

Abstract
In this issue of Cancer Cell, Rubin et al. (2011) describe using various conditional mouse models to trace the developmental origin and genetic basis of rhabdomyosarcomas. Their work provides a genetic dissection underlying rhabdomyosarcomas development and unveils unexpected relationship between various soft-tissue tumor types.
Rhabdomyosarcomas (RMSs) are the predominant soft-tissue tumors of children and adolescents (Arndt and Crist, 1999). These tumors are generally divided into alveolar rhabdomyosarcoma (aRMS) and embryonal rhabdomyosarcoma (eRMS) subtypes. Most aRMSs are the result of chromosomal translocations between PAX3 or PAX7 and FOXO1A genes, resulting in Pax3-FKHR and Pax7-FKHR fusion proteins (Tiffin et al., 2003). These fusion proteins have potent transcriptional activity leading to cellular transformation and oncogenesis into aRMS. On the other hand, a wide range of causative mutations have been implicated in eRMS, such as the loss of heterozygosity at 11p15.5 locus (Anderson et al., 1999), mutations in tumor suppressor p53 (Felix et al., 1992), retinoblastoma (Rb1) (Kohashi et al., 2008), N- and K-ras genes (Stratton et al., 1989), and PTCH1 haploinsuficiency (Hahn et al., 1998).
It is thought that eRMSs develop from cells residing within the muscle tissue, partly because eRMSs express markers of muscle cells such as MyoD, Myogenin, and Desmin. Furthermore, these tumors can also occur where muscle tissue resides. However, muscle tissue contains a heterogeneous population of muscle stem cells and downstream myogenic progenitors as well as nonmyogenic cells (Kuang et al., 2007). To study the potentials of individual subpopulations of muscle cells in eRMS development, Rubin et al. (2011) deleted p53 either with or without Ptch1 haploinsuficiency. They then used various Cre drivers to inactivate these genes in muscle stem cells and in proliferating and maturing myoblasts. In a 600 day follow-up period, they observed that all mouse Cre lines developed tumors at the penetrance rate of 13%–56%. Upon histological examination of these tumors, they found a spectrum of malignancies ranging from alveolar and embryonal RMS to undifferentiated pleomorphic sarcomas (UPSs). They observed that rhabdomyosarcomas developed from all subpopulations of muscle cells, including muscle satellite cells and differentiating myoblasts (Figure 1). More importantly, they found that the cell of origin and the mutational profile of the tumors were important in determining the proportion of rhabdomyosarcomas versus undifferentiated spindle cell sarcomas (i.e., UPSs). Loss of p53 in maturing myoblasts (Myf6+ cells) gave rise to the highest percentage of eRMSs. These tumors showed the highest degree of myogenic differentiation potential, while those derived from satellite cells (Pax7+ cells) had the lowest rate of myogenic differentiation in in vitro differentiation assays.
Figure 1. The Developmental Origin and Mutational Profile of the Tumor Determine the Proportions of RMS.
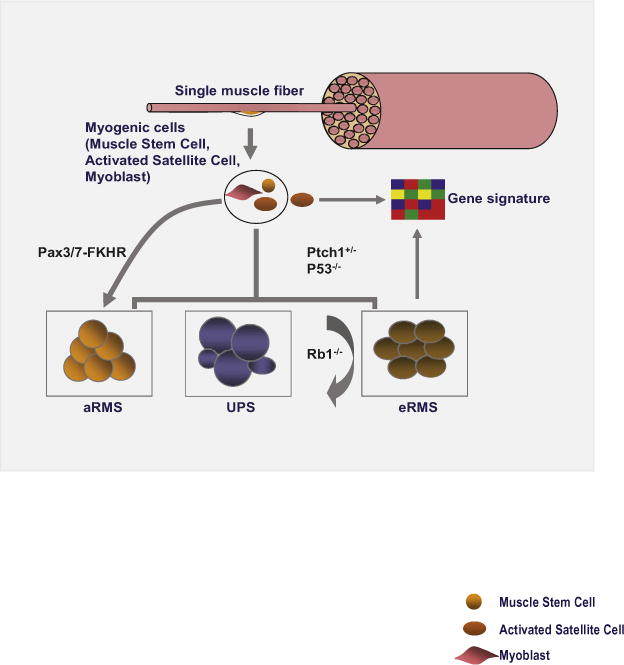
Different subpopulations of myogenic precursor cells give rise to RMS. While p53 deletion and Ptch1 haploinsufficiency are important players in cellular transformation and myodifferentiation potential of the resulting tumor, Rb1 deletion acts as a modifier of tumor phenotype in that context. Tumors (i.e., eRMSs) arising from different cells of origin exhibit the same gene expression profile as that of the activated muscle satellite cells.
To study the effect of retinoblastoma (Rb1) mutation on eRMS development in combination with the loss of p53, Rubin et al. (2011) inactivated both p53 and Rb1 with or without Ptch1 haploinsuficiancy. Loss of Rb1 alone did not lead to tumor initiation. However, unlike Rb1 loss, Ptch1 haploinsuficiency contributed to tumor initiation at every level of cellular differentiation. To further explore the role of Rb1 in rhabdomyosarcomas development, they inactivated both Rb1 and p53 in various subpopulations of muscle progenitor cells using different Cre drivers. Surprisingly, they observed that combination of Rb1 and p53 loss was generally associated with an undifferentiated phenotype in the resulting tumors. Rb1 deletion reduced the myodifferentiation potentials of p53 null tumor cells. Based on these observations, Rb1 seems to act as a modifier of tumor phenotype, in part by regulating the proliferation rate of sarcoma cells. Interestingly, analysis of the global gene expression profiling showed a marked difference between tumors with intact versus mutant Rb1. These findings further support the conclusion that Rb1 may act as modifier in sarcoma development, potentially by regulating a broad range of genetic and transcriptional networks.
Perhaps one of the most intriguing aspects of this study by Rubin et al. (2011) is that at least a subset of eRMS and UPS tumors seem to share a common cell of origin. This is interesting, as UPS tumors, a broad range of heterogeneous neoplasms including malignant fibrous histiocytomas or undifferentiated spindle cell sarcomas have a poorly defined etiology. These tumors also show no obvious signs of differentiation by immunohistochemical and molecular criteria. On the other hand, eRMSs express a broad range of muscle cell markers and possess myodifferentiation potentials. Their data further show that while maturing myoblasts are more prone to giving rise to eRMS tumors, UPS tumors are more likely to develop from Pax7 expressing muscle satellite cells. Furthermore, irrespective of the cell of origin, Rb1 modifies tumor phenotype to mimic UPS (Figure 1). Therefore, UPSs and eRMSs may constitute a continuum of the same disease.
By comparative analysis and global gene expression profiling, Rubin et al. (2011) delineate gene expression signature for UPS and eRMS and show that eRMSs have a similar gene signature with that of the activated muscle satellite cell (Figure 1). This finding is interesting for two reasons: first, it shows that gene expression pattern is not a predictor of the cell of origin, as eRMSs develop from a range of muscle cells, including muscle satellite cells and downstream myogenic progenitors such as maturing myoblasts, as shown in this study. Second, it implies that the cohort of mutations giving rise to eRMS likely results in a broad reprogramming of the transcriptional network of the transformed cells, making it to resemble gene signature of the activated satellite cells.
The study by Rubin et al. (2011) provides important new insight into the genetic basis of rhabdomyosarcomas in the context of p53, Rb1, and Ptch1 mutational pathways, and shows the potential of various subpopulations of muscle stem cells and downstream myogenic precursors in rhabdomyosarcomas development. Importantly, this study opens a forum for addressing other fundamental questions in the future. For example, to assess the relevance of their mouse sarcoma models to the human disease, Rubin et al. (2011) studied the gene expression profile of 111 primary human fusion-negative rhabdomyosarcomas from public databases and found that in at least 29% of cases they were unable to identify a gene signature in line with their mouse sarcoma models. The authors rightly argue that there might be additional mutations involving other tumor suppressors that may be involved in rhabdomyosarcomas development. Indeed, there is recent evidence indicating that other tumor suppressor genes such as PTEN may also play a role in sarcoma development (Gibault et al., 2011). In addition, given the observation that muscle stem cells and the downstream myogenic precursors can give rise to eRMS as demonstrated in this study does not preclude the possibility of other nonmuscle cells to contribute to the disease, as also emphasized by the authors. The conclusions from this study and previous work suggest that the genetic basis of rhabdomyosarcomas, especially that of the eRMS, is complex and is likely defined by a wide range of genetic and epigenetic factors. The heterogeneity in rhabdomyosarcomas phenotype may therefore be the result of the balance between the mutational profile of the tumor and the cell of origin. The involvement of tumor modifiers such as Rb1 in changing sarcoma phenotype as shown in this study raises many interesting questions about the possibility of yet other unknown modifiers and the genetic context in which these modifiers exert their effect on shaping the tumor phenotype. Future studies involving comparative genetic and epigenetic analysis of these tumors may provide a more concrete understanding of a cohort of potential players in rhabdomyosarcomas development. The feasibility and relative affordability of large scale genomic sequencing platforms provide opportunities to perform comparative genome-wide analysis in large sets of tumor samples in search of these genetics or epigenetic factors. In addition, a role for posttranscriptional gene regulation by microRNAs in rhabdomyosarcomas development has also been demonstrated (Wang et al., 2008). Further studies on the role of microRNAs in sarcomas development are another avenue that is important to pursue.
References
- Anderson J, Gordon A, Pritchard-Jones K, Shipley J. Genes Chromosomes Cancer. 1999;26:275–285. [PubMed] [Google Scholar]
- Arndt CA, Crist WM. N Engl J Med. 1999;341:342–352. doi: 10.1056/NEJM199907293410507. [DOI] [PubMed] [Google Scholar]
- Felix CA, Kappel CC, Mitsudomi T, Nau MM, Tsokos M, Crouch GD, Nisen PD, Winick NJ, Helman LJ. Cancer Res. 1992;52:2243–2247. [PubMed] [Google Scholar]
- Gibault L, Perot G, Chibon F, Bonnin S, Lagarde P, Terrier P, Coindre JM, Aurias A. J Pathol. 2011;223:64–71. doi: 10.1002/path.2787. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Nat Med. 1998;4:619–622. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- Kohashi K, Oda Y, Yamamoto H, Tamiya S, Takahira T, Takahashi Y, Tajiri T, Taguchi T, Suita S, Tsuneyoshi M. J Cancer Res Clin Oncol. 2008;134:1097–1103. doi: 10.1007/s00432-008-0385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Cell. 2007;129:999–1010. doi: 10.1016/j.cell.2007.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin BP, Nishijo K, Chen HIH, Yi X, Scheutze DP, Pal R, Prajapati SI, Abraham J, Arenkiel BR, Chen QR, et al. Cancer Cell. 2011;19:177–191. doi: 10.1016/j.ccr.2010.12.023. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton MR, Fisher C, Gusterson BA, Cooper CS. Cancer Res. 1989;49:6324–6327. [PubMed] [Google Scholar]
- Tiffin N, Williams RD, Shipley J, Pritchard-Jones K. Br J Cancer. 2003;89:327–332. doi: 10.1038/sj.bjc.6601040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Garzon R, Sun H, Ladner KJ, Singh R, Dahlman J, Cheng A, Hall BM, Qualman SJ, Chandler DS, et al. Cancer Cell. 2008;14:369–381. doi: 10.1016/j.ccr.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]