

Abstract
Herbaceous peony (Paeonia lactiflora Pall.), one of the world’s most important ornamental plants, is highly susceptible to Botrytis cinerea, and improving resistance to this pathogenic fungus is a problem yet to be solved. MicroRNAs (miRNAs) play an essential role in resistance to B. cinerea, but until now, no studies have been reported concerning miRNAs induction in P. lactiflora. Here, we constructed and sequenced two small RNA (sRNA) libraries from two B. cinerea-infected P. lactiflora cultivars (“Zifengyu” and “Dafugui”) with significantly different levels of resistance to B. cinerea, using the Illumina HiSeq 2000 platform. From the raw reads generated, 4,592,881 and 5,809,796 sRNAs were obtained, and 280 and 306 miRNAs were identified from “Zifengyu” and “Dafugui”, respectively. A total of 237 conserved and 7 novel sequences of miRNAs were differentially expressed between the two cultivars, and we predicted and annotated their potential target genes. Subsequently, 7 differentially expressed candidate miRNAs were screened according to their target genes annotated in KEGG pathways, and the expression patterns of miRNAs and corresponding target genes were elucidated. We found that miR5254, miR165a-3p, miR3897-3p and miR6450a might be involved in the P. lactiflora response to B. cinerea infection. These results provide insight into the molecular mechanisms responsible for resistance to B. cinerea in P. lactiflora.
Keywords: herbaceous peony, Botrytis cinerea, miRNAs, target genes
1. Introduction
Herbaceous peony (Paeonia lactiflora Pall.) is a perennial root flower belonging to the Paeoniaceae family. It is popular worldwide due to its high ornamental values in locations including New Zealand, Australia, Europe, Asia, and North America [1]. In China, P. lactiflora has been widely applied in urban and rural landscaping due to its high levels of resistance to infection, tolerance to environmental factors, and easy maintenance, making it ideal for a variety of specialized gardens, flower beds, and perennial borders [2]. However in practice, P. lactiflora cultivated in a landscape greenbelt in the middle and lower reaches of the Chinese Yangtze River region was highly susceptible to Botrytis cinerea after flowering. This infection mainly damaged the P. lactiflora leaf and resulted in rot and led to the death of the entire plant, which was consistent with the previous findings in Nanjing, Shanghai and Hangzhou [3]. Lan [3] reported that the optimum temperature for B. cinerea growth and sporulation was 20 °C–24 °C and the optimum pH was 4–5, and it could be controlled by many chemical treatments. However, chemical spraying is expensive, requires manual labor, pollutes the environment, and leaves white stains on the leaves, ultimately reducing the ornamental value of P. lactiflora while not necessarily preventing B. cinerea infection. Conversely, breeding resistant cultivars is the best method to solve this problem, and the rapid development of genetic engineering technology in recent years has laid the foundation for effective development of this field. Recent studies have provided compelling evidence demonstrating that microRNAs (miRNAs) are essential endogenous regulatory factors and play an important role in plant response to biotic stress [4,5]. Analysis of miRNAs thus provides a novel method to study the molecular mechanism of resistance in P. lactiflora B. cinerea.
miRNAs are approximately 21-nucleotides (nt) in length and are non-coding small RNAs (sRNAs) found in animals and plants, typically encoded by endogenous genes. miRNAs could play an important regulatory role at the post-transcriptional level by targeting mRNA degradation and leading to translation repression [6]. Since they were first found in Caenorhabditis elegans [7], a large number of miRNAs have been continually identified in higher plants, including Arabidopsis thaliana [5], Zea mays [8], Triticum aestivum [9] and Oryza sativa [10]. The latest version of the miRNA database (miRBase 21.0, http://www.mirbase.org/) contains 28,645 entries of hairpin precursor miRNAs (pre-miRNAs), expressing 35,828 mature miRNA products over a range of 223 species [11]. Moreover, numerous studies have revealed that miRNAs are involved in diverse biological and metabolic processes, such as the regulation of plant organ development [12,13], signal conduction of plant hormones [14,15], abiotic stress response [16,17], and pathogen defense [18]. To the best of our knowledge concerning B. cinerea, only Jin et al. [19,20] have reported that tomato miRNAs had a functional role in resistance to B. cinerea. However, there no P. lactiflora miRNAs have yet been deposited in miRBase and no related reports on this topic have been completed. In order to investigate the roles of P. lactiflora miRNAs in response to B. cinerea stress, two cultivars, “Zifengyu” and “Dafugui,” with significantly different levels of resistance to this pathogen were processed for sRNA and transcriptome sequencing. In this study, miRNAs and their target genes responsive to B. cinerea were identified in P. lactiflora, and these results provide a foundation for understanding the functions and regulatory mechanisms of miRNAs in P. lactiflora resistance to B. cinerea.
2. Experimental Section
2.1. Plant Materials
P. lactiflora was grown in the germplasm repository of Horticulture and Plant Protection College, Yangzhou University, Jiangsu Province, China (32°30' N, 119°25' E). Under field conditions, two cultivars, “Zifengyu” and “Dafugui”, with significantly different levels of resistance to B. cinerea (Figure 1, in the later stage infected by B. cinerea, the resistant cultivar “Zifengyu” grew well with few disease spots, while the susceptible cultivar “Dafugui” became weak and almost entirely withered) were used as the experimental materials for sRNA and transcriptome sequencing. After flowering, P. lactiflora was naturally infected by B. cinerea, and the infected and fully expanded leaves from the fourth apical node in four developmental stages (S1, late May; S2, mid June; S3, early July; and S4, late July) were taken from May to July 2013. The sample population of each cultivar were sixty plants (four stages, fifteen plants each stage). The leaves collected from four stages each cultivar were then equally mixed to prepare for two independent sRNA libraries (i.e., “Zifengyu” and “Dafugui”). All samples were immediately frozen in liquid nitrogen and stored at −80 °C until further analysis.
Figure 1.

The infection status of the cultivars “Zifengyu” and “Dafugui” under field conditions by B. cinerea. S1, late May; S2, mid June; S3, early July; and S4, late July.
2.2. Small RNA Library Construction and Sequencing
Total RNA was extracted according to a modified CTAB extraction protocol [21]. Prior to sRNA library construction, RNA samples were examined by a spectrophotometer (Eppendorf, Hamburg, Germany) and 1% agarose gel electrophoresis. Moreover, RNA fragments of 18–30 nt long were separated from total RNA using polyacrylamide gel electrophoresis. The Solexa adaptors were added to the fragments at both 5'- and 3'-ends, and they were converted to cDNA according to a reverse transcription PCR kit (Invitrogen, Carlsbad, CA, USA). The purified fragments were sent for sequencing at the Beijing Genomic Institute (Shenzhen, China) using the Illumina HiSeq 2000 platform (Illumina Inc., San Diego, CA, USA). The data from “Zifengyu” and “Dafugui” were submitted to the National Center for Biotechnology Information (NCBI) under accession numbers SRS926009 and SRS926051, respectively.
2.3. miRNA Identification and Corresponding Target Gene Prediction
After getting rid of low-quality sequences (reads with 5' primer contaminants, reads without 3' primer, reads without the insert tag, reads with poly A and reads shorter than 18 nt), unique reads were screened against GenBank as well as Rfam (http://rfam.sanger.ac.uk) to remove ribosomal RNA (rRNA), transfer RNA (tRNA), small nuclear RNA (snRNA), and small nucleolar RNA (snoRNA). Moreover, the common and specific sequences between two libraries were identified through the comparison of corresponding genes annotated by transcripts. After these, the merged unique reads were also screened against the miRBase 21.0 using a nucleotide-nucleotide Basic Local Alignment Search Tool (BLASTn) to identify the conserved miRNAs [11]. Meanwhile, in order to identify the novel miRNAs, all candidate precursors with hairpin-like structures were obtained using the Mireap program (http://sourceforge.net/projects/mireap). Additionally, the unigene sequences of “Zifengyu” and “Dafugui” transcriptomes submitted to the NCBI with accession numbers SRS774325 and SRS774327 were used to predict the target genes of miRNAs by the psRNA Target program (http://plantgrn.noble.org/psRNATarget) with the default parameters. The specific methods were from Allen et al. [22] and Schwab et al. [23].
2.4. Differentially Expressed miRNAs and Their Target Gene Annotation
miRNAs expression levels were calculated according the value of reads per million reads (RPM). Differentially expressed miRNAs were defined based on strict criteria (p value ≤ 0.05 and differential expression fold > 2). Functional annotation of target genes was performed using various bioinformatics procedures, including GO and KEGG. The specific methods were referred to in Gong et al. [24].
2.5. Digital Gene Expression Analysis
Firstly, eight cDNA libraries from “Zifengyu” and “Dafugui” in four developmental stages were constructed. After quantification and qualification of the sample library using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster, California, CA, USA), the samples were also sequenced using the Illumina HiSeq 2000 platform (Illumina, San Diego, CA, USA). The gene expression level was compared with S1, respectively (S1/S1, S2/S1, S3/S1, S4/S1), and calculated using the reads per kb per million reads (RPKM) method [25] based on the numbers of reads uniquely mapped to the specific gene and the total number of uniquely mapped reads in the sample.
2.6. Expression Analysis of Target Genes by qRT-PCR
Expression levels of selected target genes were analyzed by quantitative real-time PCR (qRT-PCR) with three biological replications each sample via a CFX96 Real-Time System (Bio-Rad, Hercules, CA, USA). The specific methods were referred to in Zhao et al. [26]. The cDNA was synthesized from 1 g RNA using PrimeScript RT reagent Kit With gDNA Eraser (TaKaRa, Kyoto, Japan). P. lactiflora Actin (JN105299) had been used as an internal control in this study [27]. Gene-specific primers were designed using PRIMER5.0 software and listed in Table S1. Two µL of the cDNAs of each sample were used for ordinary PCR to test the amplification specificity of the corresponding primer pairs. qRT-PCR was performed using the SYBR Premix Ex Taq (Perfect Real Time) (TaKaRa). The amplification system consisted of an initial denaturation of 95 °C/30 s, followed by 40 cycles of 95 °C/5 s, 51 °C/30 s, 72 °C/30 s. Gene relative expression levels were calculated by the 2−∆∆Ct comparative threshold cycle (Ct) method [28]. The Ct values of the triplicate reactions were gathered using the Bio-Rad CFX Manager V1.6.541.1028 software (Bio-Rad, Hercules, CA, USA, 2008).
3. Results
3.1. Sequence Analysis of sRNAs
To identify miRNAs responsive to B. cinerea in P. lactiflora, two independent sRNA libraries were sequenced from different cultivars collected at four developmental stages and equally mixed, both subject to B. cinerea infection, but one of which (“Zifengyu”) is resistant to the pathogen. A total of 24,008,974 and 22,108,093 reads were generated from “Zifengyu” and “Dafugui”, respectively. The low quality sequences were removed including 5' contaminants, those missing the 3' primer or insert tag, sequences with a poly A tail, and finally those shorter than 18 nt. The final data sets consisted of 23,520,582 and 21,452,306 clean reads in “Zifengyu” and “Dafugui”, respectively, which was in both cases more than 97.97% of the total original reads. Subsequent analysis revealed the number of total sRNAs (i.e., sum of sRNAs) was 4,592,881 in “Zifengyu” and 5,809,796 in “Dafugui” (Figure 2A), while the number of unique sRNAs (i.e., variety of sRNAs) was 2,960,307 and 3,033,015, respectively (Figure 2B). Moreover, the length distribution of sRNA was similar between the two libraries, where the sRNAs ranging from 21 to 24 nt was the most frequent length (more than 80%) identified, c (more than 47%), followed by 22 nt, 20 nt, and 24 nt (Figure 2C). These sRNAs were classified into different categories after screening against GenBank and Rfam using BLAST searches, and the results from these analyses are presented in Table 1.
Figure 2.

Venn diagrams for analysis of total (A) and unique (B) sRNAs from “Zifengyu” and “Dafugui” libraries, and the length distribution of sRNAs (C).
Table 1.
Distribution of sRNAs among different categories in “Zifengyu” and “Dafugui”.
| Type | Unique sRNAs | Total sRNAs | ||
|---|---|---|---|---|
| “Zifengyu” | “Dafugui” | “Zifengyu” | “Dafugui” | |
| rRNA | 60653 | 77278 | 1047570 | 1338595 |
| tRNA | 16257 | 19165 | 1180850 | 1206549 |
| snRNA | 4057 | 4310 | 16627 | 15686 |
| snoRNA | 1031 | 1033 | 3289 | 3180 |
| Unannotated | 3496291 | 3540283 | 19428764 | 17158589 |
| miRNA | 20136 | 29064 | 1843482 | 1729707 |
| Total | 3598425 | 3671133 | 23520582 | 21452306 |
3.2. Identification of Conserved miRNAs
To identify conserved miRNAs, all mappable sRNA sequences were compared with the known plant miRNAs in miRBase. A total of 271 and 298 conserved miRNA sequences were identified in “Zifengyu” and “Dafugu”, respectively (Tables S2 and S3). The read counts among different miRNA families were markedly different. A few conserved miRNA families such as miR5078, miR166a, miR167a, miR157a, miR6113, miR718, miR1882e-3p, miR3353 and miR8019-5p were enriched in both of the libraries. The secondary structures of some conserved pre-miRNAs in “Zifengy” and “Dafugu” are listed in Figures S1 and S2, respectively. Moreover, putative target genes of conserved miRNAs were predicted, with 406 and 458 potential target genes from 133 (out of 271) and 136 (out of 298) conserved miRNAs identified in “Zifengy” and “Dafugu”, respectively (Tables S4 and S5).
3.3. Identification of Novel miRNAs
After searching for potential pre-miRNAs and predicting their hairpin-like structures, 9 and 8 unique sequences were identified as novel miRNAs in “Zifengyu” and “Dafugui”, respectively (Table 2 and Table 3), only 2 of which (pla-MIR11601 and pla-MIR11605) were shared by both cultivars. The novel miRNA sequences were 20 to 23 nt in length, and among these 21 nt reads were the most abundant. The pre-miRNAs ranged from 76 to 262 nt in “Zifengyu” and 76 to 274 nt in “Dafugui” (Table 2 and Table 3) in length. We also obtained the secondary structures of select novel pre-miRNAs (Figure S3). The average minimum free energy values were −41.31 kcal/mol in “Zifengyu” and −42.35 kcal/mol in “Dafugui”. Furthermore, we predicted the putative target genes of novel miRNAs were predicted, with 12 and 40 potential target genes from 8 (out of 9) and 7 (out of 8) novel miRNAs identified in “Zifengyu” and “Dafugui”, respectively (Tables S6 and S7).
Table 2.
Novel miRNA candidates in “Zifengyu”.
| Name in miRBase | Sequence | Length (nt) | Start/End Precursor | Length of Precursor (nt) | Arm | MFE (kcal/mol) | Count |
|---|---|---|---|---|---|---|---|
| pla-MIR11601 | TGCTCTAAAAGATCGTAGTTC | 21 | 1–262 | 262 | 5' | −34.80 | 12 |
| pla-MIR11605 | TTGAGGCGGCATATTCTCAAT | 21 | 73–178 | 106 | 3' | −29.82 | 18 |
| pla-MIR11606 | TGGTGGACTCCAATTCGCATA | 21 | 1563–1694 | 132 | 3' | −49.60 | 1113 |
| pla-MIR11607 | GATCACTCGGTTGTCTGACACAC | 23 | 169–297 | 129 | 5' | −27.50 | 1258 |
| pla-MIR11608 | TCGCTTAGGGGTTGTTGAAGCGC | 23 | 119–222 | 104 | 5' | −38.60 | 29 |
| pla-MIR11609 | TAGCTTGGTGTGAGGTCAACTT | 22 | 132–309 | 178 | 5' | −43.50 | 11 |
| pla-MIR11610 | ACAGATATGGTAGGGGGCACA | 21 | 10,914–10,989 | 76 | 3' | −36.70 | 222 |
| pla-MIR11611 | TAGGCAACCGTGGTAAAATGTC | 22 | 945–1045 | 101 | 3' | −48.10 | 6 |
| pla-MIR11612 | AAGACGGTCCAAAACGCCCAC | 21 | 151–283 | 133 | 3' | −63.20 | 490 |
Table 3.
Novel miRNA candidates in “Dafugui”.
| Name in miRBase | Sequence | Length (nt) | Start/End Precursor | Length of Precursor (nt) | Arm | MFE (kcal/mol) | Count |
|---|---|---|---|---|---|---|---|
| pla-MIR11598 | TCGTTCAAAGTAGGTTGTCAA | 21 | 113–349 | 237 | 5' | −45.60 | 16 |
| pla-MIR11599 | TTCAACCGTGGTAGATGTTAA | 21 | 95–306 | 212 | 5' | −92.70 | 13 |
| pla-MIR11600 | AACGTTCCTCGATTTCGCGAT | 21 | 471–546 | 76 | 3' | −24.90 | 6 |
| pla-MIR11601 | TGCTCTAAAAGATCGTAGTTC | 21 | 1–262 | 262 | 5' | −34.80 | 15 |
| pla-MIR11602 | TCTAACGGAACGCTATTGGATC | 22 | 6884–6998 | 115 | 3' | −22.10 | 8 |
| pla-MIR11603 | TTATAATTAGGTTGAGCGGAC | 21 | 40–313 | 274 | 5' | −62.00 | 2979 |
| pla-MIR11604 | TCCAGAGGGAGAACGTGGCGA | 21 | 328–406 | 79 | 3' | −26.90 | 9 |
| pla-MIR11605 | TTGAGGCGGCATATTCTCAAT | 21 | 73–178 | 106 | 3' | −29.82 | 41 |
3.4. Comparative Analysis of miRNAs and Corresponding Target Genes between Two Libraries
To identify key miRNAs between the two P. lactiflora libraries, their conserved miRNAs were comparatively analyzed based on “Zifengyu” as the control group. A total of 237 differentially expressed miRNAs were obtained (Figure 3A and Table S8), and 136 miRNAs were up-regulated, whereas 101 miRNAs were down-regulated. We found that 436 potential target genes were identified from 115 (out of 237) differentially expressed miRNAs. In order to further evaluate the potential functions of these target genes, a Kyoto Encyclopedia of Genes and Genomes (KEGG) functional classification was performed. In the KEGG functional classification analysis, only 130 target genes could be assigned to 12 KEGG pathways, among which metabolic pathways demonstrated the largest number of target genes, followed by biosynthesis of secondary metabolites, endocytosis and ether lipid metabolism. In addition, some pathways closely related to plant resistance were found including plant hormone signal transduction, phenylpropanoid biosynthesis, and plant-pathogen interaction (Table S9).
Figure 3.

Differentially expressed conserved (A) and novel (B) miRNAs from “Zifengyu” and “Dafugui” libraries. Red scatters indicate up-regulated miRNAs, green scatters indicate down-regulated miRNAs, and blue scatters indicate no difference miRNAs in expression between “Zifengyu” and “Dafugui”.
Similarly, the novel miRNAs were also comparatively analyzed, and 9 differentially expressed miRNAs were identified, including 2 up-regulated and 5 down-regulated miRNAs (Figure 3B and Table S10). Among these sequences, 8 potential target genes were identified from 6 (out of 7) differentially expressed miRNAs. After KEGG functional classification, only 2 target genes could be assigned to 1 KEGG pathways (Table S11).
3.5. Analysis of the Candidate B. cinerea-Responsive miRNAs
In order to identify the miRNAs responsive to B. cinerea stress, the analyses of differentially expressed miRNAs and their target genes in two libraries were performed. According to the target genes annotated in KEGG pathways, 9 genes were screened that were linked to phenylpropanoid biosynthesis (beta-glucosidase, BGLU), diterpenoid biosynthesis (gibberellin 2-oxidase, GA2OX), plant hormone signal transduction (ethylene receptor, ETR), plant-pathogen interaction (nonhost1, NHO1; peroxin-10, PEX10; serine/threonine-protein kinase PBS1, PBS1; WRKY transcription factor 29, WRKY29; calcium-dependent protein kinase, CDPK). The corresponding miRNAs were miR5254, miR846, miR6450a, miR165a-3p, miR6432, miR6450a, miR5558-3p and miR3897-3p, respectively (Table 4). When “Zifengyu” was regarded as the control group, the expression levels of the majority of the candidate miRNAs in “Dafugui” were up-regulated, except for miR6450a and miR5558-3p. Furthermore, the expression patterns of 9 target genes were analyzed in four developmental stages using digital gene expression. The results showed that these genes were essentially up-regulated after infection by B. cinerea. Additionally, BGLU, NHO1, CDPK and WRKY29 were highly expressed in “Zifengyu”, whereas the opposite trend was observed in ETR, which harbored expression levels negatively correlated with corresponding miRNAs (miR5254, miR165a-3p, miR3897-3p and miR6450a).
Table 4.
Candidate B. cinerea-responsive miRNAs and their target genes.
| miR-Name | Fold-Change (log2 Dafugui/Zifengyu) | Target Gene | Annotation | Expression Pattern of Target Gene (log2 Sn/S1) |
|---|---|---|---|---|
| miR5254 | 9.02815472 | CL2508.Contig1_All | beta-glucosidase | 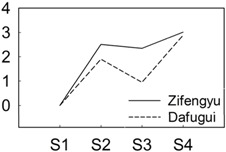 |
| miR846 | 12.12015362 | Unigene17406_All | gibberellin 2-oxidase | 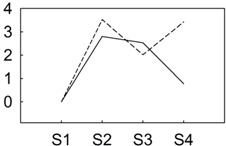 |
| miR165a-3p | 1.39849113 | CL10731.Contig2_All | nonhost1 | 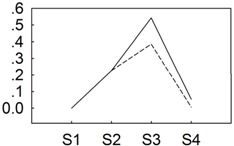 |
| miR6432 | 8.82072147 | CL7438.Contig1_All | peroxin-10 | 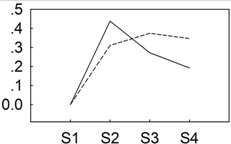 |
| miR6450a | −11.19652845 | Unigene9181_All | ethylene receptor |  |
| CL6799.Contig1_All | serine/threonine-protein kinase PBS1 | 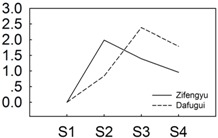 |
||
| miR5558-3p | −2.35248612 | CL8576.Contig1_All | serine/threonine-protein kinase PBS1 | 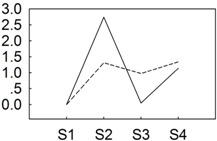 |
| miR3897-3p | 7.50620839 | Unigene17579_All | calcium-dependent protein kinase |  |
| Unigene3137_All | WRKY transcription factor 29 | 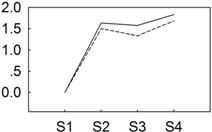 |
3.6. qRT-PCR Analysis of Target Genes
To verify the results of the digital gene expression analysis, expression levels of eight target genes including BGLU (CL2508.Contig1_All), GA2OX (Unigene17406_All), NHO1 (CL10731.Contig2_All), ETR (Unigene9181_All), PBS1 (CL6799.Contig1_All), PBS1 (CL8576.Contig1_All), CDPK (Unigene17579_All), and WRKY29 (Unigene3137_All) were evaluated by qRT-PCR (Figure 4). Overall, the relative expression of the examined target genes in “Zifengyu” and “Dafugui” at four different stages showed similar expression patterns according to the digital gene expression profiles, which indicated a correspondence of the results from qRT-PCR with the digital gene expression sequencing.
Figure 4.

qRT-PCR validations of expression levels of target genes from digital gene expression analysis. Expression levels by qRT-PCR of selected target genes of P. lactiflora cultivars “Dafugui” and “Zifengyu” were validated from the levels of digital gene expression data. The corresponding genes are specified above each map. The Y axis represents the normalized log2 value of gene expression levels. The X axis represents the comparisons of different stages. “S2/S1” indicates a comparison of gene expression levels between S1 and S2. “S3/S1” and “S4/S1” indicate analogous comparisons. S1: late May; S2: mid June; S3: early July; S4: late July.
4. Discussion
It has been recently found that miRNAs play important roles in physiological processes as a non-coding gene expression and regulating factor [29]. With the dramatic changes in the environment, more attention has been paid to the protective role of miRNAs in plants. Response to adversity stress in plants includes the differential expression of miRNAs, causing the accumulation of select substances and altering metabolic pathways [4,5,30]. Plant adversity stress is divided into abiotic and biotic categories, with more in-depth studies focused on plant miRNAs in response to abiotic stress, which includes stresses related to changes in nutrition [31], water [32], temperature [33], and heavy metals [34]. Meanwhile, miRNAs also play an important role in response to biotic stress; in particular, when plants are infected by pathogens, the expression of a variety of miRNAs and related target genes will change [35,36]. In the present study, high-throughput sequencing technology was used to identify miRNAs responsive to B. cinerea infection in P. lactiflora for the first time. Two cultivars, “Zifengyu” and “Dafugui”, with significantly different levels of resistance to B. cinerea were selected as the materials. When they were subjected to B. cinerea infection, “Zifengyu” was resistant to the pathogen. The two plant cultivars with different genotypes were used as experimental materials and compared in terms of many stress responses, such as temperature stress response [32] and Verticillium dahliae and Sporisorium reilianum infection [37,38]. In B. cinerea infection, our group compared the digital gene expression (DGE) of “Zifengyu” and “Dafugui” subjected to B. cinerea infection, and a great deal of disease resistance-relevant genes were successfully screened [24]. Thus, these two cultivars might be good materials to study the functions and molecular mechanisms of miRNAs in P. lactiflora resistance to B. cinerea. Through comparing two independent sRNA libraries, 23,520,582 and 21,452,306 sRNAs in “Zifengyu” and “Dafugui” were obtained, respectively. The length distribution of sRNAs in the libraries was very similar with the majority ranging from 20 to 24 nt. Among these sequences, the 21 nt sRNAs were the most abundant, followed by 22 nt, which is consistent with reports for Chinese yew [39] and Norway spruce [40]. However, in reports for other plants, such as celery [33], trifoliate oranges [41], and olives [42], the 24 nt sRNAs were the most abundant. These differences are mainly because the length of sRNAs is dependent on specific enzymes. For example, the sRNAs are 21 nt in length when processed by DCL1, while they are 24 nt when processed by DCL2 [43]. These sRNAs were classified into different categories, with the rRNA and miRNA groups enriched, which is consistent with what has been found in winter wheat [44], suggesting that sRNAs have been conserved in plant evolution. In the comparative analysis of miRNAs between “Zifengyu” and “Dafugui”, 237 conserved and 7 novel differentially expressed miRNAs were obtained. We examined the KEGG pathways with which they were associated in order to distinguish those likely to be involved in resistance to B. cinerea from those related to other phenotypic differences between the two cultivars.
The molecular mechanistic changes in plants infected by B. cinerea are complex; however, previous studies mainly focused on the transcriptional level of this response. De Cremer et al. [45] analyzed the transcriptome of lettuce infected by B. cinerea, and identified a complex network of genes, including the induction of those related to the phenylpropanoid pathway, terpenoid biosynthesis, and photosynthesis. At the same time, through sequencing the tomato transcriptome, Smith et al. [46] found that almost immediately after B. cinerea infection, a variety of processes were suppressed including photosynthesis as well as pathways involved in growth, energy generation, and response to stimuli. Simultaneously, some processes were also induced such as various defense-related genes, including pathogenesis-related protein 1 (PR1), a beta-1, 3-glucanase (glucanase), and a subtilisin-like protease. At the post-translational level, the relevant reports concerning B. cinerea infection have been limited to the tomato. Firstly, Jin et al. [20] identified three B. cinerea stress-responsive miRNAs using microarray analysis, which regulated metabolic, morphological, and physiological adaptations of tomato seedlings at the post-transcriptional level. Subsequently, Jin and Wu [19] investigated the miRNA expression patterns in tomato in response to B. cinerea stress using high-throughput sequencing, and found that 57 conserved miRNAs and one novel miRNA were differentially expressed in B. cinerea-infected leaves, and additionally that miR319, miR394 and miRn1 might be involved in the tomato leaf response to B. cinerea infection. In the present study, 7 candidate differentially expressed miRNAs related to adversity stress were screened, and the 9 corresponding target genes were annotated in phenylpropanoid biosynthesis, diterpenoid biosynthesis, plant hormone signal transduction, and plant-pathogen interaction using KEGG. Among the candidate miRNAs, miR5254, miR846, miR165a-3p, miR6432 and miR3897-3p revealed higher expression levels in “Dafugui” than “Zifengyu”, while the conserved miRNAs, including miR6450a and miR5558-3p, had the opposite expression pattern. For the target genes analyzed, the expression levels of BGLU, NHO1, CDPK, and WRKY29 in “Zifengyu” were always higher than those in “Dafugui”, whereas that of ETR was always lower. The expression levels of PEX10 and PBS1 were higher in “Zifengyu” than “Dafugui” at an early stage of infection (S2), but lower later during the infection (S3 and S4), while that of GA2OX was lower in “Zifengyu” than “Dafugui” at S2 and S4, but higher at S3. In previous studies, BGLU was associated with many biological processes in plants that could resist abiotic and biotic stresses by activating plant hormones and resistant pathways [47]; for example, overexpression of an Arabidopsis BGLU gene enhanced drought resistance in creeping bentgrass [48]. Lu et al. [49] found NHO1 was required for general resistance against Pseudomonas bacteria in Arabidopsis. CDPK has multiple functions in plant disease resistance, such as enhancing the production of active oxygen species (AOS) by stimulating NADPH oxidase activity [50]. Additionally, as a member of the complex family of WRKY transcription factors, WRKY29 has been proved to be a positive regulator of disease resistance [51], while ETR negatively regulated ethylene responses [52]. The expression levels of these 5 target genes in the present study were consistent with previous studies, which indicated that they might be involved in the P. lactiflora response to B. cinerea infection at the transcriptional level. Moreover, these 5 target genes were negatively correlated with their corresponding miRNAs (miR5254, miR165a-3p, miR3897-3p and miR6450a), which also suggested that they might be involved in the P. lactiflora response to B. cinerea infection at the post-transcriptional level. However, to the best of our knowledge concerning miRNAs, only miR165a was previously reported in response to abiotic stress [53], meaning the roles of miR5254, miR3897-3p and miR6450a in response to B. cinerea infection require further study. These results would further the understanding of miRNA regulation in response to B. cinerea stress in P. lactiflora.
5. Conclusions
In this study, high-throughput sequencing technology was used to characterize the miRNAs in the B. cinerea-infected two P. lactiflora cultivars “Zifengyu” and “Dafugui” with significantly different resistance to B. cinerea. After stringent quality checking and data cleaning, a total of 23,520,582 and 21,452,306 clean reads were obtained. Differential expression analysis revealed 237 conserved and 7 novel miRNAs between “Zifengyu” and “Dafugui” might be associated with B. cinerea stress resistance. Among screening, miR5254, miR165a-3p, miR3897-3p and miR6450a might be involved in the P. lactiflora response to B. cinerea infection. Our work was useful for breeding new cultivars of B. cinerea stress resistance.
Acknowledgments
This work was supported by the Agricultural Science & Technology Independent Innovation Fund of Jiangsu Province (CX[13]2014), Agricultural Science & Technology Support Project of Jiangsu Province (BE2013389), Postgraduate Science Research Innovation Project of Jiangsu province (CXLX_1426) and the Priority Academic Program Development from Jiangsu Government.
Supplementary Files
Author Contributions
Sample preparation, performing the experiments, production of results and drafting the manuscript: Daqiu Zhao, Saijie Gong and Zhaojun Hao. Conceiving and designing the experiments: Daqiu Zhao and Jun Tao. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Stevens S., Stevens A.B., Gast K.L.B., O’Mara J.A., Tisserat N.A., Bauernfeind R. Commercial Specialty Cut Flower Production, Peonies. Cooperative Extension Service, Kansas State University; Manhattan, KS, USA: 1993. [Google Scholar]
- 2.Wang Y., Jiang W., Wei J., Weng M., Han J. Cultural implication and landscape application of Chinese herbaceous peony. Guangdong Agric. Sci. 2013;20:58–61. [Google Scholar]
- 3.Lan Y. Pathogenic identification and biological research on the gray-mold disease of peony. J. Nanjing For. Univ. 1987;1:8–14. [Google Scholar]
- 4.Jones-Rhoades M.W., Bartel D.P. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol. Cell. 2004;14:787–799. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 5.Sunkar R., Zhu J.K. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001–2019. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lai E.C. Two decades of miRNA biology: Lessons and challenges. RNA. 2015;21:675–677. doi: 10.1261/rna.051193.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee R.C., Feinbaum R.L., Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-Y. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B.H., Pan X.P., Anderson T.A. Identification of 188 conserved maize microRNAs and their targets. FEBS Lett. 2006;580:3753–3762. doi: 10.1016/j.febslet.2006.05.063. [DOI] [PubMed] [Google Scholar]
- 9.Yao Y., Guo G., Ni Z., Sunkar R., Du J., Zhu J.K., Sun Q. Cloning and characterization of microRNAs from wheat (Triticum aestivum L.) Genome Biol. 2007 doi: 10.1186/gb-2007-8-6-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jian X.Y., Zhang L., Li G.L., Zhang L., Wang X.J., Cao X.F., Fang X., Chen F. Identification of novel stress-regulated microRNAs from Oryza sativa L. Genomics. 2010;95:47–55. doi: 10.1016/j.ygeno.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 11.Kozomara A., Griffiths-Jones S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu N., Cai W.J., Wang S., Shan C.M., Wang L.J., Chen X.Y. Temporal control of trichome distribution by microRNA156-targeted SPL genes in Arabidopsis thaliana. Plant Cell. 2010;22:2322–2335. doi: 10.1105/tpc.109.072579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carlsbecker A., Lee J.Y., Roberts C.J., Dettmer J., Lehesranta S., Zhou J., Lindgren O., Moreno-Risueno M.A., Vatén A., Thitamadee S., et al. Cell signalling by microRNA165/6 directs gene dose-dependent root cell fate. Nature. 2010;465:316–321. doi: 10.1038/nature08977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen R.S., Li J.Y., Stahle M.L., Dubroué A., Gubler F., Millar A.A. Genetic analysis reveals functional redundancy and the major target genes of the Arabidopsis miR159 family. Proc. Natl. Acad. Sci. USA. 2007;104:16371–16376. doi: 10.1073/pnas.0707653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu Q., Zhang Y.C., Wang C.Y., Luo Y.C., Huang Q.J., Chen S.Y., Zhou H., Qu L.H., Chen Y.Q. Expression analysis of phytohormone regulated microRNAs in rice, implying their regulation roles in plant hormone signaling. FEBS Lett. 2009;583:723–728. doi: 10.1016/j.febslet.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 16.Carnavale Bottino M., Rosario S., Grativol C., Thiebaut F., Rojas C.A., Farrineli L., Hemerly A.S., Ferreira P.C. High-throughput sequencing of small RNA transcriptome reveals salt stress regulated microRNAs in sugarcane. PLoS ONE. 2014;8:e59423. doi: 10.1371/journal.pone.0059423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiba Y., Mineta K., Hirai M.Y., Suzuki Y., Kanaya S., Takahashi H., Onouchi H., Yamaguchi J., Naito S. Changes in mRNA stability associated with cold stress in Arabidopsis cells. Plant Cell Physiol. 2013;54:180–194. doi: 10.1093/pcp/pcs164. [DOI] [PubMed] [Google Scholar]
- 18.Yin X., Wang J., Cheng H., Wang X., Yu D. Detection and evolutionary analysis of soybean miRNAs responsive to soybean mosaic virus. Planta. 2013;237:1213–1225. doi: 10.1007/s00425-012-1835-3. [DOI] [PubMed] [Google Scholar]
- 19.Jin W.B., Wu F.L. Characterization of miRNAs associated with Botrytis cinerea infection of tomato leaves. BMC Plant Biol. 2015 doi: 10.1186/s12870-014-0410-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin W.B., Wu F.L., Xiao L., Liang G.W., Zhen Y.X., Guo Z.K., Guo A.G. Microarray-based analysis of tomato miRNA regulated by Botrytis cinerea. J. Plant Growth Regul. 2012;31:38–46. doi: 10.1007/s00344-011-9217-9. [DOI] [Google Scholar]
- 21.Zhao D.Q., Zhou C.H., Tao J. Carotenoid accumulation and carotenogenic genes expression during two types of persimmon fruit (Diospyros kaki L.) development. Plant Mol. Biol. Rep. 2011;29:646–654. doi: 10.1007/s11105-010-0272-3. [DOI] [Google Scholar]
- 22.Allen E., Xie Z.X., Gustafson A.M., Carrington J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 23.Schwab R., Palatnik J.F., Riester M., Schommer C., Schmid M., Weigel D. Specific effects of microRNAs on the plant transcriptome. Dev. Cell. 2005;8:517–527. doi: 10.1016/j.devcel.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 24.Gong S.J., Hao Z.J., Meng J.S., Liu D., Wei M.R., Tao J. Digital gene expression analysis to screen disease resistance-relevant genes from leaves of herbaceous peony (Paeonia lactiflora Pall.) infected by Botrytis cinerea. PLoS ONE. 2015;10:e0133305. doi: 10.1371/journal.pone.0133305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortazavi A., Williams B.A., McCue K., Schaeffer L., Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 26.Zhao D.Q., Jiang Y., Ning C.L., Meng J.S., Lin S.S., Ding W. Transcriptome sequencing of a chimaera reveals coordinated expression of anthocyanin biosynthetic genes mediating yellow formation in herbaceous peony (Paeonia lactiflora Pall.) BMC Genomics. 2014;15:689. doi: 10.1186/1471-2164-15-689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao D., Tao J., Han C., Ge J. Actin as an alternative internal control gene for gene expression analysis in herbaceous peony (Paeonia lactiflora Pall.) Afr. J. Agric. Res. 2012;7:2153–2159. [Google Scholar]
- 28.Schmittgen T.D., Livak K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008;36:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 29.Mallory A.C., Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nat. Genet. 2006;38:S31–S36. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- 30.Fujii H., Chiou T.J., Lin S.I., Aung K., Zhu J.K. A miRNA involved in phosphate starvation response in Arabidopsis. Curr. Biol. 2005;15:2038–2043. doi: 10.1016/j.cub.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 31.Song A., Wang L., Chen S., Jiang J., Guan Z., Li P., Chen F. Identification of nitrogen starvation-responsive microRNAs in Chrysanthemum nankingense. Plant Physiol. Biochem. 2015;91:41–48. doi: 10.1016/j.plaphy.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Li D., Wang L., Liu X., Cui D., Chen T., Zhang H., Jiang C., Xu C., Li P., Li S., et al. Deep sequencing of maize small RNAs reveals a diverse set of microRNA in dry and imbibed seeds. PLoS ONE. 2013;8:e55107. doi: 10.1371/journal.pone.0055107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li M.Y., Wang F., Xu Z.S., Jiang Q., Ma J., Tan G.F., Xiong A.S. High throughput sequencing of two celery cultivars small RNAs identifies microRNAs involved in temperature stress response. BMC Genomics. 2014 doi: 10.1186/1471-2164-15-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ding Y.F., Zhu C. The role of microRNAs in copper and cadmium homeostasis. Biochem. Biophys. Res. Commun. 2009;386:6–10. doi: 10.1016/j.bbrc.2009.05.137. [DOI] [PubMed] [Google Scholar]
- 35.Gal-On A., Kaplan I., Palukaitis P. Differential effects of satellite RNA on the accumulation of cucumber mosaic virus RNAs and their encoded proteins in tobacco vs. zucchini squash with two strains of CMV helper virus. Virology. 1995;208:58–66. doi: 10.1006/viro.1995.1129. [DOI] [PubMed] [Google Scholar]
- 36.Diermann N., Matoušek J., Junge M., Riesner D., Steger G. Characterization of plant miRNAs and small RNAs derived from potato spindle tuber viroid (PSTVd) in infected tomato. Biol. Chem. 2010;391:1379–1390. doi: 10.1515/bc.2010.148. [DOI] [PubMed] [Google Scholar]
- 37.Zhang S.P., Xiao Y.N., Zhao J.R. Digital gene expression analysis of early root infection resistance to Sporisorium reilianum f. sp. zeae in maize. Mol. Genet. Genomics. 2013;288:21–37. doi: 10.1007/s00438-012-0727-3. [DOI] [PubMed] [Google Scholar]
- 38.Sun Q., Jiang H.Z., Zhu X.Y., Wang W.N., He X.H., Shi Y.Z. Analysis of sea-island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genomics. 2013 doi: 10.1186/1471-2164-14-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qiu D., Pan X., Wilson I.W., Li F., Liu M., Teng W., Zhang B. High throughput sequencing technology reveals that the taxoid elicitor methyl jasmonate regulates microRNA expression in Chinese yew (Taxus chinensis) Gene. 2009;436:37–44. doi: 10.1016/j.gene.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 40.Yakovlev I.A., Fossdal C.G., Johnsen Ø. MicroRNAs, the epigenetic memory and climatic adaptation in Norway spruce. New Phytol. 2010;187:1154–1169. doi: 10.1111/j.1469-8137.2010.03341.x. [DOI] [PubMed] [Google Scholar]
- 41.Song C., Wang C., Zhang C., Korir N.K., Yu H., Ma Z., Fang J. Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata) BMC Genomics. 2010 doi: 10.1186/1471-2164-11-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donaire L., Pedrola L., Rosa Rde L., Llave C. High-throughput sequencing of RNA silencing-associated small RNAs in olive (Olea europaea L.) PLoS ONE. 2011;6:e27916. doi: 10.1371/journal.pone.0027916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao T., Li G., Mi S., Li S., Hannon G.J., Wang X.J., Qi Y. A complex system of small RNAs in the unicellular green alga Chlamydomonas reinhardtii. Genes Dev. 2007;21:1190–1203. doi: 10.1101/gad.1543507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang R., Cang J., Yu J., Lu B.W., Liu L.J., Wang J.F. Solexa sequencing and bioinformatics analysis of small RNA in winter wheat. Chin. Bull. Bot. 2014;49:8–18. [Google Scholar]
- 45.De Cremer K., Mathys J., Vos C., Froenicke L., Michelmore R.W., Cammue B.P., de Coninck B. RNAseq-based transcriptome analysis of Lactuca sativa infected by the fungal necrotroph Botrytis cinerea. Plant Cell Environ. 2013;36:1992–2007. doi: 10.1111/pce.12106. [DOI] [PubMed] [Google Scholar]
- 46.Smith J.E., Mengesha B., Tang H., Mengiste T., Bluhm B.H. Resistance to Botrytis cinerea in Solanum lycopersicoides involves widespread transcriptional reprogramming. BMC Genomics. 2014 doi: 10.1186/1471-2164-15-334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gomez-Anduro G., Ceniceros-Ojeda E.A., Casados-Vazquez L.E. Genome-wide analysis of the beta-glucosidase gene family in maize (Zea mays L. var B73) Plant Mol. Biol. 2011;77:159–183. doi: 10.1007/s11103-011-9800-2. [DOI] [PubMed] [Google Scholar]
- 48.Han Y.J., Cho K.C., Hwang O.J., Choi Y.S., Shin A.Y., Hwang I., Kim J.I. Overexpression of an Arabidopsis β-glucosidase gene enhances drought resistance with dwarf phenotype in creeping bentgrass. Plant Cell Rep. 2012;31:1677–1686. doi: 10.1007/s00299-012-1280-6. [DOI] [PubMed] [Google Scholar]
- 49.Lu M., Tang X.Y., Zhou J.M. Arabidopsis NHO1 is required for general resistance against Pseudomonas bacteria. Plant Cell. 2001;13:437–447. doi: 10.1105/tpc.13.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xing T., Wang X.J., Malik K., Miki B.L. Ectopic expression of an Arabidopsis Calmodulin-like domain protein kinase-enhanced NADPH oxidase activity and oxidative burst in tomato protoplasts. Mol. Plant Microbe Interact. 2001;14:1261–1264. doi: 10.1094/MPMI.2001.14.10.1261. [DOI] [PubMed] [Google Scholar]
- 51.Eulgem T., Somssich I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007;10:366–371. doi: 10.1016/j.pbi.2007.04.020. [DOI] [PubMed] [Google Scholar]
- 52.Hua J., Meyerowitz E.M. Ethylene responses are negatively regulated by a receptor gene family in Arabidopsis thaliana. Cell. 1998;94:261–271. doi: 10.1016/S0092-8674(00)81425-7. [DOI] [PubMed] [Google Scholar]
- 53.Zhu J.F., Li W.F., Yang W.H. Identification of microRNAs in Caragana intermedia by high-throughput sequencing and expression analysis of 12 microRNAs and their targets under salt stress. Plant Cell Rep. 2013;32:1339–1349. doi: 10.1007/s00299-013-1446-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.