. J. Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer's disease. Journal of Alzheimer's Disease. 2011;24(3):547–557. doi: 10.3233/jad-2011-101782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Greengard P., Valtorta F., Czernik A. J., Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259(5096):780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 95.Cunha G. M. A., Canas P. M., Oliveira C. R., Cunha R. A. Increased density and synapto-protective effect of adenosine A2A receptors upon sub-chronic restraint stress. Neuroscience. 2006;141(4):1775–1781. doi: 10.1016/j.neuroscience.2006.05.024. [DOI] [PubMed] [Google Scholar]
- 96.Rapp S., Baader M., Hu M., Jennen-Steinmetz C., Henn F. A., Thome J. Differential regulation of synaptic vesicle proteins by antidepressant drugs. Pharmacogenomics Journal. 2004;4(2):110–113. doi: 10.1038/sj.tpj.6500229. [DOI] [PubMed] [Google Scholar]
- 97.Toker A., Cantley L. C. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387(6634):673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 98.Yamaguchi A., Tamatani M., Matsuzaki H., et al. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. Journal of Biological Chemistry. 2001;276(7):5256–5264. doi: 10.1074/jbc.m008552200. [DOI] [PubMed] [Google Scholar]
- 99.Zhang Y., Park T. S., Gidday J. M. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. American Journal of Physiology—Heart and Circulatory Physiology. 2007;292(6):H2573–H2581. doi: 10.1152/ajpheart.01098.2006. [DOI] [PubMed] [Google Scholar]
- 100.Dimmeler S., Assmus B., Hermann C., Haendeler J., Zeiher A. M. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circulation Research. 1998;83(3):334–341. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 101.Kato Y., Ozaki N., Yamada T., Miura Y., Oiso Y. H-89 potentiates adipogenesis in 3T3-L1 cells by activating insulin signaling independently of protein kinase A. Life Sciences. 2007;80(5):476–483. doi: 10.1016/j.lfs.2006.09.029. [DOI] [PubMed] [Google Scholar]