

Abstract
The G protein-coupled receptor (GPCR) superfamily constitutes the largest collection of cell surface signaling proteins with approximately 800 members in the human genome. GPCRs regulate virtually all aspects of physiology and they are an important class of drug targets with ~30% of drugs on the market targeting a GPCR. Breakthroughs in GPCR structural biology in recent years have significantly expanded our understanding of GPCR structure and function and ushered in a new era of structure-based drug design for GPCRs. Crystal structures for nearly thirty distinct GPCRs are now available including receptors from each of the major classes, A, B, C, and F. These structures provide a foundation for understanding the molecular basis of GPCR pharmacology. Here, we review structural mechanisms of ligand recognition and selectivity of GPCRs with a focus on selected examples from classes A, B, and C, and we highlight major unresolved questions for future structural studies.
Keywords: GPCR structure, GPCR subtype, X-ray crystallography, Ligand recognition, Subtype selectivity
1. GPCR structural revolution
G protein-coupled receptors (GPCRs) are cell surface receptors that mediate biological responses to external stimuli by transducing signals across the plasma membrane to heterotrimeric G proteins and arrestins, which in turn activate cellular signaling cascades. GPCRs regulate many physiological processes and respond to a diverse array of endogenous ligands including neurotransmitters, peptide and protein hormones, amino acids, ions, lipids, and various other bioactive molecules (Lagerstrom and Schioth, 2008). In addition, chemosensory GPCRs are responsible for our senses of sight, smell, and taste by responding to photons, odorants, and tastants. GPCRs are important drug targets for many diseases because of their fundamental roles in critical processes and inherent druggability as cell surface proteins. It is estimated that ~ 30% of drugs on the market target a GPCR and yet a relatively small number of GPCRs are currently targeted by marketed drugs (Overington et al., 2006). There remains an immense opportunity for development of drugs targeting many other GPCRs. High-resolution protein structures, such as those obtained from X-ray crystallography, reveal the molecular basis for protein function and facilitate rational drug design. In this review we highlight recent progress on structural studies of GPCRs that have revolutionized the field by greatly expanding our understanding of the molecular basis of GPCR pharmacology and enabling structure-based drug design to be applied to this important protein family. Several excellent recent reviews cover GPCR structural topics including orthosteric and allosteric ligand binding (Katritch et al., 2012, 2013; Kruse et al., 2014), ligand efficacy (Warne and Tate, 2013), receptor activation (Audet and Bouvier, 2012; Tehan et al., 2014), and structure-based drug design (Congreve et al., 2014). Here, we focus on structural mechanisms of ligand recognition and selectivity with selected examples from three major GPCR classes.
The human genome encodes approximately 800 GPCRs, which are grouped as four classes, A, B, C, and F (Kolakowski, 1994) or as five families, Rhodopsin, Adhesion, Secretin, Glutamate, and Frizzled (Fredriksson et al., 2003). The class A/Rhodopsin family comprises the largest number of GPCRs and our structural understanding of these receptors is most advanced. Rhodopsin was the first GPCR to have its structure determined by X-ray crystallography in 2000 (Palczewski et al., 2000), followed by the β2-adrenergic receptor structure in 2007 (Cherezov et al., 2007; Rosenbaum et al., 2007) and reports of other class A GPCR structures continually appearing since (reviewed in (Katritch et al., 2013)). We now have structures of over twenty distinct class A GPCRs including the landmark achievement of the ternary agonist-bound β2-adrenergic receptor-Gs complex structure (Rasmussen et al., 2011b). The year 2013 saw reports of the first 7-transmembrane (7TM) domain structures for two class B/Secretin family GPCRs (Hollenstein et al., 2013; Siu et al., 2013) and of the class F/Frizzled family smoothened GPCR (Wang et al., 2013b). In 2014 the first 7TM domain structures for two class C/Glutamate family GPCRs were reported (Dore et al., 2014; Wu et al., 2014). We do not yet have a high-resolution crystal structure of a GPCR-β-arrestin complex, but we are beginning to understand the architecture of this complex as revealed at low resolution by electron microscopy (Shukla et al., 2014).
The explosion of GPCR crystal structures was enabled by several technological breakthroughs that made possible the crystallization of these dynamic integral membrane proteins that were once thought “uncrystallizable”. Most of the structures utilized recombinant protein expressed in insect cells and codon optimization technology enabled increased expression levels. Development of novel detergents (Rasmussen et al., 2011b), lipid cubic phase crystallization (Cherezov, 2011), and X-ray microdiffraction beamlines (Riekel et al., 2005) all contributed to making GPCR crystallography a reality. Perhaps the most important was the development of methods to stabilize the receptors for crystallization including the use of fusion protein approaches such as T4 lysozyme insertion into loops to facilitate crystal packing combined with lipid cubic phase crystallization (Cherezov et al., 2007; Rosenbaum et al., 2007) and the generation of thermostabilized receptors by mutagenesis (Tate, 2012). A recent twist on the thermostabilization approach utilizing elegant directed evolution methods in E. coli (Scott and Pluckthun, 2013) resulted in the first crystal structure of a GPCR utilizing recombinant protein produced in E. coli (Egloff et al., 2014), a feat once thought impossible. Most GPCR structures are inactive antagonist- or inverse agonist-bound states. Crystallizing active state GPCRs is still a challenge, but this obstacle has been overcome by using nanobodies to stabilize the active state by mimicking G proteins (Kruse et al., 2013; Rasmussen et al., 2011a).
2. Class A GPCR structure and ligand binding
Class A/Rhodopsin family GPCRs make up the vast majority of the GPCR repertoire in the human genome with hundreds of receptors. Class A receptors consist of a 7-transmembrane (7TM) helical bundle in the membrane with three extracellular and three intracellular loops connecting the individual helices and an extracellular N-terminus and intracellular C-terminus (Fig. 1A) (Cherezov et al., 2007; Rosenbaum et al., 2007). A few class A GPCRs such as the glycoprotein hormone receptors have large N-terminal extracellular domains (ECDs) that bind ligands, but these will not be discussed here. The seven TM helices form a bundle; when viewed from the extracellular side helices I–VII are arranged in a counter-clockwise fashion (Fig. 1B). A short helix VIII that runs parallel to the membrane is often found near the C-terminus and includes sites of palmitoylation (Fig. 1A). TM helix III is a central hub critical to GPCR structure, ligand binding, and receptor activation (Venkatakrishnan et al., 2013). The orthosteric ligand binding pocket is positioned in the extracellular half of the bundle and is formed by residues from helices III, VI, and VII (Fig. 1A, B). In some cases residues from other helices also make specific ligand contacts such as with helix V for the β-adrenoceptors, which is an important determinant for ligand efficacy (Warne and Tate, 2013). In the β2-adrenergic receptor access to the deep orthosteric pocket is available from the extracellular milieu because the extracellular loops do not completely cover the top of the receptor (Fig. 1B). An extracellular vestibule surrounded by the tops of helices V, VI, and VII and ECL2 and ECL3 is present above the orthosteric site. Molecular dynamics simulations detailed the pathway by which ligands first contact the extracellular vestibule and subsequently enter the deep orthosteric pocket (Dror et al., 2011).
Fig. 1.

Structures of representative class A GPCRs and positions of orthosteric ligand binding pockets. (A) Structure of the β2-adrenergic receptor with a bound inverse agonist carazolol (PDB: 2RH1). (B) The β2-adrenergic receptor with bound carazolol viewed from the extracellular side. (C) Structure of the sphingosine-1-phosphate receptor with a bound antagonist lipid-mimic ML056 (PDB: 3V2Y). (D) Structure of the NTSR1 neurotensin receptor with a bound agonist peptide neurotensin8–13 (PDB: 4GRV). In all panels the receptor is color coded from N- to C-termini and the ligand is shown in stick representation and space-filling spheres. Extracellular (ECL) and intracellular (ICL) loops and the seven transmembrane helices are labeled. Disulfide bonds, palmitoylation at the C-terminus, and glycans at the N-terminus are also shown in stick representation. All figures were prepared with PyMol (Schrodinger).
The numerous class A GPCR structures available highlight the diversity in ligand binding sites and extracellular loops (Katritch et al., 2012). Although the orthosteric ligand binding sites are all in a similar position in the extracellular half of the helical bundle, the precise architectures and depth of the binding sites as well as access to the sites vary greatly. The β2-adrenergic receptor ligand binding site is representative of the aminergic GPCRs (Fig. 1A, B). The lipid-binding sphingosine-1-phosphate (S1P) receptor has its orthosteric ligand binding pocket positioned very similar to the aminergic receptors, but access to the pocket from the extracellular milieu is blocked by an extended N-terminal α-helix that covers the top of the receptor (Fig. 1C) (Hanson et al., 2012). A lateral opening between helices I and VII is presumed to allow the hydrophobic lipid ligand to access the orthosteric site from within the lipid bilayer (Fig. 1C). Peptide-binding class A GPCRs often have an ECL2 with a β-hairpin structural element and relatively open access at the top of the receptor (Fig. 1D). The neurotensin peptide binds the neurotensin type 1 receptor in an extended conformation and does not penetrate as deep into the receptor as observed for ligands of many other class A GPCRs; as a consequence, the neurotensin peptide makes several contacts with the receptor extracellular loops (Fig. 1D) (Egloff et al., 2014; White et al., 2012).
3. Class A GPCR ligand selectivity
Crystal structures of several subtypes of class A GPCRs were recently revealed: serotonin 5-HT1B and 5-HT2B receptors, muscarinic acetylcholine M2 and M3 receptors, and four subtypes of opioid receptors (Fenalti et al., 2014; Granier et al., 2012; Haga et al., 2012; Kruse et al., 2012; Manglik et al., 2012; Thompson et al., 2012; Wacker et al., 2013; Wang et al., 2013a; Wu et al., 2012). Although these receptors originate from several species (most from human, but some from rat and mouse) (Granier et al., 2012; Kruse et al., 2012; Manglik et al., 2012), structural comparisons provide insights into the mechanisms for receptor subtype selectivity. The opioid receptor crystal structures are useful to test the “message-address” hypothesis of opioid ligands that has been commonly assumed for opioid receptor subtype selectivity (Lipkowski et al., 1986; Portoghese, 1989).
3.1 Serotonin receptors
Serotonin [5-hydroxytryptamine (5-HT)] is an endogenous neurotransmitter acting on multiple organs. Physiological effects of serotonin are mediated by a large family of serotonin receptors that are divided into seven subfamily groups (5-HT1–7) based on sequence homology and signaling pathways (Kroeze et al., 2002). The seven subfamily groups include several serotonin receptors, and total thirteen serotonin receptor subtypes have been reported in human (McCorvy and Roth, 2015). All the serotonin receptors are G protein-coupled receptors with one exception of the 5-HT3 receptor, which is an ion channel. Recently, two subtype 5-HT1B and 5-HT2B receptors bound with an antimigraine drug ergotamine were reported (Wacker et al., 2013; Wang et al., 2013a). Although the main target of ergotamine is 5-HT1B and 5-HT1D receptors (Tfelt-Hansen et al., 2000; Villalon et al., 1999), its usage is very limited due to off-target actions on the other 5-HT subtype receptors particularly on the 5-HT2B receptor that causes life-threatening valvulopathy on heart (Elangbam et al., 2008; Nebigil et al., 2001).
The orthosteric binding pockets of 5-HT1B and 5-HT2B receptors are located deeply in the transmembrane core, and they conserve key amino acids for ligand interactions (Wang et al., 2013a). One difference in the orthosteric binding pocket is that Y2085.38 in the 5-HT1B receptor is unable to interact with ergotamine, whereas the corresponding F2175.38 in the 5-HT2B receptor provides a hydrophobic cap (Fig. 2A, upper letters refer to Ballesteros-Weinstein nomenclature to indicate the amino acids in transmembrane domains (Ballesteros and Weinstein, 1995)). A 3.0 Å distance is observed between the α carbons of T2095.39 in the 5-HT1B receptor and M2185.39 in the 5-HT2B receptor (Fig. 2A). This shift provides a narrower opening of the extended binding pocket for the 5-HT2B receptor, compared to the 5-HT1B receptor. In addition, the side chain of M2185.39 in the 5-HT2B receptor is bulkier than that of T2095.39 in the 5-HT1B receptor. The narrower binding pocket in the 5-HT2B receptor appears to play a role in subtype selectivity. Docking simulations with a selective 5-HT1B agonist sumatriptan support this idea (Wang et al., 2013a). Although mutagenesis results with sumatriptan are unavailable, an M2185.39A mutation markedly increased the potency of other triptans (electriptan and donitriptan) by removing the methionine side chain and expanding the binding pocket (Wang et al., 2013a). These mutagenesis results suggest that the opening of helix V in the serotonin receptor contributes to subtype selectivity.
Fig. 2.
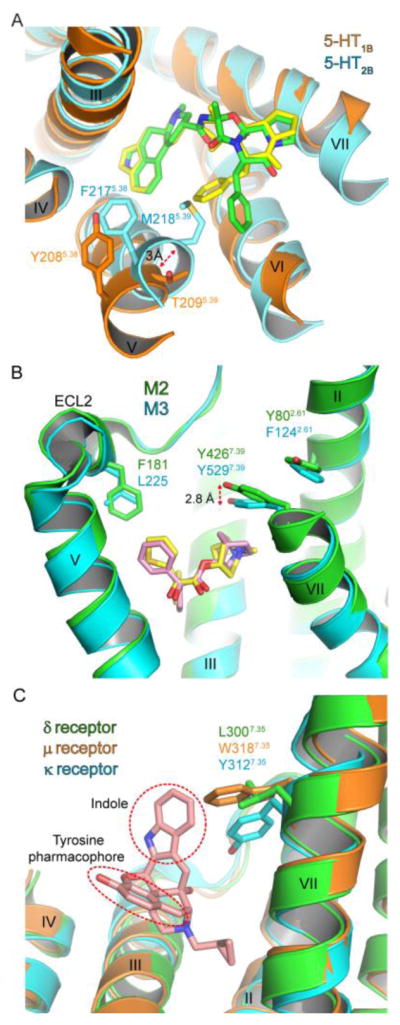
Structural determinants for class A GPCR subtype selectivity. (A) Comparison of the serotonin 5-HT1B (PDB: 4IAR) and 5-HT2B (PDB: 4IB4) receptors viewed from the extracellular side. 5-HT1B and 5-HT2B receptors are colored orange and cyan, respectively. Ergotamine in 5-HT1B and 5-HT2B receptors is colored yellow and green, respectively. The top of helix V and the ECL3 are omitted for clarity. (B) Ligand binding pockets of the M2 (PDB: 3UON) and M3 (PDB: 4DAJ) muscarinic acetylcholine receptors. M2 and M3 receptors are colored green and cyan, respectively. QNB and tiotropium is colored pink and yellow, respectively. The top of helix VII and the entire helix VI are omitted for clarity. (C) Naltrindole selectivity for the δ receptor. The indole moiety and tyrosine pharmacophore of naltrindole are shown in red-dotted circles. The δ receptor (PDB: 4N6H) is colored green, the μ receptor (PDB: 4DKL) orange, the κ receptor (PDB: 4DJH) cyan, and naltrindole pink. The top of helix IV and the entire helices V and VI are eliminated for clarity.
Docking simulations of norfenfluramine with the serotonin receptors also suggest the mechanism for subtype selectivity (Wang et al., 2013a). Norfenfluramine is the active metabolite of fenfluramine that was once used as an antiobesity drug but now withdrawn due to severe side effects caused by the 5-HT2B receptor activation (e.g., valvulopathy) (Connolly et al., 1997). Although the antiobesity effects of norfenfluramine are mediated by the 5-HT2C receptor (Vickers et al., 2001), norfenfluramine also interacts with 5-HT1B and 5-HT2B receptors. Norfenfluramine activates the 5-HT2B receptor with higher potency and efficacy than those at the 5-HT1B receptor (Fitzgerald et al., 2000). Docking simulations with norfenfluramine suggested that F2175.38 and M2185.39 in the 5-HT2B receptor provide a hydrophobic cap and interact with norfenfluramine. F2175.38A and M2185.39V mutations that remove the hydrophobic components greatly reduced the potency of norfenfluramine (Setola et al., 2005; Wang et al., 2013a). In contrast, the corresponding Y2085.38 and T2095.39 in the 5-HT1B receptor are positioned away from norfenfluramine, and this appears to cause weak affinity of norfenfluramine for the 5-HT1B receptor. Together with results of sumatriptan, the simulations and mutagenesis results of norfenfluramine suggest that the extracellular end of helix V contributes to the opening of the binding pocket and that the consequential change of molecular interactions may mediate subtype selectivity of the 5-HT receptors.
3.2 Muscarinic acetylcholine receptors
Acetylcholine is a neurotransmitter that mediates multiple physiological actions by activating a family of GPCRs, muscarinic acetylcholine receptors (mAChRs). mAChRs are classified into five subtypes (M1-M5) that share a high degree of sequence homology. The high sequence conservation of mAChRs makes it challenging to design subtype selective ligands. Although there may be species-originated differences, the crystal structures of human M2 and rat M3 receptors provide clues to the mechanisms of Class A GPCR subtype selectivity.
The M2 receptor was crystallized with a non-subtype selective mAChR antagonist R-(–)-3-quinuclidinyl benzilate (QNB), and the M3 receptor was in complex with a muscarinic receptor inverse agonist tiotropium that is prescribed for chronic obstructive pulmonary disease (COPD) (Haga et al., 2012; Kruse et al., 2012). The orthosteric binding sites of the M2 and M3 receptors are well conserved, and the extracellular loops of M2 and M3 receptors also show high conformational similarity in the backbone fold (Haga et al., 2012; Kruse et al., 2012). Despite the high sequence similarity, the orthosteric and extended binding sites do have displacements that may be useful for subtype selectivity. One apparent difference is the positions of ECL2 F181 in the extended binding pocket at M2 and the corresponding L225 at M3 (Fig. 2B). A small side chain of L225 at M3 produces a more spacious binding pocket than that of M2. Another difference is a 2.8 Å upward shift of Y4267.39 in the orthosteric binding pocket at M2 compared to its corresponding Y5297.39 at M3. Slightly different conformations of the position 2.61 (Y802.61 in M2 and F1242.61 in M3) are accompanied with the shift of Y7.39 (Fig. 2B). These differences may help with the design of subtype selective muscarinic receptor ligands targeting the orthosteric and extended binding sites.
The crystal structure of the M2 receptor was also reported with a positive allosteric modulator LY2119620 highlighting the allosteric binding site in the extracellular vestibule (Kruse et al., 2013). Positive allosteric modulators potentiate the orthosteric ligand interaction, and this can occur in a subtype selective manner with muscarinic acetylcholine receptors (Lazareno et al., 2004). Thus, allosteric interactions may be exploited for muscarinic receptor subtype selectivity. Several bitopic molecules that retain moieties that bind both orthosteric and allosteric sites also showed the selective interactions with muscarinic receptor subtypes (Disingrini et al., 2006; Steinfeld et al., 2007).
3.3 Opioid receptors
Opioid receptors interact with endogenous peptides such as endorphins, enkephalins, and dynorphins and opioid ligands including natural products and small molecules. Opioids were traditionally used for reducing pain as natural products, and the opioid receptors have been a drug target for alleviating severe pain. Crystal structures of all four opioid receptors (δ receptor, μ receptor, κ receptor, and nociceptin receptor) have been revealed (Fenalti et al., 2014; Granier et al., 2012; Manglik et al., 2012; Thompson et al., 2012; Wu et al., 2012). Comparison of these receptors provides structural information on receptor subtype selectivity and further confers evidence to validate the “message-address” hypothesis that has been used to explain the interactions between opioid receptors and ligands.
The ligand binding pockets of δ, μ, and κ receptors are well conserved. Although chemically diverse opioid ligands can interact with these receptors, a phenolic hydroxyl group and a nearby positively charged amine are found in most ligands. This pharmacophore appears to mimic the tyrosine terminal of endogenous opioid peptides and is thereby a key interaction site with the opioid receptor (Bryant et al., 2003; Mosberg, 1999). Superposition of δ, μ, and κ receptors suggests that the position 7.35 may be critical for opioid receptor subtype selectivity (Fig. 2C). L3007.35 in the δ receptor forms hydrophobic interactions with the indole group of a selective δ receptor antagonist naltrindole (Granier et al., 2012). The corresponding residues in μ and κ receptors are W3187.35 and Y3127.35 that contain a much larger aromatic side chain than L3007.35 in the δ receptor. Thus, the indole ring of naltrindole would produce steric hindrance with the bulky aromatic side chain at the position 7.35 in the μ and κ receptors. Mutations of μ receptor W3187.35 to leucine and lysine greatly increased affinity of δ receptor-selective ligands but considerably lowered affinity of μ receptor-selective ligands (Bonner et al., 2000). These results support the critical role of the position 7.35 in opioid receptor subtype selectivity.
The nociceptin receptor has been classified into the opioid receptor family due to its high sequence homology with the other three opioid receptors. However, most opioid alkaloids show very low affinity to the nociceptin receptor (Mogil and Pasternak, 2001). Although the mechanism for the low affinity is unclear, mutations on V276, Q277, and V278 of TM VI helix, and A213 in the ECL2/TM V interface of the nociceptin receptor markedly increased the binding affinity of opioid ligands (Meng et al., 1998). This study introduced the bulky residues that are conserved among δ, μ, and κ receptors to the nociception receptor. Thus, the nociception receptor retain a relatively greater ligand binding pocket than those of δ, μ, and κ receptors, and this may result in the low affinity of opioid ligands for the nociception receptor.
Opioid ligands have been proposed to contain two chemical components: one part for efficacy (message) and the other for subtype selectivity (address) (Dondio et al., 1997; Lipkowski et al., 1986; Portoghese, 1989). The message-address concept has been widely adopted to explain the pharmacology of opioid ligands. Crystal structures of the opioid receptors indicate that the lower part of the ligand binding pocket is highly conserved, receives the “message” of the ligand, and mediates receptor efficacy. In contrast, the upper part of the pocket shows sequence variability where the “address” part of the ligand elicits subtype selectivity. For instance, the morphinan group and tyrosine pharmacophore of naltrindole fit well in the lower binding pocket that accepts the “message” of the ligand (Granier et al., 2012). In addition, the upper binding pocket of the δ receptor is markedly favorable for the interaction of the indole group in naltrindole versus μ and κ receptors as exemplified with the position 7.35 (Fig. 2C). Although other factors may be involved in selective subtype interactions, the “message-address” concept is consistent with the crystal structures of the opioid receptors.
4. Class B GPCR structure and ligand binding
The class B/Secretin family GPCRs comprise fifteen receptors in humans that are activated by peptide endocrine hormones, peptide paracrine factors, and neuropeptides (Hoare, 2005). The topology of class B receptors is similar to that of the class A receptors except that class B receptors have an N-terminal extracellular domain (ECD) of ~120 amino acids in addition to the 7TM domain. Peptide agonist binding to these receptors follows a two-domain model. The peptides are ~ 30–40 amino acids in length and their C-terminal region binds to the ECD to confer affinity and specificity whereas their N-terminal region binds to the 7TM helical bundle to activate the receptor. N-terminally truncated peptides typically act as antagonists. Several class B receptors are validated drug targets with drugs either on the market or in clinical trials for diseases including diabetes, osteoporosis, and migraine headache (Bortolato et al., 2014). In most cases the drugs are peptide agonists. For some class B receptors the desired drugs are antagonists, such as for the corticotropin releasing factor (CRF) 1 receptor for depression or the calcitonin gene-related peptide (CGRP) receptor for migraine headache, and small-molecule antagonists have been developed that target the 7TM of the CRF1 receptor or ECD of the CGRP receptor, respectively. Our structural understanding of ligand binding to class B GPCRs has increased with recent reports of structures of the isolated domains, but no structure of a full-length class B GPCR is available. The orientation of the ECD relative to the 7TM domain in the intact receptor is unclear and how peptides bind the 7TM domain remains poorly understood.
Crystal and NMR structures of several class B GPCR ECDs determined in complex with bound C-terminal peptide fragments confirmed a conserved ECD fold and mode of peptide binding (Grace et al., 2007; Grace et al., 2010; Pal et al., 2010; Parthier et al., 2007; Pioszak et al., 2009; Pioszak et al., 2008; Pioszak and Xu, 2008; Runge et al., 2008; Underwood et al., 2010). The ECD has an N-terminal α-helix and a set of four β-strands that form a short consensus repeat or sushi fold; these secondary structure elements are held together by three conserved disulfide bonds (Fig. 3A). Most of the class B receptor ECDs have a long N-terminal α-helix, but this helix is significantly shortened in the CRF1 receptor. The peptide ligands form continuous α-helices that occupy a shallow binding groove situated between the N- and C-termini of the ECD. This groove is generally hydrophobic, but specific polar contacts are also observed in the various structures. Most of the peptide-bound ECD structures show peptides in a position similar to that of parathyroid hormone (PTH), but CRF and the related urocortin (Ucn) peptides occupy a binding site that is slightly shifted compared to the other receptors (Fig. 3A). The position of the peptides in the ECD structures is such that their N-terminal regions would be directed towards the 7TM bundle.
Fig. 3.
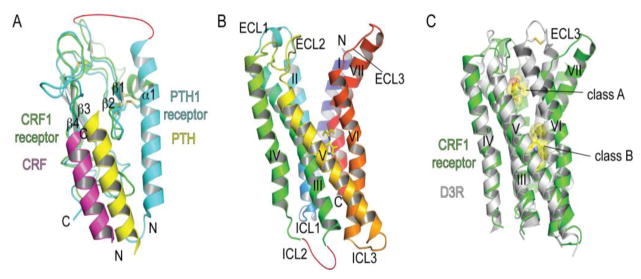
Structures of class B GPCR domains and positions of ligand binding surfaces and pockets. (A) N-terminal extracellular domain structures of the parathyroid hormone PTH1 receptor and corticotropin releasing factor CRF1 receptor with bound PTH (PDB: 3C4M) and CRF (PDB: 3EHU) peptide fragments, respectively. (B) The CRF1 receptor 7TM structure with a bound small molecule antagonist CP-376395 (PDB: 4K5Y) shown in stick representation. (C) Superposition of the class B CRF1 receptor 7TM and class A dopamine D3 receptor with a bound antagonist eticlopride (PDB: 3PBL) highlighting the difference in positions of the antagonist drug binding pockets and the open cleft in the CRF1 receptor. Ligands are shown in stick and space-filling representation.
Crystal structures of the isolated 7TM domain of the CRF1 receptor with a bound small molecule antagonist and of the ligand-free glucagon receptor 7TM domain were reported in 2013 (Hollenstein et al., 2013; Siu et al., 2013). These structures provided the first views of the class B 7TM bundle and allowed comparison to the class A 7TM bundle. The intracellular half of the class B 7TM bundle is similar to that of the class A receptors as expected for proteins that couple to the same G proteins. The class B and class A receptors are most different in their extracellular halves. The CRF1 receptor 7TM bundle is V-shaped with a prominent open cleft between ECL1/2 and ECL3 (Fig. 3B) (Hollenstein et al., 2013). The cleft presumably enables occupancy by the large N-terminal portion of the peptide ligand. There is considerable evidence for the N-terminal portion of the peptide forming α-helical structure and contacting all three extracellular loops (Dong et al., 2014). Comparison of the CRF1 receptor structure to that of the antagonist-bound class A dopamine D3 receptor (Chien et al., 2010) reveals similar positioning of helices I, II, III, IV, and V, but a very different position of helices VI and VII and ECL3 that gives rise to the open cleft (Fig. 3C). The CRF1 receptor structure also revealed a dramatically different position of the antagonist-binding pocket as compared to class A receptors (Fig. 3B, C). The small-molecule CRF1 receptor antagonist occupies a pocket in the intracellular half of the 7TM bundle comprised of residues from helices III, V, and VI (Fig. 3B). The pocket is largely hydrophobic in nature, but there is a single hydrogen bond formed between the receptor and the ligand.
5. Class B GPCR peptide ligand selectivity
The structural basis for ligand selectivity is still poorly understood for class B receptors because of the lack of peptide-bound 7TM or full-length structures. Nonetheless, the ECD is sufficient to confer peptide selectivity for several class B receptors including the CRF1 and CRF2 receptors and the CGRP and adrenomedullin receptors (Moad and Pioszak, 2013; Pal et al., 2010). ECD-mediated selectivity is understood structurally for the CRF receptors, for which we have crystal structures of both subtype ECDs with bound CRF and Ucn1, -2, and -3 peptides (Pal et al., 2010; Pioszak et al., 2008). The related CRF and Ucn peptides regulate central stress responses, the cardiovascular system, and metabolism via activation of CRF1 and CRF2 receptors (Bale and Vale, 2004). CRF and Ucn1 activate both receptors whereas Ucn2 and -3 are selective for the CRF2 receptor. Comparison of the CRF-bound CRF1 receptor ECD and Ucn3-bound CRF2 receptor ECD structures indicated that the two α-helical peptides occupy identical positions on the receptors and that peptide binding site architectures of the CRF1and CRF2 receptors are very similar other than a single crucial amino acid difference, CRF1 receptor E104 and CRF2 receptor P100, which is near peptide residues that differ, CRF/Ucn1 R35 and Ucn2/3 A35 (Fig. 4A). The electrostatic surface potential of the CRF1 receptor is considerably more negative near E104 as compared to the equivalent region near the nonpolar P100 in the CRF2 receptor (Fig. 4B, C). The nonpolar Ucn2/3 A35 appears to be incompatible with the negatively charged surface of the CRF1 receptor ECD, whereas CRF/Ucn1 R35 is electrostatically compatible with the negatively charged region of the CRF1 receptor and also the nonpolar P100 of the CRF2 receptor via the aliphatic portion of the R35 side chain. The charge compatibility mechanism is supported by peptide binding experiments showing that Ucn1 R35A abolished binding to the CRF1 receptor ECD with relatively minor impact on CRF2 receptor ECD binding and that Ucn3 A35R conferred binding ability to CRF1 receptor ECD while having a relatively minor effect on CRF2 receptor ECD binding (Pal et al., 2010).
Fig. 4.
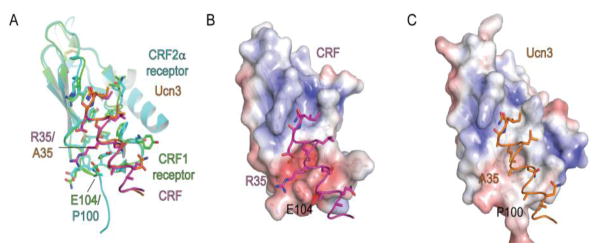
Structural determinants for ECD-mediated class B GPCR subtype selectivity. (A) Superposition of the CRF1 receptor and CRF2α receptor ECDs with bound CRF (PDB: 3EHU) and Ucn3 (PDB: 3N93) peptide fragments, respectively. Receptor and peptide residues involved in selectivity are labeled. (B, C) Electrostatic surface potentials [from −5 (red) to +5 (blue) kT/e] of the CRF1 receptor ECD highlighting the negative area near CRF1 receptor E104 (B) and of the CRF2α receptor ECD highlighting the neutral area near CRF2α receptor P100 (C).
6. Class C GPCR structure, ligand binding and selectivity
The class C/Glutamate family GPCRs comprise fifteen receptors in humans that are activated by small molecules such as amino acids and ions (Fredriksson et al., 2003). This family includes eight metabotropic receptors for the neurotransmitter glutamate, the GABA receptors, the calcium sensing receptor, and some taste receptors. Similar to the class B receptors, the class C receptors have an N-terminal ECD in addition to the 7TM domain, but the class C ECD is very large and it contains the entire orthosteric ligand binding site. The class C “venus flytrap domain” (VFT) ECD forms a bi-lobed structure with the orthosteric ligand binding pocket situated in a central cleft (Fig. 5A) (Kunishima et al., 2000). The VFT domain fluctuates between open and closed conformations; agonists generally stabilize the closed conformation whereas antagonists maintain the open conformation (Geng et al., 2013; Muto et al., 2007; Tsuchiya et al., 2002). In all cases except the GABAB receptor, a small cysteine-rich domain links the VFT domain to the 7TM domain. Class C GPCRs exist as heteromers, either as homodimers or in the case of the GABAB receptor as a heterodimer of GABAB receptor 1 and 2 (Pin et al., 2004). The VFT domain mediates dimerization (Fig. 5A). The mechanism by which agonist binding to the VFT results in activation of the 7TM domain is complex and not yet fully understood (Pin et al., 2004).
Fig. 5.

Class C GPCR structure, ligand binding sites and ligand selectivity. (A) Structure of the homodimeric extracellular venus flytrap domain of mGlu1 with a bound agonist glutamate shown in stick and space-filling representation in the orthosteric binding site (PDB: 1EWK). Glycans and disulfide bonds are shown in stick representation. (B) Structure of the mGlu1 7TM domain with a bound negative allosteric modulator FITM shown in stick and space-filling representation (PDB: 4OR2). Cholesterol molecules at the interface of the 7TM “homodimer” and disulfide bonds are shown in stick representation. (C) Superposition of the class C mGlu1 7TM domain and the class A dopamine D3 receptor with a bound antagonist eticlopride (PDB: 3PBL) viewed from the extracellular side. FITM is colored yellow and eticlopride magenta. FITM, eticlopride, and disulfide bonds are shown in stick representation. (D) Superposition of the mGlu1 and mGlu5 (PDB: 4OO9) 7TM structures highlighting a subtype selectivity determinant for negative allosteric modulator binding. FITM is colored green and mavoglurant is slate blue.
Drugs targeting several class C receptors are available in the market or in clinical trials for diseases including hyperparathyroidism, anxiety, schizophrenia, Fragile X syndrome, and Parkinson’s (Urwyler, 2011). The orthosteric binding sites of the VFTs are generally well conserved and thus create challenges for designing selective drugs. Most of the drugs in development are allosteric modulators that bind to the 7TM domain at a site similar to the orthosteric site of class A GPCRs; the allosteric 7TM sites are generally less conserved and therefore enable subtype selectivity. In 2014 crystal structures of the 7TM domains of the metabotropic glutamate receptors mGlu1 and mGlu5 were reported, both with bound negative allosteric modulators (NAMs) (Dore et al., 2014; Wu et al., 2014). The mGlu1 7TM domain intriguingly crystallized as a homodimer with the dimer interface mediated by TM I interactions and a cluster of cholesterol molecules (Wu et al., 2014), but the physiological relevance of this dimer remains to be determined (Fig. 5B). The mGlu5 7TM structure did not form a dimer (Dore et al., 2014). The NAM binding sites were similar in both structures and positioned equivalent to the orthosteric binding site of class A GPCRs in the extracellular half of the receptors near helices III, VI, and VII (Fig. 5B, C). The helical bundle of the class C GPCRs is more similar to that of the class A GPCRs than the class B GPCRs. The extracellular half of the class C helical bundle is compact with TM V and VII situated more towards the core of the receptor than in the class A receptors making the NAM binding pocket relatively small (Fig. 5C). The mGlu1 structure provided a clear explanation for the selectivity of the NAM FITM for mGlu1 over mGlu5. Residue T815 of mGlu1 on helix VII makes a hydrogen bond with FITM whereas the equivalent position in mGlu5 is M802, which lacks hydrogen bonding ability and would sterically clash with FITM (Fig. 5D). Consistent with the structures, the mGlu1 T815M mutant exhibited significantly reduced FITM binding highlighting that this position is an important selectivity determinant (Wu et al., 2014).
7. Conclusions and future perspectives
The large number of GPCR crystal structures accumulated over the last several years is striking when considering that in early 2007 only one GPCR structure, that of Rhodopsin, was available. Representative structures for each of the major GPCR classes (other than the adhesion family) are now available and these have been invaluable for understanding ligand binding and selectivity as reviewed here. The surge in structural information has dramatically changed the field. We now have an atomic resolution structural understanding of orthosteric and allosteric ligand binding, ligand selectivity, efficacy, receptor activation, and G protein binding, and we are beginning to understand biased agonism (Liu et al., 2012; Wacker et al., 2013). The wealth of structural data has significantly expanded our understanding of GPCR pharmacology and opened up new avenues for GPCR structure-based drug design, which has enormous potential to yield drugs targeting GPCRs with optimal selectivity, potency, and efficacy profiles. Despite all of these advances, several areas remain in need of a structural explanation. A wish list of structures, in no particular order, includes a peptide bound class B GPCR 7TM domain and/or full-length receptor, full-length class C and class F GPCRs, active state structures for class B, C, and F receptors, GPCRs in complex with various G proteins, a GPCR-arrestin complex, and a GPCR-G protein-coupled receptor kinase (GRK) complex. In addition, there are many accessory/regulatory proteins that interact with GPCRs and we still understand very little about these interactions at the structural level. Lastly, despite the technological breakthroughs GPCR crystallography is still very expensive and labor-intensive. There is a need for methodological advances that make obtaining these structures easier and cheaper. These tasks will keep structural biologists busy for some time in the GPCR field.
Acknowledgments
Research in the Pioszak laboratory is supported by a grant from the National Institutes of Health [R01GM104251].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Audet M, Bouvier M. Restructuring G-protein- coupled receptor activation. Cell. 2012;151:14–23. doi: 10.1016/j.cell.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annual review of pharmacology and toxicology. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neurosciences. 1995;25:366–428. [Google Scholar]
- Bonner G, Meng F, Akil H. Selectivity of mu-opioid receptor determined by interfacial residues near third extracellular loop. European journal of pharmacology. 2000;403:37–44. doi: 10.1016/s0014-2999(00)00578-1. [DOI] [PubMed] [Google Scholar]
- Bortolato A, Dore AS, Hollenstein K, Tehan BG, Mason JS, Marshall FH. Structure of Class B GPCRs: new horizons for drug discovery. British journal of pharmacology. 2014;171:3132–3145. doi: 10.1111/bph.12689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Dmt and opioid peptides: a potent alliance. Biopolymers. 2003;71:86–102. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
- Cherezov V. Lipidic cubic phase technologies for membrane protein structural studies. Current opinion in structural biology. 2011;21:559–566. doi: 10.1016/j.sbi.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, Shi L, Newman AH, Javitch JA, Cherezov V, Stevens RC. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–1095. doi: 10.1126/science.1197410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M, Dias JM, Marshall FH. Structure-based drug design for G protein-coupled receptors. Progress in medicinal chemistry. 2014;53:1–63. doi: 10.1016/B978-0-444-63380-4.00001-9. [DOI] [PubMed] [Google Scholar]
- Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, Schaff HV. Valvular heart disease associated with fenfluramine-phentermine. The New England journal of medicine. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- Disingrini T, Muth M, Dallanoce C, Barocelli E, Bertoni S, Kellershohn K, Mohr K, De Amici M, Holzgrabe U. Design, synthesis, and action of oxotremorine-related hybrid-type allosteric modulators of muscarinic acetylcholine receptors. Journal of medicinal chemistry. 2006;49:366–372. doi: 10.1021/jm050769s. [DOI] [PubMed] [Google Scholar]
- Dondio G, Ronzoni S, Eggleston DS, Artico M, Petrillo P, Petrone G, Visentin L, Farina C, Vecchietti V, Clarke GD. Discovery of a novel class of substituted pyrrolooctahydroisoquinolines as potent and selective delta opioid agonists, based on an extension of the message-address concept. Journal of medicinal chemistry. 1997;40:3192–3198. doi: 10.1021/jm9608218. [DOI] [PubMed] [Google Scholar]
- Dong M, Koole C, Wootten D, Sexton PM, Miller LJ. Structural and functional insights into the juxtamembranous amino-terminal tail and extracellular loop regions of class B GPCRs. British journal of pharmacology. 2014;171:1085–1101. doi: 10.1111/bph.12293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore AS, Okrasa K, Patel JC, Serrano-Vega M, Bennett K, Cooke RM, Errey JC, Jazayeri A, Khan S, Tehan B, Weir M, Wiggin GR, Marshall FH. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, Xu H, Shaw DE. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egloff P, Hillenbrand M, Klenk C, Batyuk A, Heine P, Balada S, Schlinkmann KM, Scott DJ, Schutz M, Pluckthun A. Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E655–662. doi: 10.1073/pnas.1317903111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elangbam CS, Job LE, Zadrozny LM, Barton JC, Yoon LW, Gates LD, Slocum N. 5-hydroxytryptamine (5HT)-induced valvulopathy: compositional valvular alterations are associated with 5HT2B receptor and 5HT transporter transcript changes in Sprague-Dawley rats. Experimental and toxicologic pathology : official journal of the Gesellschaft fur Toxikologische Pathologie. 2008;60:253–262. doi: 10.1016/j.etp.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of delta-opioid receptor signalling. Nature. 2014;506:191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald LW, Burn TC, Brown BS, Patterson JP, Corjay MH, Valentine PA, Sun JH, Link JR, Abbaszade I, Hollis JM, Largent BL, Hartig PR, Hollis GF, Meunier PC, Robichaud AJ, Robertson DW. Possible role of valvular serotonin 5-HT(2B) receptors in the cardiopathy associated with fenfluramine. Molecular pharmacology. 2000;57:75–81. [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Molecular pharmacology. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Geng Y, Bush M, Mosyak L, Wang F, Fan QR. Structural mechanism of ligand activation in human GABA(B) receptor. Nature. 2013;504:254–259. doi: 10.1038/nature12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace CR, Perrin MH, Gulyas J, Digruccio MR, Cantle JP, Rivier JE, Vale WW, Riek R. Structure of the N-terminal domain of a type B1 G protein-coupled receptor in complex with a peptide ligand. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4858–4863. doi: 10.1073/pnas.0700682104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace CR, Perrin MH, Gulyas J, Rivier JE, Vale WW, Riek R. NMR structure of the first extracellular domain of corticotropin-releasing factor receptor 1 (ECD1-CRF-R1) complexed with a high affinity agonist. The Journal of biological chemistry. 2010;285:38580–38589. doi: 10.1074/jbc.M110.121897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, Kobilka BK. Structure of the delta-opioid receptor bound to naltrindole. Nature. 2012;485:400–404. doi: 10.1038/nature11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, Kobayashi T. Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature. 2012;482:547–551. doi: 10.1038/nature10753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, Stevens RC. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoare SR. Mechanisms of peptide and nonpeptide ligand binding to Class B G-protein-coupled receptors. Drug Discov Today. 2005;10:417–427. doi: 10.1016/S1359-6446(05)03370-2. [DOI] [PubMed] [Google Scholar]
- Hollenstein K, Kean J, Bortolato A, Cheng RK, Dore AS, Jazayeri A, Cooke RM, Weir M, Marshall FH. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Diversity and modularity of G protein-coupled receptor structures. Trends in pharmacological sciences. 2012;33:17–27. doi: 10.1016/j.tips.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annual review of pharmacology and toxicology. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski LF., Jr GCRDb: a G-protein-coupled receptor database. Receptors & channels. 1994;2:1–7. [PubMed] [Google Scholar]
- Kroeze WK, Kristiansen K, Roth BL. Molecular biology of serotonin receptors structure and function at the molecular level. Current topics in medicinal chemistry. 2002;2:507–528. doi: 10.2174/1568026023393796. [DOI] [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, Shaw DE, Weis WI, Wess J, Kobilka BK. Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature. 2012;482:552–556. doi: 10.1038/nature10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Muscarinic acetylcholine receptors: novel opportunities for drug development. Nature reviews Drug discovery. 2014;13:549–560. doi: 10.1038/nrd4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Ring AM, Manglik A, Hu J, Hu K, Eitel K, Hubner H, Pardon E, Valant C, Sexton PM, Christopoulos A, Felder CC, Gmeiner P, Steyaert J, Weis WI, Garcia KC, Wess J, Kobilka BK. Activation and allosteric modulation of a muscarinic acetylcholine receptor. Nature. 2013;504:101–106. doi: 10.1038/nature12735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nature reviews Drug discovery. 2008;7:339–357. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Dolezal V, Popham A, Birdsall NJ. Thiochrome enhances acetylcholine affinity at muscarinic M4 receptors: receptor subtype selectivity via cooperativity rather than affinity. Molecular pharmacology. 2004;65:257–266. doi: 10.1124/mol.65.1.257. [DOI] [PubMed] [Google Scholar]
- Lipkowski AW, Tam SW, Portoghese PS. Peptides as receptor selectivity modulators of opiate pharmacophores. Journal of medicinal chemistry. 1986;29:1222–1225. doi: 10.1021/jm00157a018. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science. 2012;335:1106–1110. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Kruse AC, Kobilka TS, Thian FS, Mathiesen JM, Sunahara RK, Pardo L, Weis WI, Kobilka BK, Granier S. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012;485:321–326. doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorvy JD, Roth BL. Structure and Function of Serotonin G protein Coupled Receptors. Pharmacology & therapeutics. 2015 doi: 10.1016/j.pharmthera.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F, Ueda Y, Hoversten MT, Taylor LP, Reinscheid RK, Monsma FJ, Watson SJ, Civelli O, Akil H. Creating a functional opioid alkaloid binding site in the orphanin FQ receptor through site-directed mutagenesis. Molecular pharmacology. 1998;53:772–777. doi: 10.1124/mol.53.4.772. [DOI] [PubMed] [Google Scholar]
- Moad HE, Pioszak AA. Selective CGRP and adrenomedullin peptide binding by tethered RAMP-calcitonin receptor-like receptor extracellular domain fusion proteins. Protein science : a publication of the Protein Society. 2013;22:1775–1785. doi: 10.1002/pro.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, Pasternak GW. The molecular and behavioral pharmacology of the orphanin FQ/nociceptin peptide and receptor family. Pharmacological reviews. 2001;53:381–415. [PubMed] [Google Scholar]
- Mosberg HI. Complementarity of delta opioid ligand pharmacophore and receptor models. Biopolymers. 1999;51:426–439. doi: 10.1002/(SICI)1097-0282(1999)51:6<426::AID-BIP5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Muto T, Tsuchiya D, Morikawa K, Jingami H. Structures of the extracellular regions of the group II/III metabotropic glutamate receptors. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3759–3764. doi: 10.1073/pnas.0611577104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebigil CG, Hickel P, Messaddeq N, Vonesch JL, Douchet MP, Monassier L, Gyorgy K, Matz R, Andriantsitohaina R, Manivet P, Launay JM, Maroteaux L. Ablation of serotonin 5-HT(2B) receptors in mice leads to abnormal cardiac structure and function. Circulation. 2001;103:2973–2979. doi: 10.1161/01.cir.103.24.2973. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nature reviews. Drug discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Pal K, Swaminathan K, Xu HE, Pioszak AA. Structural basis for hormone recognition by the Human CRFR2{alpha} G protein-coupled receptor. The Journal of biological chemistry. 2010;285:40351–40361. doi: 10.1074/jbc.M110.186072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE, Yamamoto M, Miyano M. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Parthier C, Kleinschmidt M, Neumann P, Rudolph R, Manhart S, Schlenzig D, Fanghanel J, Rahfeld JU, Demuth HU, Stubbs MT. Crystal structure of the incretin-bound extracellular domain of a G protein-coupled receptor. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:13942–13947. doi: 10.1073/pnas.0706404104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Kniazeff J, Goudet C, Bessis AS, Liu J, Galvez T, Acher F, Rondard P, Prezeau L. The activation mechanism of class-C G-protein coupled receptors. Biology of the cell / under the auspices of the European Cell Biology Organization. 2004;96:335–342. doi: 10.1016/j.biolcel.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Gardella TJ, Xu HE. Structural basis for parathyroid hormone-related protein binding to the parathyroid hormone receptor and design of conformation-selective peptides. The Journal of biological chemistry. 2009;284:28382–28391. doi: 10.1074/jbc.M109.022905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. The Journal of biological chemistry. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portoghese PS. Bivalent ligands and the message-address concept in the design of selective opioid receptor antagonists. Trends in pharmacological sciences. 1989;10:230–235. doi: 10.1016/0165-6147(89)90267-8. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Fung JJ, Pardon E, Casarosa P, Chae PS, Devree BT, Rosenbaum DM, Thian FS, Kobilka TS, Schnapp A, Konetzki I, Sunahara RK, Gellman SH, Pautsch A, Steyaert J, Weis WI, Kobilka BK. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature. 2011a;469:175–180. doi: 10.1038/nature09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, Mathiesen JM, Shah ST, Lyons JA, Caffrey M, Gellman SH, Steyaert J, Skiniotis G, Weis WI, Sunahara RK, Kobilka BK. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011b;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riekel C, Burghammer M, Schertler G. Protein crystallography microdiffraction. Current opinion in structural biology. 2005;15:556–562. doi: 10.1016/j.sbi.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Runge S, Thogersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. The Journal of biological chemistry. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Scott DJ, Pluckthun A. Direct molecular evolution of detergent-stable G protein-coupled receptors using polymer encapsulated cells. Journal of molecular biology. 2013;425:662–677. doi: 10.1016/j.jmb.2012.11.015. [DOI] [PubMed] [Google Scholar]
- Setola V, Dukat M, Glennon RA, Roth BL. Molecular determinants for the interaction of the valvulopathic anorexigen norfenfluramine with the 5-HT2B receptor. Molecular pharmacology. 2005;68:20–33. doi: 10.1124/mol.104.009266. [DOI] [PubMed] [Google Scholar]
- Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, Qian J, Li S, Blanc A, Oleskie AN, Dosey AM, Su M, Liang CR, Gu LL, Shan JM, Chen X, Hanna R, Choi M, Yao XJ, Klink BU, Kahsai AW, Sidhu SS, Koide S, Penczek PA, Kossiakoff AA, Woods VL, Jr, Kobilka BK, Skiniotis G, Lefkowitz RJ. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu FY, He M, de Graaf C, Han GW, Yang D, Zhang Z, Zhou C, Xu Q, Wacker D, Joseph JS, Liu W, Lau J, Cherezov V, Katritch V, Wang MW, Stevens RC. Structure of the human glucagon class B G-protein-coupled receptor. Nature. 2013;499:444–449. doi: 10.1038/nature12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinfeld T, Mammen M, Smith JA, Wilson RD, Jasper JR. A novel multivalent ligand that bridges the allosteric and orthosteric binding sites of the M2 muscarinic receptor. Molecular pharmacology. 2007;72:291–302. doi: 10.1124/mol.106.033746. [DOI] [PubMed] [Google Scholar]
- Tate CG. A crystal clear solution for determining G-protein-coupled receptor structures. Trends in biochemical sciences. 2012;37:343–352. doi: 10.1016/j.tibs.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Tehan BG, Bortolato A, Blaney FE, Weir MP, Mason JS. Unifying family A GPCR theories of activation. Pharmacology & therapeutics. 2014;143:51–60. doi: 10.1016/j.pharmthera.2014.02.004. [DOI] [PubMed] [Google Scholar]
- Tfelt-Hansen P, Saxena PR, Dahlof C, Pascual J, Lainez M, Henry P, Diener H, Schoenen J, Ferrari MD, Goadsby PJ. Ergotamine in the acute treatment of migraine: a review and European consensus. Brain : a journal of neurology. 2000;123 ( Pt 1):9–18. doi: 10.1093/brain/123.1.9. [DOI] [PubMed] [Google Scholar]
- Thompson AA, Liu W, Chun E, Katritch V, Wu H, Vardy E, Huang XP, Trapella C, Guerrini R, Calo G, Roth BL, Cherezov V, Stevens RC. Structure of the nociceptin/orphanin FQ receptor in complex with a peptide mimetic. Nature. 2012;485:395–399. doi: 10.1038/nature11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K. Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+ Proceedings of the National Academy of Sciences of the United States of America. 2002;99:2660–2665. doi: 10.1073/pnas.052708599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood CR, Garibay P, Knudsen LB, Hastrup S, Peters GH, Rudolph R, Reedtz-Runge S. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. The Journal of biological chemistry. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwyler S. Allosteric modulation of family C G-protein-coupled receptors: from molecular insights to therapeutic perspectives. Pharmacological reviews. 2011;63:59–126. doi: 10.1124/pr.109.002501. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Vickers SP, Dourish CT, Kennett GA. Evidence that hypophagia induced by d-fenfluramine and d-norfenfluramine in the rat is mediated by 5-HT2C receptors. Neuropharmacology. 2001;41:200–209. doi: 10.1016/s0028-3908(01)00063-6. [DOI] [PubMed] [Google Scholar]
- Villalon CM, De Vries P, Rabelo G, Centurion D, Sanchez-Lopez A, Saxena P. Canine external carotid vasoconstriction to methysergide, ergotamine and dihydroergotamine: role of 5-HT1B/1D receptors and alpha2-adrenoceptors. British journal of pharmacology. 1999;126:585–594. doi: 10.1038/sj.bjp.0702324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D, Wang C, Katritch V, Han GW, Huang XP, Vardy E, McCorvy JD, Jiang Y, Chu M, Siu FY, Liu W, Xu HE, Cherezov V, Roth BL, Stevens RC. Structural features for functional selectivity at serotonin receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Jiang Y, Ma J, Wu H, Wacker D, Katritch V, Han GW, Liu W, Huang XP, Vardy E, McCorvy JD, Gao X, Zhou XE, Melcher K, Zhang C, Bai F, Yang H, Yang L, Jiang H, Roth BL, Cherezov V, Stevens RC, Xu HE. Structural basis for molecular recognition at serotonin receptors. Science. 2013a;340:610–614. doi: 10.1126/science.1232807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013b;497:338–343. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T, Tate CG. The importance of interactions with helix 5 in determining the efficacy of beta-adrenoceptor ligands. Biochemical Society transactions. 2013;41:159–165. doi: 10.1042/BST20120228. [DOI] [PubMed] [Google Scholar]
- White JF, Noinaj N, Shibata Y, Love J, Kloss B, Xu F, Gvozdenovic-Jeremic J, Shah P, Shiloach J, Tate CG, Grisshammer R. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang XP, Carroll FI, Mascarella SW, Westkaemper RB, Mosier PD, Roth BL, Cherezov V, Stevens RC. Structure of the human kappa-opioid receptor in complex with JDTic. Nature. 2012;485:327–332. doi: 10.1038/nature10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Wang C, Gregory KJ, Han GW, Cho HP, Xia Y, Niswender CM, Katritch V, Meiler J, Cherezov V, Conn PJ, Stevens RC. Structure of a class C GPCR metabotropic glutamate receptor 1 bound to an allosteric modulator. Science. 2014;344:58–64. doi: 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]