

Abstract
Rationale: Pulmonary arterial hypertension is characterized by endothelial dysregulation, but global changes in gene expression have not been related to perturbations in function.
Objectives: RNA sequencing was used to discriminate changes in transcriptomes of endothelial cells cultured from lungs of patients with idiopathic pulmonary arterial hypertension versus control subjects and to assess the functional significance of major differentially expressed transcripts.
Methods: The endothelial transcriptomes from the lungs of seven control subjects and six patients with idiopathic pulmonary arterial hypertension were analyzed. Differentially expressed genes were related to bone morphogenetic protein type 2 receptor (BMPR2) signaling. Those down-regulated were assessed for function in cultured cells and in a transgenic mouse.
Measurements and Main Results: Fold differences in 10 genes were significant (P < 0.05), four increased and six decreased in patients versus control subjects. No patient was mutant for BMPR2. However, knockdown of BMPR2 by siRNA in control pulmonary arterial endothelial cells recapitulated 6 of 10 patient-related gene changes, including decreased collagen IV (COL4A1, COL4A2) and ephrinA1 (EFNA1). Reduction of BMPR2-regulated transcripts was related to decreased β-catenin. Reducing COL4A1, COL4A2, and EFNA1 by siRNA inhibited pulmonary endothelial adhesion, migration, and tube formation. In mice null for the EFNA1 receptor, EphA2, versus control animals, vascular endothelial growth factor receptor blockade and hypoxia caused more severe pulmonary hypertension, judged by elevated right ventricular systolic pressure, right ventricular hypertrophy, and loss of small arteries.
Conclusions: The novel relationship between BMPR2 dysfunction and reduced expression of endothelial COL4 and EFNA1 may underlie vulnerability to injury in pulmonary arterial hypertension.
Keywords: pulmonary hypertension, vascular endothelium, transcriptome, ephrin, collagen IV
At a Glance Commentary
Scientific Knowledge on the Subject
The endothelium is an early site of injury in the pathogenesis of pulmonary arterial hypertension, and loss of endothelial control of pulmonary vascular tone and structure contributes to the adverse remodeling and to the increased pulmonary artery pressure and resistance that define the disease. Transcriptomic approaches allow for broad comparisons of the gene expression profiles of diseased and healthy tissues and shed light on novel pathways that are relevant to pathogenesis.
What This Study Adds to the Field
This is the first RNAseq transcriptome comparison of pulmonary arterial endothelial cells from patients with idiopathic pulmonary arterial hypertension versus controls, and a novel set of genes is now implicated in disease pathogenesis. Although the search was unbiased, it is particularly of interest that most of those genes differentially expressed could be reproduced in control cells by reducing levels of bone morphogenetic protein receptor 2, which is mutated in familial and dysfunctional in other forms of pulmonary arterial hypertension. The down-regulated genes further pursued, collagen IV (A1 and A2) and ephrin A1, support cell adhesion and angiogenesis and are implicated in restoration and regeneration of damaged blood vessels. Thus, reduced levels of these genes may underlie the inability of the pulmonary vasculature to respond to and repair the damage observed in pulmonary hypertension. Consistent with observations in cultured cells, we also provide evidence that mice lacking the main receptor for ephrin A1 (EphA2) develop more severe pulmonary hypertension under conditions where there is endothelial vulnerability to apoptosis.
Idiopathic pulmonary arterial hypertension (IPAH) is a rare but debilitating disease characterized by endothelial cell (EC) dysfunction and pulmonary vascular obliteration and loss (for details, see Reference 1). These features cause a progressive increase in resistance to flow that can culminate in right heart failure and in the patient’s demise. Several lines of evidence pinpoint the pulmonary arterial (PA) endothelium as playing a critical role in the pathogenesis of IPAH and pulmonary arterial hypertension (PAH) associated with other primary conditions (APAH). Pulmonary arterial ECs (PAEC) cultured from patients with PAH display impaired angiogenesis (2–5), altered bioenergetics (6), and chromosomal/genetic instability not present in other cell types (7). These features influence PAH pathogenesis and the potential for reversibility of this condition. Moreover, PAECs are the major site of expression of bone morphogenetic protein type 2 receptor (BMPR2) in the lung (8), and mutations in BMPR2 underlie most heritable cases and a small proportion of sporadic cases of IPAH (9, 10). Reduced BMPR2 expression and function is also described in patients with IPAH or with APAH who do not have a mutation (8). Loss of BMPR2 impairs PAEC survival (2–5) and barrier function (11). Our previous studies demonstrated that BMPR2 signaling activates β-catenin/peroxisome proliferator-activated receptor (PPAR) γ–mediated transcription of PAEC genes, such as apelin (3, 4). In PAH PAECs, we have shown that BMPR2 deficiency and the resultant reduction in apelin can lead to apoptosis, and an inability to form vascular networks and to control smooth muscle cell proliferation (4, 5).
We reasoned that a comprehensive search for genes that are dysregulated in PAH ECs could improve our understanding of the mechanisms underlying the dysfunction of these cells and might facilitate development of targeted therapies. Such unbiased approaches to gene expression analysis in PAH have been performed on peripheral blood mononuclear cells (12), whole lung tissue (13), and microdissected vessels (14). A recent study investigated proteomic changes in blood outgrowth ECs from four control subjects and four heritable patients with PAH (15) and another compared metabolomic and transcriptomic changes in PAECs engineered with a BMPR2 mutation (16), but no studies to date have specifically addressed changes in gene expression and the functional significance of those features in PAECs derived from patients with IPAH versus those from control lungs. This opportunity has been made possible through the Pulmonary Hypertension Breakthrough Initiative Network, which collects lung tissue from patients with IPAH and APAH and from unused donor control lungs (see the online supplement).
Moreover, a new opportunity to sensitively detect specific changes in gene expression is available through RNA-sequencing analysis (RNAseq) (17). We therefore applied RNAseq to compare gene expression profiles of ECs derived from the lungs of patients with IPAH with those obtained from unused donor lungs as control subjects. Significant changes in gene expression were related to reduced BMPR2 and a downstream effector, β-catenin. The functional significance of three transcripts that were highly down-regulated in IPAH PAECs (collagen IV-A1 and A2 [COL4A1, COL4A2] and ephrinA1 [EFNA1]) were investigated in cultured cells, and one (EFNA1) was pursued in a transgenic mouse with loss of the cognate receptor EphA2.
Methods
An expanded section is included in the online supplement.
Cell Culture
PAECs from patients with IPAH and unused donor control PAECs were harvested and cultured according to previous protocols (4) from lungs obtained through the Pulmonary Hypertension Breakthrough Initiative Network of transplant centers described in the Acknowledgment section. Demographic information regarding patients and unused donor control lungs is provided in Table 1.
Table 1.
Characteristics of Patients with IPAH and Unused Donor Control PAECs Used in RNAseq Experiments
| Patient ID | Age/Sex | Race | Cell Type | Diagnosis/Cause of Death | PAP S/D/M (mm Hg) | PVR (Wood Units) | Total PAH Medications |
|---|---|---|---|---|---|---|---|
|
Cells used in RNAseq experiment 1 | |||||||
| Con-1 | 57/F | White | MV | Intracranial hemorrhage/stroke | |||
| Con-2 | 41/F | White | MV | Grade IV subarachnoid hemorrhage, ruptured anterior cerebral artery aneurysm | |||
| Con-3 | 19/M | White | MV | Brain anoxia | |||
| PAH-1 | 56/F | White | MV | FPAH, no BMPR2 mutation | 110/55/75 | 29.6 | Epoprostenol, bosentan, ambrisentan, sildenafil |
| PAH-2 | 49/F | White | MV | IPAH | 100/50/75 | 16.8 | Ambrisentan, sildenafil, epoprostenol |
| PAH-3 | 55/F | African American | MV | IPAH | 89/41/53 | 12.3 | Sildenafil, bosentan, epoprostenol |
| | |||||||
|
Cells used in RNAseq experiment 2 | |||||||
| Con-4 | 40/F | White | MV | Motor vehicle accident: extensive subarachnoid hemorrhage | |||
| Con-5 | 52/F | White | MV | Hypoxic brain death | |||
| Con-6 | 55/M | White | MV | Anoxia | |||
| Con-7 | 14/M | White | MV | Gunshot wound to head | |||
| PAH-4 | 54/F | White | MV | IPAH | 100/45/60 | 12.0 | Sildenafil, epoprostenol, ambrisentan, bosentan |
| PAH-5 | 56/F | White | MV | IPAH | 83/39/57 | 11.4 | Sildenafil, ambrisentan, treprostinil |
| PAH-6 | 25/M | White | SPA | IPAH | 65/15/36 | 9.0 | Epoprostenol, sildenafil, treprostinil |
Definition of abbreviations: BMPR2 = bone morphogenetic protein type 2 receptor; Con = control; FPAH = familial pulmonary arterial hypertension; ID = identification; IPAH = idiopathic pulmonary arterial hypertension; MV = microvasculature; PAEC = pulmonary arterial endothelial cell; PAH = pulmonary arterial hypertension; PAP = pulmonary artery pressure; PVR = pulmonary vascular resistance; S/D/M = systolic/diastolic/mean; SPA = small pulmonary arteries <1 mm by dissection.
PAH medications are listed according to total drug exposure during treatment period of patient, not necessarily in combination.
Commercially available human PAECs (PromoCell, Heidelberg, Germany) were used for studies not involving patient-derived cell lines. PAECs were cultured in EC medium containing growth factors, 5% fetal bovine serum, and penicillin/streptomycin (ScienCell, Carlsbad, CA).
RNAseq Analysis of PAECs
We used PAECs from control subjects and subjects with IPAH at similar passage 3–5 for RNAseq and reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) experiments. RNA was extracted from 90–100% confluent PAECs in growth medium and RNAseq libraries were prepared. The libraries were sequenced on 1–2 HiSeq 2000 lanes to obtain an average of approximately 100 and 150 million uniquely mapped reads for control and IPAH groups, respectively.
For all control and IPAH samples, raw expression (RPKM) data were filtered to exclude any genes that did not meet the prespecified criteria of more than five RPKM. To allow comparisons across samples, the RPKM data distributions were quantile-normalized using the R package “preprocessCore” (Benjamin Milo Bolstad Preprocess Core: A collection of pre-processing functions, v1.18.0).
Transcriptome data (FASTQ files) are being submitted to the NIH database of Genotypes and Phenotypes (dbGaP).
Quantitative PCR
RNA was extracted using spin column-based kits and RT-PCR performed as per the manufacturer’s guidelines. qPCR was performed with primer sequences, designed using National Center for Biotechnology (NCBI) (Bethesda, MD) Primer-BLAST function or taken from Reference 18 (see Table E1 in the online supplement). Identification of specific MiRs and housekeeping MiRs were from Life Technologies (Carlsbad, CA), indicated in the online supplement.
Immunoblotting
Nitrocellulose membranes were incubated with rabbit anti-COL4, rabbit anti-EFNA1, or mouse anti–β-actin overnight at 4°C, then incubated with secondary antibodies and imaged using enhanced chemiluminescence reagent.
siRNA and DNA Transfections
Control, BMPR2, EFNA1, COL4A1, COL4A2, β-catenin, PPARγ, ID1 siRNA, and SMAD1 and SMAD3 mutant constructs were transfected into subconfluent PAECs.
Modified Boyden Chamber PAEC Migration Assay
Migration was assessed 8 hours after seeding the cells using the modified Boyden Chamber Assay previously described (4).
Cell Adhesion and Tube Formation Assays
Cell adhesion to gelatin or collagen was assessed at 40 minutes, and tube formation assays in Matrigel were carried out as previously described (19).
Studies in Transgenic Mice
The Animal Care Committee at Stanford University approved all the experimental protocols used in this study. Wild-type C57BL/6J and EphA2 knockout mice (Jackson Laboratory, Bar Harbor, ME) were given a subcutaneous injection of the vascular endothelial growth factor (VEGF) receptor blocker Sugen 5416 (20 mg/kg) followed by 4-week exposure to chronic hypoxia (air at 10% O2). Cardiac function and output were assessed by echocardiography and right ventricular (RV) systolic pressure was measured by closed chest technique (20). RV hypertrophy was assessed as the weight of the RV relative to left ventricle (LV) plus septum. Lungs were perfused with normal saline and fixed for routine histology, and morphometric analysis performed as previously described (5). Mouse lung tissue sections were stained with fluorescent antibodies for von Willebrand factor and α-smooth muscle actin as previously described (20).
Statistical Analysis
Genes analyzed by RNAseq and qPCR were compared in control and IPAH cells by expression level using average fold differences and an unpaired t test to establish significance. Correlations were assessed by Spearman rank test. Comparisons between two cohorts were assessed by t test; between three or more cohorts by one-way analysis of variance with Dunnett; and when combining more than one condition, a two-way analysis of variance with Bonferroni-corrected post hoc testing was used. All cell culture experiments were conducted three or more times with two to four replicates per condition.
Results
RNAseq Analysis of PAECs from Patients with IPAH
RNAseq transcriptomic profiling was performed on RNA isolates from PAECs from two separate groups of control subjects (unused donor, n = 3 in RNAseq1 and n = 4 in RNAseq2) and patients (IPAH, n = 3 in RNAseq1 and n = 3 in RNAseq2) (Table 1, Figure 1A). Genes with average fold differences greater than or equal to 50% were selected for comparison. When combining the two sets of control and IPAH PAECs, 23 genes were differentially expressed by greater than or equal to 50% (Table 2). In 12 of these, the difference between IPAH and donor control gene expression was P less than 0.1, and in 9 of 12 P less than 0.05. These gene expression differences were selected for further investigation.
Figure 1.

RNAseq analysis of pulmonary arterial endothelial cells (PAECs) from patients with idiopathic pulmonary arterial hypertension (IPAH) and unused donor controls (Con) reveals differences in gene expression. (A) Volcano plots illustrate fold differences in individual gene expression in IPAH versus control subjects, and associated P values (negative log10), in two RNAseq experiments. (B) Mean ± SEM fold differences (e.g., +1 = 100% higher, +0.5 = 50% higher) in gene candidates as measured by quantitative polymerase chain reaction (qPCR) and RNAseq. *P < 0.05, **P < 0.01, and ***P < 0.001 versus control value, by t test. (C) Comparison of RNAseq and qPCR measurements in individual IPAH and control cells for COL4A1 and COL4A2. Data are arbitrary units normalized to the mean value in each experiment. (D) Representative immunoblot for COL4 protein in PAEC lysates, with β-actin as a loading control, and densitometry analysis below. Bars represent mean ± SEM of n = 3 with ****P < 0.0001. COL4A = collagen IV-A.
Table 2.
Patients with IPAH versus Control Subjects: Gene Expression Fold Differences by RNAseq and PCR
| Gene | RNAseq1 | RNAseq2 | Combined Data | P Value | PCR | P Value |
|---|---|---|---|---|---|---|
|
Genes increased in IPAH PAECs in both experiments | ||||||
| DNAJB4 | +0.57 | +0.67 | +0.62 | 0.0001 | +0.37 | 0.08 |
| NOS3 | +0.73 | +1.63 | +1.18 | 0.012 | +1.50 | 0.006 |
| HMGA1 | +0.69 | +0.71 | +0.70 | 0.04 | +0.63 | 0.020 |
| PIR | +1.20 | +0.72 | +0.96 | 0.055 | +1.10 | 0.023 |
| PTGR1 | +0.57 | +0.55 | +0.56 | 0.13 | ||
| MT1E | +0.89 | +0.61 | +0.75 | 0.16 | ||
| MT2A | +0.69 | +0.73 | +0.71 | 0.19 | ||
| | ||||||
|
Genes decreased in IPAH PAECs in both experiments | ||||||
| COL4A1 | −0.62 | −3.47 | −1.37 | 0.016 | −1.65 | 0.010 |
| COL4A2 | −0.62 | −2.23 | −1.16 | 0.016 | −1.39 | 0.004 |
| ISG20 | −2.40 | −0.59 | −1.17 | 0.016 | −2.19 | 0.016 |
| EFNA1 | −0.91 | −0.83 | −0.87 | 0.023 | −1.06 | 0.061 |
| GPRC5B | −0.68 | −0.65 | −0.66 | 0.026 | −0.88 | 0.025 |
| GADD45B | −0.72 | −0.90 | −0.80 | 0.050 | −1.01 | 0.011 |
| PTPRE | −0.64 | −0.62 | −0.63 | 0.056 | −1.00 | 0.061 |
| IQSEC1 | −0.65 | −0.89 | −0.76 | 0.091 | −0.59 | 0.189 |
| EPHX1 | −0.53 | −0.63 | −0.58 | 0.12 | ||
| SOCS3 | −0.52 | −0.86 | −0.67 | 0.12 | ||
| JUNB | −2.06 | −0.65 | −1.14 | 0.14 | ||
| TIMP1 | −1.17 | −1.03 | −1.10 | 0.16 | ||
| VWF | −0.51 | −0.57 | −0.54 | 0.17 | ||
| CCL2 | −0.57 | −0.57 | −0.57 | 0.19 | ||
| LOX | −0.78 | −1.54 | −1.09 | 0.20 | ||
| TGFBI | −0.87 | −0.86 | −0.87 | 0.21 | ||
Definition of abbreviations: IPAH = idiopathic pulmonary arterial hypertension; PAEC = pulmonary arterial endothelial cell; PCR = polymerase chain reaction.
Genes with the most consistent differences in combined data (P < 0.1) were selected for further study by quantitative PCR analysis. Fold differences in quantitative PCR are normalized to β-actin expression.
qPCR analysis of these 12 genes confirmed similar fold changes detected by RNAseq analysis (Spearman ρ = 0.97; P < 0.0001) (Figure 1B). In 10 genes we established a significant difference (P < 0.05) between IPAH and control PAECs, either by RNAseq and/or by qPCR. Correlations for the two alpha chains of collagen IV (COL4A1 and A2) between RNAseq and qPCR further illustrate the close agreement using RNAseq and qPCR to measure gene expression (Figure 1C). Immunoblot analysis confirmed that COL4 protein levels were decreased in cell lysates obtained from IPAH versus control PAECs (P < 0.0001) (Figure 1D). Both collagen chains were also down-regulated along with BMPR2 expression in a separate cohort of six patients and five control subjects (see Figure E1). Levels of EFNA1 also tended to be lower in these patients but did not reach statistical significance (P = 0.059) (see Figure E2).
Reduced BMP Signaling and Gene Expression Changes in IPAH PAECs
Dysfunction in signaling of BMPR2 underlies most heritable cases of PAH and also has been demonstrated in patients with IPAH lacking a mutation (8). To investigate the relevance of BMPR2 dysfunction to the gene expression changes detected in our RNAseq analyses, we reduced levels of BMPR2 with targeted siRNA in commercially available control large PAECs (see Figure E2). We reproduced the same significant changes in expression in 6 of 10 genes seen in IPAH versus control PAECs by qPCR or RNAseq (Figure 2A). That is, with BMPR2 siRNA versus control siRNA, we recapitulated changes in three of four genes that were significantly up-regulated and three of six that were down-regulated in IPAH versus control PAECs. EFNA1 can regulate the release of COL4 from ECs (21) and therefore was selected along with COL4 for further functional analysis.
Figure 2.
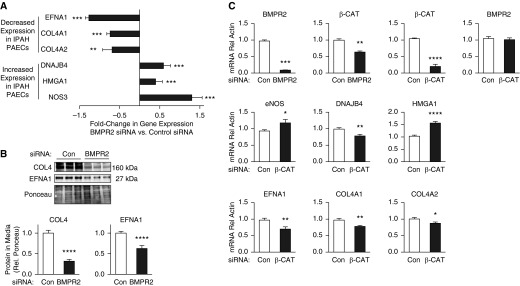
Reduced bone morphogenetic protein (BMP) signaling recapitulates many of the changes in transcripts identified by RNAseq analysis in idiopathic pulmonary arterial hypertension (IPAH) versus control pulmonary arterial endothelial cells (PAECs). (A) Fold change in gene expression of siBMPR2-transfected PAECs versus scrambled small interfering RNA (siRNA)-transfected cells calculated as in Figure 1B. Bars represent mean ± SEM of n = 3, **P < 0.01 and ***P < 0.001 by t test. (B) Immunoblot analysis of collagen IV (COL4) and ephrinA1 (EFNA1) protein in media from PAECs transfected with scrambled (Con) or BMPR2 targeting siRNA. Ponceau indicates loading. Bars represent mean ± SEM. ****P < 0.0001 by t test versus control siRNA. (C) Quantitative polymerase chain reaction of transcripts altered by BMPR2 siRNA or β-catenin (β-CAT) siRNA versus nontargeting control siRNA. Bars represent mean ± SEM of n = 3 different PAEC harvests. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way analysis of variance and post hoc Bonferroni tests. BMPR2 = bone morphogenetic protein type 2 receptor; eNOS = endothelial nitric oxide synthase; HMGA1 = high-mobility group AT-hook 1; mRNA = messenger RNA.
Protein levels of both COL4A1/A2 and EFNA1 were reduced in the media of PAECs transfected with BMPR2 targeting siRNA (both P < 0.0001) (Figure 2B). In contrast to down-regulation of the receptor, BMP9 stimulation of PAECs up-regulated COL4A1 and COL4A2, in association with increasing mRNA expression levels of BMPR2, ALK3, and ALK6. However, BMP9 stimulation actually reduced mRNA expression of EFNA1 and ALK1 in a manner similar to loss of BMPR2 (see Figure E3).
To address the mechanism leading to the reduction in gene and protein expression of COL4A1/A2 and EFNA1 observed with loss of BMPR2, we monitored levels of microRNAs that have been implicated in PAH, BMPR2 function, or regulation of COL4A1/A2; miRs-145 (22), -130a (23), -21 (24), -29a (25), -137 (26), and -9 (27) in PAECs from control subjects and patients, but no differences were found that could explain the changes in transcripts identified by loss of BMPR2 (data not shown). We also transfected control PAECs with dominant negative constructs of downstream effectors of BMPR2 signaling, SMAD1 or SMAD3 (3), or siRNA against ID1 (a downstream effector of pSMAD1/5) (28) but suppressing this axis could not reproduce any of the changes in the transcripts differentially expressed by loss of BMPR2 (see Figure E4).
Having shown in PAEC that BMPR2 activation induces a transcriptionally active complex between PPARγ and β-catenin and that these factors independently regulate genes downstream of BMPR2 signaling (4), we assessed gene expression in PAECs following knockdown of PPARγ or β-catenin. COL4A2 was down-regulated by both PPARγ and β-catenin siRNAs as it was by BMPR2 siRNA. However, all of the other differentially expressed genes were altered only by β-catenin siRNA in a manner similar to loss of BMPR2 (Figure 2C). Loss of BMPR2 and loss of β-catenin similarly down-regulated DNAJB4 (Figure 2), in contrast to the up-regulation of DNAJB4 observed in IPAH PAECs.
Functional Effects of Loss of COL4 and EFNA1 Expression on PAECs
COL4A1/2 and EFNA1 increase adhesion and migration of systemic ECs (21), therefore we evaluated these properties in PAECs using siRNA. In PAECs, COL4A1/2 and EFNA1 siRNA reduced levels of the proteins more than 50% (see Figure E5) and this resulted in a similar inhibition of both adhesion and migration of PAECs (all P < 0.001) (Figures 3A and 3B). Combined knockdown of EFNA1 and COL4A1/A2 did not result in a further decrease in adhesion or migration when compared with reducing levels of the individual transcripts, suggesting that these proteins may interact through the same mechanism. Coating wells with collagen IV significantly improved the adhesion of PAECs in cells transfected with siRNA targeting EFNA1 or COL4 to levels observed in control cells on gelatin (Figure 3A). However, there was still reduced adhesion when compared with PAECs transfected with scrambled siRNA on collagen-coated wells. This suggested that endogenous production of COL4 is necessary for restoration of the normal phenotype.
Figure 3.

The functional consequences of loss of collagen IV (COL4) and ephrinA1 (EFNA1) expression in pulmonary arterial endothelial cells (PAECs). Representative fields are shown above and quantitative data below. (A) PAECs transfected with EFNA1 and/or COL4 small interfering RNA (siRNA), normalized to scrambled siRNA-transfected cells (Con) were adherent to gelatin or collagen-IV-coated wells after 40 minutes, assessed by semiautomated counting of 4′6-diamidino-2-phenylindole–positive nuclei. (B) PAECs transfected with EFNA1 and/or COL4 siRNA versus control siRNA and migration assessed at 8 hours by modified Boyden Chamber assay. (C) Matrigel assays in cells transfected with EFNA1, COL4 siRNA, or both versus scrambled (Con) siRNA, over 19 hours, in response to 0.2% FBS + 25 ng/ml vascular endothelial growth factor. Scale bars = 100 μm. Bars represent mean ± SEM of n = 3. ***P < 0.001, **P < 0.01, and *P < 0.05 versus Con siRNA, ##P < 0.01 and ###P < 0.001 versus gelatin coated condition by one-way analysis of variance or two-way analysis of variance when comparing collagen with gelatin and siRNA condition, and post hoc Bonferroni test. FBS = fetal bovine serum.
Tube formation on reduced growth factor Matrigel, induced by 25 ng/ml VEGF in 0.2% fetal bovine serum, was significantly decreased by loss of either EFNA1 or COL4A1/2 or both (all P < 0.001) (Figure 3C). Coating wells or inserts with collagen IV also significantly improved adhesion (P < 0.05) and migration (P < 0.001) of PAECs from patients with IPAH (see Figure E6).
EphA2 Knockout Mice versus Control Animals and Severity of Pulmonary Hypertension
Because EFNA1 and COL4 seem to act through the same pathway in promoting adhesion or migration of PAECs, we investigated whether reducing EFNA1 signaling in mice lacking the EFNA1 receptor (EphA2−/−) would increase endothelial dysfunction and augment pulmonary hypertension during hypoxia by promoting a greater loss of small arteries. There were no differences at baseline in normoxia when comparing pulmonary hemodynamic data in EphA2−/− mice versus control animals, and no differences in the response to a 3-week period of hypoxia. However, the addition of the VEGF receptor blocker Sugen 5416 by subcutaneous injection followed by a 4-week period of hypoxia (10% oxygen) elicited more severe pulmonary hypertension in EphA2−/− when compared with control mice. EphA2−/− mice demonstrated greater elevation in RV systolic pressure (P < 0.001) and RV hypertrophy judged by the weight of the RV/LV and septum (LV+S) (P < 0.01) (Figures 4A and 4B). Cardiac output and muscularization of distal vessels was similar (Figures 4C and 4D) but morphometric analyses revealed a greater loss of small arteries relative to alveoli in EphA2−/− mice when compared with wild-type control animals (P < 0.0001) (Figure 4E). The number of alveoli per square millimeter was similar in the EphA2−/− and wild-type mice (Figure 4F).
Figure 4.

Mice with deletion of EphA2 develop exaggerated pulmonary hypertension (PH). EphA2−/− (KO) and wild-type (WT) mice were exposed to room air (normoxia), 10% O2 (hypoxia), or subcutaneous injection of the vascular endothelial growth factor receptor blocker Sugen 5416 (Su) in addition to hypoxia for 4 weeks and development of PH assessed. n = 5 (3 males, 2 females) for each group. Bars indicate mean ± SEM. (A) Right ventricular systolic pressure (RVSP). (B) Right ventricular hypertrophy (RVH) given by the Fulton index (weight of right ventricle [RV]/left ventricle [LV] and septum [S]). **P < 0.01, ***P < 0.001 versus WT by two-way analysis of variance with Bonferroni post test. (C) Representative histology of distal pulmonary arteries (DPA) from mice exposed to hypoxia following Sugen injection; scale bars = 50 μm. The photomicrographs show similar muscularization of DPAs in the two genotypes but a greater reduction in the number of DPAs in the EphA2−/− versus WT. Left images show high magnification of α-smooth muscle actin–immunostained vessels, and right images lower-magnification hematoxylin and eosin–stained tissue sections. (D) Muscularization of distal pulmonary arteries. FM = fully muscularized; NM = nonmuscularized; PM = partially muscularized. (E) Number of vessels (DPA) per 100 alveoli. (F) Alveoli per square millimeter. Bars represent mean ± SEM, ****P < 0.0001 versus normoxia, ####P < 0.0001 versus WT, by two-way analysis of variance and post hoc Bonferroni test.
Discussion
Endothelial dysfunction is a key feature in the pathogenesis of PAH. In this study, we identified disease-related differences in gene expression not previously reported, including reduced COL4 and EFNA1. The latter have important functional consequences on angiogenesis and on the PA response to injury that relate directly to the development of PAH. We focused on COL4 and EFNA1 in particular, because both genes were down-regulated in the RNAseq analysis, and both have important, intertwining roles in endothelial biology; COL4 is a major component of vascular basement membranes and EFNA1 is an EC guidance molecule (29) that induces the release of COL4 from these cells (21). Loss of BMPR2 signaling also impaired the ability of PAECs to produce COL4 and EFNA1 by a mechanism that was reproduced by reducing β-catenin, a previously reported downstream effector of BMPR2 gene regulation (4). Repressing the expression of these proteins had negative effects on PAEC adhesion and migration, with a loss of the receptor for EFNA1 in transgenic mice worsening the severity of pulmonary hypertension in association with a greater loss of distal arteries.
In addition to the reduction in EFNA1 and COL4 expression, RNAseq analyses identified IQSEC1 and ISG20 as transcripts decreased in patients with IPAH versus control subjects. IQSEC1 (IQ motif and Sec7 domain 1) is a guanidine nucleotide exchange protein recruited by EGFR/VEGFR2. It stimulates angiogenesis and is important in cell adhesion by controlling the turnover of β-integrins (30). Dominant-negative ISG20 (interferon-stimulated exonuclease gene 20 kD) inhibits angiogenic function in human umbilical vein ECs by a less well-defined mechanism (31). Other transcripts that were reduced included GADD45B, the DNA repair protein (reviewed in Reference 32); GPCR5B, best studied in differentiation of macrophages (33); and PTPRE, a phosphatase that can up-regulate EGF receptor signaling (34). Transcripts that were increased in IPAH versus control PAECs by RNAseq include high-mobility group AT-hook 1 (HMGA1) and endothelial nitric oxide synthase (NOS3). HMGA1 has been extensively studied as a transforming factor linking viral infection and other perturbations to cancer (reviewed in Reference 35) and HMGA1 is also highly proinflammatory in ECs in response to viral infection (36). The increase in NOS3 expression may reflect uncoupling of NO and peroxynitrite production seen with elevated thromboxane (37) and with inflammation (38). Up-regulation of the NOS3 transcript may represent an attempt to make up for reduced levels of the NOS3 protein in PAH as has been recently shown (39). DNAJB4 and PIR were also increased in IPAH versus control PAECs. DNAJB4 is a chaperone protein induced by DNA damage that stabilizes E-cadherin (40). PIR is an iron-binding protein driven by the antioxidant transcription factor NRF2 that could modulate the proinflammatory effects of nuclear factor-κB (41). Interestingly, none of these transcripts matched proteins identified in blood outgrowth ECs previously studied in patients with heritable PAH, perhaps related to differences in cell type and patient cohort (15).
RNAseq was chosen as a methodology in this analysis because it is highly sensitive and enables precise measurement of thousands of distinct RNAs. For example, in our study around 6,000 RNAs met the strict inclusion criteria of detection above five RPKM, although it is likely that thresholds lower than this will produce useful data in the future as total number of reads per lane increases. The cell lines analyzed are obtained from patients undergoing transplantation and therefore represent the most severe cases of end-stage disease. Because in our experience it is technically difficult to separate PAECs from neointimal cells in PAH arteries for laser capture microscopy, prospective studies using single-cell transcriptomic analysis may hold promise in distinguishing the subset of PAECs that express low COL4A1/2 and EFNA1 (42). It would be of great interest to see if these changes were replicated in IPAH ECs at disease initiation, or whether they represented changes observed in endothelial progenitor cells or in ECs derived from induced pluripotent stem cells.
The three transcripts significantly increased in response to BMPR2 siRNA were HMGA1, NOS3, and DNAJB4. Those down-regulated in response to reduction of BMPR2 by siRNA included COL4A1, COL4A2, and EFNA1. Interestingly, dasatinib, a SRC inhibitor recently linked to the development of PAH (43), reduces the phosphorylation state of the receptor EphA2 (44); it is possible that through this mechanism dasatinib mimics loss of EFNA1 and its ability to regulate COL4 production, thus disturbing the function of PAECs and leading to the development of pulmonary vascular disease (43). It is intriguing that BMP9 can up-regulate COL4 in association with an increase in expression of BMPR2, and its coreceptors ALK3 and ALK6. BMP9 causes a reduction in EFNA1 in association with a reduction in ALK1, indicating that the loss of the BMPR2 coreceptor ALK1 may be as important as the loss of BMPR2 in regulating EFNA1 expression. This pathway may therefore be especially relevant in patients who develop PAH related to ALK1 mutations, with or without associated hereditary hemorrhagic telangiectasia (45).
Concomitant knockdown of both EFNA1 and COL4 had no additional inhibitory effect on the ability of PAECs to adhere or migrate, suggesting that they function by activating the same downstream pathway. For example, COL4 is required to activate focal adhesion kinase (FAK) in ECs; EphA2, the preferred receptor for EFNA1, interacts directly with FAK (46). We have observed that knockdown of either EFNA1 or COL4 leads to increased expression of both activated and total FAK (data not shown). Because EFNA1 can down-regulate its receptor and FAK, it is possible that loss of the ligand up-regulates the receptor and FAK, in concert with p130cas (47), but the absence of EFNA1 or COL4 precludes an effective signal.
Mice lacking expression of COL4 die with vascular abnormalities at E10.5–11.5 (48). Mice lacking expression of EFNA1 develop abnormal heart valves (49) but their vasculature has not been studied. Mice lacking the receptor crucial for the response of PAECs to EFNA1, EphA2, develop normally with no overt phenotype, but are protected from tumor development because of impaired angiogenesis (50). This emphasizes the importance of both COL4 and EFNA1 in vascular development and homeostasis in the intact animal. We were able to elicit a greater loss of distal PAs associated with more severe pulmonary hypertension in mice with loss of EphA2 following VEGF receptor blockade and chronic hypoxia.
In conclusion, we have applied RNAseq for the first time to cohorts of PAECs derived from patients with IPAH and control unused donor lungs, and have identified a pathway related to reduced BMPR2 in patients without mutations that indicates additional ways in which the endothelium becomes dysfunctional. The new data suggest that optimizing BMPR2 receptor function (5, 20) may allow for normalization of a wide range of targets.
Acknowledgments
Acknowledgment
Lung tissues from patients with idiopathic pulmonary arterial hypertension and control subjects were provided by the Pulmonary Hypertension Breakthrough Initiative, which is funded by the Cardiovascular Medical Research and Education Fund. The tissues were procured at the Transplant Procurement Centers at Allegheny Hospital (Pittsburgh), Baylor University, The Cleveland Clinic, Stanford University, University of California–San Diego, Vanderbilt University, and the University of Alabama at Birmingham, and deidentified patient data were obtained via the Data Coordinating Center at the University of Michigan. The authors thank Dr. Michal Bental Roof for assistance with preparation of the manuscript.
Footnotes
Supported by National Institutes of Health/NHLBI grants U01 HL107393 (M.R., M.P.S., and J.C.W.) and R24 HL123767; Cardiovascular Medical Research and Education Fund Grant UL1RR024986 (M.R.); an endowed Dwight and Vera Dunlevie Chair in Pediatric Cardiology (M.R.); Deutsche Forschungsgemeinschaft grants Ni 1456/1-1 (N.P.N.) and He 6855/1-1 (J.K.H.); Japan Heart Foundation/Bayer Yakuhin Research Grant Abroad (K.M.); the Pediatric Heart Center Research Program, Lucile Packard Children's Hospital, Lucile Packard Foundation for Children's Health, and Stanford CTSA UL1 RR025744 (R.K.H.); an AHA postdoctoral fellowship (M.G.); and a Summer Undergraduate Research Fellowship from the California Institute of Technology (Z.X.). E.S. is currently supported by NIH/NHLBI grant K08 HL107450.
Author Contributions: Conception and design of work, C.J.R., H.I., J.C.W., M.P.S., and M.R. Data acquisition and analysis, C.J.R., H.I., A.C., J.K.H., L.W., N.P.N., R.K.H., S.S., N.F.T., C.G.L., E.S., Z.X., R.C., M.Z., D.B., M.K., K.M., P.-I.C., M.G., P.A.d.R., and R.T.Z. Interpretation of data, C.J.R., H.I., R.C., A.C., and M.R. Drafting the manuscript, C.J.R. and M.R. All authors critically revised and gave their final approval of the manuscript version to be published and agree to be accountable for all aspects of the work.
Originally Published in Press as DOI: 10.1164/rccm.201408-1528OC on June 1, 2015
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest. 2012;122:4306–4313. doi: 10.1172/JCI60658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J, Stewart DJ. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98:209–217. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 3.de Jesus Perez VA, Alastalo TP, Wu JC, Axelrod JD, Cooke JP, Amieva M, Rabinovitch M. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J Cell Biol. 2009;184:83–99. doi: 10.1083/jcb.200806049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alastalo TP, Li M, Perez VdeJ, Pham D, Sawada H, Wang JK, Koskenvuo M, Wang L, Freeman BA, Chang HY, et al. Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest. 2011;121:3735–3746. doi: 10.1172/JCI43382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, Li CG, El-Bizri N, Sawada H, Haghighat R, Chan R, et al. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest. 2013;123:3600–3613. doi: 10.1172/JCI65592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA. 2007;104:1342–1347. doi: 10.1073/pnas.0605080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, Thistlethwaite PA, Tuder RM, Erzurum SC, Geraci MW, et al. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:1153–1160. doi: 10.1164/rccm.201003-0491OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, Morrell NW. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105:1672–1678. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 9.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, III, Loyd JE, Nichols WC, Trembath RC International PPH Consortium. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26:81–84. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 10.Deng Z, Haghighi F, Helleby L, Vanterpool K, Horn EM, Barst RJ, Hodge SE, Morse JH, Knowles JA. Fine mapping of PPH1, a gene for familial primary pulmonary hypertension, to a 3-cM region on chromosome 2q33. Am J Respir Crit Care Med. 2000;161:1055–1059. doi: 10.1164/ajrccm.161.3.9906051. [DOI] [PubMed] [Google Scholar]
- 11.Burton VJ, Ciuclan LI, Holmes AM, Rodman DM, Walker C, Budd DC. Bone morphogenetic protein receptor II regulates pulmonary artery endothelial cell barrier function. Blood. 2011;117:333–341. doi: 10.1182/blood-2010-05-285973. [DOI] [PubMed] [Google Scholar]
- 12.Cheadle C, Berger AE, Mathai SC, Grigoryev DN, Watkins TN, Sugawara Y, Barkataki S, Fan J, Boorgula M, Hummers L, et al. Erythroid-specific transcriptional changes in PBMCs from pulmonary hypertension patients. PLoS ONE. 2012;7:e34951. doi: 10.1371/journal.pone.0034951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geraci MW, Gao B, Hoshikawa Y, Yeager ME, Tuder RM, Voelkel NF. Genomic approaches to research in pulmonary hypertension. Respir Res. 2001;2:210–215. doi: 10.1186/rr59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63:783–794. doi: 10.1002/art.30159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavoie JR, Ormiston ML, Perez-Iratxeta C, Courtman DW, Jiang B, Ferrer E, Caruso P, Southwood M, Foster WS, Morrell NW, et al. Proteomic analysis implicates translationally controlled tumor protein as a novel mediator of occlusive vascular remodeling in pulmonary arterial hypertension. Circulation. 2014;129:2125–2135. doi: 10.1161/CIRCULATIONAHA.114.008777. [DOI] [PubMed] [Google Scholar]
- 16.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, Tada Y, Tanabe N, Tatsumi K, Hemnes AR, West JD. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ. 2012;2:201–213. doi: 10.4103/2045-8932.97606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Upton PD, Davies RJ, Trembath RC, Morrell NW. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J Biol Chem. 2009;284:15794–15804. doi: 10.1074/jbc.M109.002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Jesus Perez VA, Yuan K, Orcholski ME, Sawada H, Zhao M, Li CG, Tojais NF, Nickel N, Rajagopalan V, Spiekerkoetter E, et al. Loss of adenomatous poliposis coli-α3 integrin interaction promotes endothelial apoptosis in mice and humans. Circ Res. 2012;111:1551–1564. doi: 10.1161/CIRCRESAHA.112.267849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nickel NP, Spiekerkoetter E, Gu M, Li CG, Li H, Kaschwich M, Diebold I, Hennigs JK, Kim KY, Miyagawa K, et al. Elafin reverses pulmonary hypertension via caveolin-1–dependent bone morphogenetic protein signaling. Am J Respir Crit Care Med. 2015;191:1273–1286. doi: 10.1164/rccm.201412-2291OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saik JE, Gould DJ, Keswani AH, Dickinson ME, West JL. Biomimetic hydrogels with immobilized ephrinA1 for therapeutic angiogenesis. Biomacromolecules. 2011;12:2715–2722. doi: 10.1021/bm200492h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A. down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem. 2011;286:28097–28110. doi: 10.1074/jbc.M111.236950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bertero T, Cottrill K, Krauszman A, Lu Y, Annis S, Hale A, Bhat B, Waxman AB, Chau BN, Kuebler WM, et al. The microRNA-130/301 family controls vasoconstriction in pulmonary hypertension. J Biol Chem. 2015;290:2069–2085. doi: 10.1074/jbc.M114.617845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parikh VN, Jin RC, Rabello S, Gulbahce N, White K, Hale A, Cottrill KA, Shaik RS, Waxman AB, Zhang YY, et al. MicroRNA-21 integrates pathogenic signaling to control pulmonary hypertension: results of a network bioinformatics approach. Circulation. 2012;125:1520–1532. doi: 10.1161/CIRCULATIONAHA.111.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du B, Ma LM, Huang MB, Zhou H, Huang HL, Shao P, Chen YQ, Qu LH. High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 2010;584:811–816. doi: 10.1016/j.febslet.2009.12.053. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, Heng X, Yu J, Su Q, Guan X, You C, Wang L, Che F. miR-137 regulates the migration of human umbilical vein endothelial cells by targeting ephrin-type A receptor 7. Mol Med Rep. 2014;10:1475–1480. doi: 10.3892/mmr.2014.2319. [DOI] [PubMed] [Google Scholar]
- 27.Shan F, Li J, Huang QY. HIF-1 alpha-induced up-regulation of miR-9 contributes to phenotypic modulation in pulmonary artery smooth muscle cells during hypoxia. J Cell Physiol. 2014;229:1511–1520. doi: 10.1002/jcp.24593. [DOI] [PubMed] [Google Scholar]
- 28.Frank DB, Abtahi A, Yamaguchi DJ, Manning S, Shyr Y, Pozzi A, Baldwin HS, Johnson JE, de Caestecker MP. Bone morphogenetic protein 4 promotes pulmonary vascular remodeling in hypoxic pulmonary hypertension. Circ Res. 2005;97:496–504. doi: 10.1161/01.RES.0000181152.65534.07. [DOI] [PubMed] [Google Scholar]
- 29.van Gils JM, Ramkhelawon B, Fernandes L, Stewart MC, Guo L, Seibert T, Menezes GB, Cara DC, Chow C, Kinane TB, et al. Endothelial expression of guidance cues in vessel wall homeostasis dysregulation under proatherosclerotic conditions. Arterioscler Thromb Vasc Biol. 2013;33:911–919. doi: 10.1161/ATVBAHA.112.301155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morishige M, Hashimoto S, Ogawa E, Toda Y, Kotani H, Hirose M, Wei S, Hashimoto A, Yamada A, Yano H, et al. GEP100 links epidermal growth factor receptor signalling to Arf6 activation to induce breast cancer invasion. Nat Cell Biol. 2008;10:85–92. doi: 10.1038/ncb1672. [DOI] [PubMed] [Google Scholar]
- 31.Taylor KL, Leaman DW, Grane R, Mechti N, Borden EC, Lindner DJ. Identification of interferon-beta-stimulated genes that inhibit angiogenesis in vitro. J Interferon Cytokine Res. 2008;28:733–740. doi: 10.1089/jir.2008.0030. [DOI] [PubMed] [Google Scholar]
- 32.Schäfer A. Gadd45 proteins: key players of repair-mediated DNA demethylation. Adv Exp Med Biol. 2013;793:35–50. doi: 10.1007/978-1-4614-8289-5_3. [DOI] [PubMed] [Google Scholar]
- 33.Hohenhaus DM, Schaale K, Le Cao KA, Seow V, Iyer A, Fairlie DP, Sweet MJ. An mRNA atlas of G protein-coupled receptor expression during primary human monocyte/macrophage differentiation and lipopolysaccharide-mediated activation identifies targetable candidate regulators of inflammation. Immunobiology. 2013;218:1345–1353. doi: 10.1016/j.imbio.2013.07.001. [DOI] [PubMed] [Google Scholar]
- 34.Nunes-Xavier CE, Elson A, Pulido R. Epidermal growth factor receptor (EGFR)-mediated positive feedback of protein-tyrosine phosphatase epsilon (PTPepsilon) on ERK1/2 and AKT protein pathways is required for survival of human breast cancer cells. J Biol Chem. 2012;287:3433–3444. doi: 10.1074/jbc.M111.293928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huso TH, Resar LM. The high mobility group A1 molecular switch: turning on cancer - can we turn it off? Expert Opin Ther Targets. 2014;18:541–553. doi: 10.1517/14728222.2014.900045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viemann D, Schmolke M, Lueken A, Boergeling Y, Friesenhagen J, Wittkowski H, Ludwig S, Roth J. H5N1 virus activates signaling pathways in human endothelial cells resulting in a specific imbalanced inflammatory response. J Immunol. 2011;186:164–173. doi: 10.4049/jimmunol.0904170. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Song P, Xu J, Zou MH. Activation of NAD(P)H oxidases by thromboxane A2 receptor uncouples endothelial nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2011;31:125–132. doi: 10.1161/ATVBAHA.110.207712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hein TW, Singh U, Vasquez-Vivar J, Devaraj S, Kuo L, Jialal I. Human C-reactive protein induces endothelial dysfunction and uncoupling of eNOS in vivo. Atherosclerosis. 2009;206:61–68. doi: 10.1016/j.atherosclerosis.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bakhshi FR, Mao M, Shajahan AN, Piegeler T, Chen Z, Chernaya O, Sharma T, Elliott WM, Szulcek R, Bogaard HJ, et al. Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ. 2013;3:816–830. doi: 10.1086/674753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simões-Correia J, Silva DI, Melo S, Figueiredo J, Caldeira J, Pinto MT, Girão H, Pereira P, Seruca R. DNAJB4 molecular chaperone distinguishes WT from mutant E-cadherin, determining their fate in vitro and in vivo. Hum Mol Genet. 2014;23:2094–2105. doi: 10.1093/hmg/ddt602. [DOI] [PubMed] [Google Scholar]
- 41.Brzóska K, Stępkowski TM, Kruszewski M. Basal PIR expression in HeLa cells is driven by NRF2 via evolutionary conserved antioxidant response element. Mol Cell Biochem. 2014;389:99–111. doi: 10.1007/s11010-013-1931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawada H, Saito T, Nickel NP, Alastalo TP, Glotzbach JP, Chan R, Haghighat L, Fuchs G, Januszyk M, Cao A, et al. Reduced BMPR2 expression induces GM-CSF translation and macrophage recruitment in humans and mice to exacerbate pulmonary hypertension. J Exp Med. 2014;211:263–280. doi: 10.1084/jem.20111741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, Bouvaist H, Canuet M, Pison C, Macro M, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125:2128–2137. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
- 44.Caccia D, Miccichè F, Cassinelli G, Mondellini P, Casalini P, Bongarzone I. Dasatinib reduces FAK phosphorylation increasing the effects of RPI-1 inhibition in a RET/PTC1-expressing cell line. Mol Cancer. 2010;9:278. doi: 10.1186/1476-4598-9-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujiwara M, Yagi H, Matsuoka R, Akimoto K, Furutani M, Imamura S, Uehara R, Nakayama T, Takao A, Nakazawa M, et al. Implications of mutations of activin receptor-like kinase 1 gene (ALK1) in addition to bone morphogenetic protein receptor II gene (BMPR2) in children with pulmonary arterial hypertension. Circ J. 2008;72:127–133. doi: 10.1253/circj.72.127. [DOI] [PubMed] [Google Scholar]
- 46.Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69. doi: 10.1038/35000008. [DOI] [PubMed] [Google Scholar]
- 47.Carter N, Nakamoto T, Hirai H, Hunter T. EphrinA1-induced cytoskeletal re-organization requires FAK and p130(cas) Nat Cell Biol. 2002;4:565–573. doi: 10.1038/ncb823. [DOI] [PubMed] [Google Scholar]
- 48.Pöschl E, Schlötzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131:1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 49.Frieden LA, Townsend TA, Vaught DB, Delaughter DM, Hwang Y, Barnett JV, Chen J. Regulation of heart valve morphogenesis by Eph receptor ligand, ephrin-A1. Dev Dyn. 2010;239:3226–3234. doi: 10.1002/dvdy.22458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brantley-Sieders DM, Fang WB, Hicks DJ, Zhuang G, Shyr Y, Chen J. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J. 2005;19:1884–1886. doi: 10.1096/fj.05-4038fje. [DOI] [PubMed] [Google Scholar]