

Abstract
Statins are widely used as anti hyperlipidemic agents. Hepatotoxicity is one of their adverse effects appearing in some patients. No protective agents have yet been developed to treat statins-induced hepatotoxicity. Different investigations have suggested L-carnitine as a hepatoprotective agent against drugs-induced toxicity. This study was designed to evaluate the effect of L-carnitine on the cytotoxic effects of statins on the freshly-isolated rat hepatocytes. Hepatocytes were isolated from male Sprague-Dawley rats by collagenase enzyme perfusion via portal vein. Cells were treated with the different concentrations of statins (simvastatin, lovastatin and atorvastatin), alone or in combination with L-carnitine. Cell death, reactive oxygen species (ROS) formation, lipid peroxidation, and mitochondrial depolarization were assessed as toxicity markers. Furthermore, the effects of statins on cellular reduced and oxidized glutathione reservoirs were evaluated. In accordance with previous studies, an elevation in ROS formation, cellular oxidized glutathione and lipid peroxidation were observed after statins administration. Moreover, a decrease in cellular reduced glutathione level and cellular mitochondrial membrane potential collapse occurred. L-carnitine co-administration decreased the intensity of aforementioned toxicity markers produced by statins treatment. This study suggests the protective role of L-carnitine against statins-induced cellular damage probably through its anti oxidative and reactive radical scavenging properties as well as its effects on sub cellular components such as mitochondria. The mechanism of L-carnitine protection may be related to its capacity to facilitate fatty acid entry into mitochondria; possibly adenosine tri-phosphate or the reducing equivalents are increased, and the toxic effects of statins toward mitochondria are encountered.
Keywords: Hepatotoxicity, Isolated rat hepatocytes, L-carnitine, Mitochondria, Statins
INTRODUCTION
Statins, known as HMG-CoA reductase inhibitors, are widely used as anti hyperlipidemic drugs (1) their clinical use might cause hepatotoxicity in some patients (2,3,4). Previous case reports have indicated that different statins could produce adverse effects against liver (5,6,7). Although the precise mechanism of liver injury induced by these drugs is not yet elucidated, but it seems that statins are metabolized to reactive metabolites in the liver (8), which might be related to the hepatotoxicity reactions. Furthermore, sub cellular targets such as mitochondria seem to be an important target of statins-induced cellular damage (9). In a recent study we showed that statins increase the reactive oxygen species (ROS) formation, lipid peroxidation and mitochondrial injury (10).
L-carnitine, an essential cofactor in mitochondrial respiration, plays an important role in the transferring of long-chain fatty acids from cytosol to mitochondria (11). L-carnitine could also improve the antioxidant status in rats and possess free radical scavenging activity (12,13). It has also been reported that L-carnitine had a protective effect on lipid peroxidation by reducing the formation of hydrogen peroxide.
In the current study, we examined the possible protective effects of L-carnitine on oxidative stress induced by statins in hepatocytes.
MATERIALS AND METHODS
Chemicals
4-(2-hydroxyethyl) 1-piperazine-ethanessul-fonic acid (HEPES) was obtained from Acros (New Jersey, USA). Albumin bovine type was purchased from Roche diagnostic corporation (Indianapolis, USA). Statins (lovastatin, simvastatin and atorvastatin), 2,7-dichlorofluorescein diacetate, rhodamine 123 and collagenase from clostridium histolyticum were obtained from Sigma Aldrich (St. Louis, USA). Trichloro acetic acid (TCA), Ethyleneglycol-bis (ρ-aminoethylether)-N,N,N′,N′-tetra acetic acid (EGTA) and Trypan blue were obtained from Merck (Darmstadt, Germany). Thiobarbituric acid (TBA) was obtained from SERVA (Heidenberg, New York, USA). All salts used for preparing buffer solutions were of analytical grade and obtained from Merck (Darmstadt, Germany).
Hepatocyte preparation
Male Sprague–Dawley rats weighing 250–300 g were kept in cages in an ambient temperature of 25 ± 3° C. Animals were fed a normal diet and water ad libitum. Collagenase perfusion method was used to isolate rat hepatocytes (14). This technique is based on liver perfusion with collagenase after removal of calcium ion (Ca2+) with a chelator (EGTA 0.5 mM).
Liver was perfused with different buffer solutions through the portal vein. Collagenase-containing buffer solution destructs liver interstitial tissue and cause hepatocyte to be easily isolated in next steps. Isolated hepatocytes (10 ml, 106 cells/ml) were incubated in Krebs-Henseleit buffer (pH 7.4) under an atmosphere of 95% O2 and 5% CO2, in 50 ml round bottom flasks rotating continuously at 37 °C in a water bath. A local ethic committee in Tabriz University of Medical Sciences, Tabriz, Iran, approved the animal procedures.
Statins (simvastatin, lovastatin and atorvastatin) were administered at different concentrations (refer to cell viability section) to assess the toxicity markers in isolated hepatocytes. L-carnitine was administered 30 min before statins in all experiments.
Cell viability
Trypan blue dye exclusion staining was used to assess the percentage of dead cells (15). Hepatocyte viability was determined at different time intervals to evaluate the effect of statins on cell viability, determining lethal concentration 50% (LC50, the dose which caused 50% cell death after 2 h) dose of the drugs and testing the protective effects of L-carnitine against cell death. Hepatocytes were at least 85% viable before statin use.
Reactive oxygen species formation
2,7-dichlorofluorescein diacetate (Sigma-Aldrich, USA)(1.6 μM) was added to the rat hepatocytes as a method for assessing ROS production in the biological systems. Briefly, 1 ml (106 cells) of hepatocytes was taken, centrifuged at 3000 rpm for 1 min and the fluorescence intensity was measured using a Jasco® FP-750 spectrofluorometer, UK with excitation and emission wavelengths of 500 and 520 nm, respectively (14).
Lipid peroxidation measurement
The amount of thiobarbituric acid reactive substances (TBARS) formed during the decomposition of lipid hydroperoxides were measured as a toxicity marker. One ml aliquots of hepatocyte suspension (106 cells/ml) were treated with TCA (70% w/v). The supernatant was then boiled with TBA (0.8% w/v) for 20 min. The absorbance was determined using an Ultrospec® 2000 UV spectrophotometer, Sweden at 532 nm (10).
Mitochondrial membrane potential
Mitochondrial membrane potential was assessed as an indicator of toxicity induced by statins. The fluorescent dye, rhodamine 123 was used as a probe to evaluate the mitochondrial membrane potential in rat hepatocytes (16).
One ml Samples were taken from the cell suspension at scheduled time points, and centrifuged at 1000 rpm for 1 min. The cell pellet was then resuspended in 2 ml of fresh incubation medium containing 1.5 μM rhodamine 123 and gently shaken at 37 °C in a thermostatic water bath for 10 min. Hepatocytes were separated by centrifugation at 3000 rpm for 1 min and the amount of rhodamine 123 appearing in the incubation medium was measured fluorimeterically at 490 nm excitation and 520 nm emission wavelengths using a Jasco® FP-750 spectrofluorometer (15).
Cellular GSH/GSSG content
The hepatocyte glutathione (GSH) content was determined by the method of Ellman (15,17). One ml of cell suspension containing 106 cells was mixed with 2 ml of 5% TCA and centrifuged. Then 0.5 ml of Ellman's reagent [0.0198% 5,5’-dithiobis-(2-nitrobenzoic acid), DTNB, in 1% sodium citrate) and 3 ml of the phosphate buffer (pH 8.0) were added to the supernatant.
The absorbance of the developed color was measured at 412 nm using a Biotech Pharmacia Ultrospec® 2000 spectrophotometer, Sweden (18). The enzymatic recycling method was used to assess the hepatocytes oxidized glutathione (GSSG) level (19), where cellular GSH content was covalently bonded to 2-vinylpyridine at first.
Then the excess of 2-vinylpyridine was neutralized with triethanolamine. GSSG was subsequently reduced to GSH using the glutathione reductase enzyme and nicotinamide adenine dinucleotide phosphate (NADPH). The amount of glutathione was measured as already described for GSH using the Ellman reagent (0.0198% DTNB in 1% sodium citrate) (17,20).
Statistical analysis
Results are shown as the Means ± SEM for at least three independent experiments. Statistical analysis was performed by one-way analysis of variance (ANOVA) followed by a Tukey's post hoc test. P<0.05 was considered as significant difference.
RESULTS
Statins-induced cytotoxicity
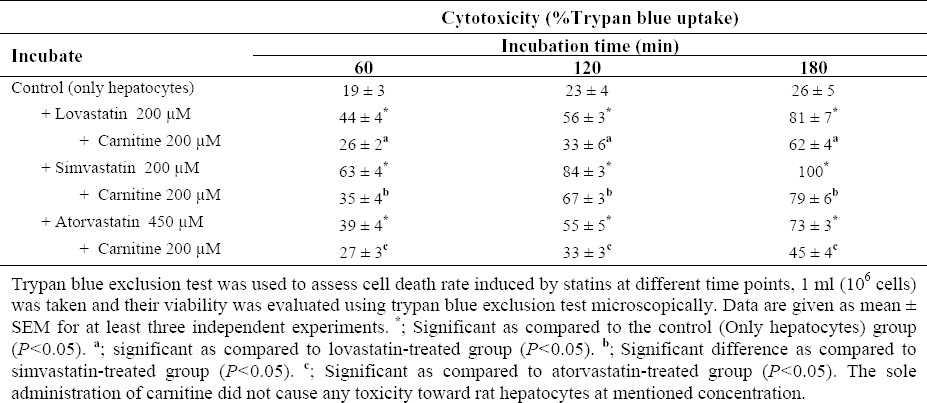
Exposure of isolated rat hepatocytes to various concentrations of statins led to cell death in statins-treated freshly isolated rat hepatocytes (Table 1). None of the chemicals used for evaluating their protective effects at tested concentrations caused significant toxicity toward hepatocytes as compared to the control cells when administered alone (Table 1). Administration of L-carnitine to statins-treated cells caused a significant decrease in cell death (Table 1).
Table 1.
Statins-induced cytotoxicity on isolated rat hepatocytes and the protective role of L-Carnitine.
Oxidative stress and lipid peroxidation
Statins caused formation of a considerable amount of ROS in isolated rat hepatocytes (Fig. 1). L-carnitine administration limited the effect of statins on ROS formation (Fig. 1) and its consequences such as lipid peroxidation (Fig. 2). Treatment with L-carnitine, in addition to decreasing the formation of free radicals (Fig. 1), significantly increased the GSH levels (Fig. 3). Moreover, the level of GSSG was decreased after L-carnitine administration in comparison with the statin-treated groups (Fig. 4).
Fig. 1.

Reactive oxygen species formation after statin and L-carnitine administration to isolated rat hepatocytes. A; atorvastatin, B; simvastatin, C; lovastatin. The fluorescent activity of dichlorofluorescin, which is directly linked to the amount of reactive oxygen species, was measured at different time points. Data are given as mean ± SEM for three experiments. ***; Significant as compared to control group (P<0.001). **; Significant as compared to statins-treated group (P<0.01). *; Significant as compared to statins-treated group (P<0.05). The sole administration of L-carnitine did not cause any toxicity toward rat hepatocytes at mentioned concentration.
Fig. 2.

Lipid peroxidation after statins administration to isolated rat hepatocytes. A; atorvastatin, B; simvastatin, C; lovastatin. Thiobarbituric acid reactive substances test was assessed in different time schedules to investigate statins-induced cytotoxicity in experimental groups. Carnitine (100 μM) caused no significant lipid peroxidation when administered alone. Data are given as mean ± SEM for three experiments. **; Significant as compared to control group (P<0.01). **; Significant as compared to statins-treated group (P<0.01). The sole administration of carnitine did not cause any toxicity toward rat hepatocytes at mentioned concentration.
Fig. 3.

Hepatocytes reduced glutathione (GSH) levels after statins administration. A; atorvastatin, B; simvastatin, C; lovastatin. Data are given as mean ± SEM for three experiments. The Ellman reagent (DTNB) test was employed to assess hepatocytes glutathione content. **; Significant as compared to control group (P<0.01). ***; Significant as compared to control group (P<0.001). *; Significant as compared to statins-treated group (P<0.05). The sole administration of carnitine did not cause any toxicity toward rat hepatocytes at mentioned concentration.
Fig. 4.

Hepatocytes oxidized glutathione levels after statins administration. A; atorvastatin, B; simvastatin, C; lovastatin. Data are given as mean ± SEM for three experiments. ***; Significant as compared to control group (P<0.001). *; Significant as compared to statins-treated group (P<0.05). **; Significant as compared to statins-treated group (P<0.01). The sole administration of carnitine did not cause any toxicity toward rat hepatocytes at mentioned concentration.
Mitochondrial membrane potential collapse
Mitochondrial membrane potential (ΔΨm), as a key parameter in evaluation of mitochondrial functionality (21), showed that the used statins caused mitochondrial depolarization (Fig. 5). L-carnitine effectively protected cellular mitochondria against statins-induced injury as revealed by an improvement in mitochondrial membrane potential in co-administrated groups (Fig. 5).
Fig. 5.

Statins-induced collapse in cellular mitochondrial potential (ΔΨm) and the role of L-carnitine administration. A; atorvastatin, B; simvastatin, C; lovastatin. Rhodamine 123 test was employed to assess the mitochondrial membrane potential. ***; Indicates P<0.001 versus control group. **; Indicates P<0.01 versus drug-treated groups. *; Indicates P<0.05 versus drug-treated groups.
According to the data obtained from current investigation, it seems that simvastatin is the most cytotoxic statin on isolated rat hepatocytes since it caused a higher rate of cytotoxicity (Table 1), higher level of ROS formation (Fig. 1), lipid peroxidation (Fig. 2), GSH depletion (Figs. 3 and 4), and mitochondrial membrane potential collapse (Fig. 5). The rank order of cytotoxic properties of investigated statins was simvastatin>lovastatin>atorvastatin (Table 1).
DISCUSSION
Reactive metabolite formation and/or oxidative stress seem to be responsible for statins-induced damage on hepatocytes (8,10,22). In addition, statins intoxication in hepatocytes impairs mitochondrial function which could lead to decrease in adenosine tri-phosphate (ATP) synthesis and also to increase in formation of free radicals (9). The experiments conducted here confirm an increase in oxidative stress induced by statins, as observed in previous investigations (9,10). Statins treatment was accompanied by an increased level of lipid peroxidation and a reduction in the cellular GSH contents. L-carnitine alleviated the toxic effects of statins on the rat hepatocytes.
Our finding is not in accordance with other studies showing that administration of antioxidants such as L-carnitine decreased the high oxidative stress and toxicity signs of drugs-induced hepatic injury (23,24). L-carnitine is a natural nutrient and essential for β-oxidation of fatty acids in mitochondria to generate ATP, and possesses antioxidant properties (25,26). Several reports showed that L-carnitine effectively protected mitochondrial injury from oxidative stress (27,28).
Mitochondria are the key organelles in energy production and cell death pathways in hepatocytes (29). Impaired mitochondrial function leads to apoptotic or necrotic cell death (30). In the case of targeting of mitochondria, reactive metabolites or parent drugs uncouple or inhibit the mitochondrial respiratory chain causing ATP depletion, increase concentrations of ROS, inhibit β-oxidation leading to steatosis (e.g. after intramitochondrial accumulation of amiodarone), damage mitochondrial DNA or interfere with its replication, or directly cause mitochondrial permeability transition pore (MPTP) (31,32). MPTP allows massive influx of protons through the inner mitochondrial membrane (decreased ΔΨm). Energy crisis (ATP depletion) resulting from MPTP (or other direct mechanisms of mitochondrial damage mentioned above) causes matrix expansion and mitochondrial outer membrane permeabilization and rupture with the release of cytochrome c and other pro-apoptotic mitochondrial proteins from the intermembrane space into the cytosol (33). Induction of mitochondrial membrane permeability transition (MPT) and the consequent cell death through apoptosis/necrosis might be another pathway through which statins induce hepatotoxicity. Future investigations are needed in order to clarify the effects of statins on MPT. For example, concurrent administration of MPT suppressing agents such as cyclosporine A, will clear the role of MPT in statins-induced cytotoxicity (34).
Our results show that both oxidative stress and hepatocytes damage were significantly higher in statins-treated groups and L-carnitine administration reduced oxidative stress and its consequences (Fig. 1). Furthermore, we found that L-carnitine may affect cellular mitochondrial damage induced by statins (Fig. 5). These findings proved another mechanism through which L-carnitine might prevent cellular damage and death. The present study extends the earlier results on L-carnitine protective properties (23,24) against xenobiotics-induced hepatotoxicity.
On the basis of present investigation we found that: (i) the blockade of oxidative stress by L-carnitine neutralizes cellular damage and improves hepatocytes health, (ii) an important part of protective effects of L-carnitine seems to be due to its effect on statins-induced mitochondrial damage.
Due to the unpredictable and idiosyncratic nature of hepatotoxicity induced by statins, it is difficult to extrapolate in vitro results to the humans. Different pharmacokinetic/dynamic factors might affect statins-induced liver injury in patients. More investigations in different animal models will promote our understanding of the mechanisms of statins-induced liver injury and consequently the ways of preventing such adverse reactions.
CONCLUSION
This study suggests that statins could cause oxidative stress and mitochondrial dysfunction in the rat hepatocytes. L-carnitine protects the rat hepatocytes against the statins toxicity probably due to its antioxidant properties and/or mitochondrial protection. However, more investigations are required to evaluate the precise mechanism by which L-carnitine protects isolated rat hepatocytes against statins toxicity.
ACKNOWLEDGMENTS
This study was funded by the School of Pharmacy of Tabriz University of Medical Sciences, Tabriz, Iran. The authors are grateful to Drug Applied Research Center for providing facilities and financial supports to carry out this study. This research was a part of Narges Abdoli's Ph.D thesis that was supported by Students Research Committee. The authors are also thankful to the Students Research Committee of Tabriz University of Medical Sciences, Tabriz, Iran, for supporting the study.
REFERENCES
- 1.Huser MA, Evans TS, Berger V. Medication adherence trends with statins. Adv Ther. 2005;22:163–171. doi: 10.1007/BF02849887. [DOI] [PubMed] [Google Scholar]
- 2.Björnsson E, Jacobsen EI, Kalaitzakis E. Hepatotoxicity associated with statins: reports of idiosyncratic liver injury post-marketing. J Hepatol. 2012;56:374–380. doi: 10.1016/j.jhep.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 3.Chitturi S, George J. Hepatotoxicity of commonly used drugs: nonsteroidal anti-inflammatory drugs, antihypertensives, antidiabetic agents, anticonvulsants, lipid-lowering agents, psychotropic drugs. Semin Liver Dis. 2002;22:169–183. doi: 10.1055/s-2002-30102. [DOI] [PubMed] [Google Scholar]
- 4.Parra JL, Reddy KR. Hepatotoxicity of hypolipidemic drugs. Clin Liver Dis. 2003;7:415–433. doi: 10.1016/s1089-3261(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 5.Horsmans Y, Desager JP, Harvengt C. Biochemical changes and morphological alterations of the liver in guinea-pigs after administration of simvastatin (hmg coa reductase-inhibitor) Pharmacol Toxicol. 1990;67:336–339. doi: 10.1111/j.1600-0773.1990.tb00840.x. [DOI] [PubMed] [Google Scholar]
- 6.Vaghasiya J, Rathod S, Bhalodia Y, Manek R, Malaviya S, Jivani N. Protective effect of polyherbal formulation on simvastatin hepatotoxicity in rats. J Young Pharm. 2009;1:57–62. [Google Scholar]
- 7.Tolman KG. The liver and lovastatin. Am J Cardiol. 2002;89:1374–1380. doi: 10.1016/s0002-9149(02)02355-x. [DOI] [PubMed] [Google Scholar]
- 8.Guengerich FP. Cytochrome P450s and other enzymes in drug metabolism and toxicity. The AAPS J. 2006;8:E101–E111. doi: 10.1208/aapsj080112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tavintharan S, Ong CN, Jeyaseelan K, Sivakumar M, Lim SC, Sum CF. Reduced mitochondrial coenzyme Q10 levels in HepG2 cells treated with high-dose simvastatin: a possible role in statininduced hepatotoxicity? Toxicol Appl Pharmacol. 2007;223:173–179. doi: 10.1016/j.taap.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Abdoli N, Heidari R, Azarmi Y, Eghbal MA. Mechanisms of the statins cytotoxicity in freshly isolated rat hepatocytes. J Biochem Mol Toxicol. 2013;27:287–294. doi: 10.1002/jbt.21485. [DOI] [PubMed] [Google Scholar]
- 11.Kelly GS. L-Carnitine: therapeutic applications of a conditionally-essential amino acid. Altern Med Rev. 1998;3:345–360. [PubMed] [Google Scholar]
- 12.Kalaiselvi T, Panneerselvam C. Effect of L-carnitine on the status of lipid peroxidation and antioxidants in aging rats. J Nutr Biochem. 1998;9:575–581. [Google Scholar]
- 13.Rani PJA, Panneerselvam C. Effect of L-carnitine on brain lipid peroxidation and antioxidant enzymes in old rats. J Gerontol A: Biol Sci Med Sci. 2002;57:B134–B137. doi: 10.1093/gerona/57.4.b134. [DOI] [PubMed] [Google Scholar]
- 14.Heidari R, Babaei H, Eghbal MA. Cytoprotective effects of organosulfur compounds against methimazole-induced toxicity in isolated rat hepatocytes. Adv Pharm Bull. 2013;3:135–142. doi: 10.5681/apb.2013.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heidari R, Babaei H, Eghbal MA. Ameliorative effects of taurine against methimazole-induced cytotoxicity in isolated rat hepatocytes. Sci Pharm. 2012;80:987–999. doi: 10.3797/scipharm.1205-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heidari R, Babaei H, Eghbal M. Mechanisms of methimazole cytotoxicity in isolated rat hepatocytes. Drug Chem Toxicol. 2012;36:403–411. doi: 10.3109/01480545.2012.749272. [DOI] [PubMed] [Google Scholar]
- 17.Nafisi S, Heidari R, Ghaffarzadeh M, Ziaee M, Hamzeiy H, Garjani A, et al. Cytoprotective effects of silafibrate, a newly-synthesised siliconated derivative of clofibrate, against acetaminophen-induced toxicity in isolated rat hepatocytes. ArhHyg Rada Toksikol. 2014;65:169–178. doi: 10.2478/10004-1254-65-2014-2434. [DOI] [PubMed] [Google Scholar]
- 18.Heidari R, Babaei H, Eghbal MA. Amodiaquine-induced toxicity in isolated rat hepatocytes and the cytoprotective effects of taurine and/or N-acetyl cysteine. Res Pharm Sci. 2014;9:97–105. [PMC free article] [PubMed] [Google Scholar]
- 19.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2007;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 20.Heidari R, Babaei H, Eghbal MA. Cytoprotective effects of taurine against toxicity induced by isoniazid and hydrazine in isolated rat hepatocytes. Arh Hyg Rada Toksikol. 2013;64:201–210. doi: 10.2478/10004-1254-64-2013-2297. [DOI] [PubMed] [Google Scholar]
- 21.Solaini G, Sgarbi G, Lenaz G, Baracca A. Evaluating mitochondrial membrane potential in cells. Biosci Rep. 2007;27:11–21. doi: 10.1007/s10540-007-9033-4. [DOI] [PubMed] [Google Scholar]
- 22.Abdoli N, Azarmi Y, Eghbal MA. Protective effects of N-acetylcysteine against the Statins cytotoxicity in freshly isolated rat hepatocytes. Adv Pharm Bull. 2014;4:249–254. doi: 10.5681/apb.2014.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J, Wang Q, Luan H, Kang Z, Wang C. Effects of L-carnitine against oxidative stress in human hepatocytes: involvement of peroxisome proliferator-activated receptor alpha. J Biomed Sci. 2012;19:32. doi: 10.1186/1423-0127-19-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yapar K, Kart A, Karapehlivan M, Atakisi O, Tunca R, Erginsoy S, et al. Hepatoprotective effect of L-carnitine against acute acetaminophen toxicity in mice. Exp Toxicol Pathol. 2007;59:121–128. doi: 10.1016/j.etp.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Bremer J. Carnitine-metabolism and functions. Physiol Rev. 1983;63:1420–1480. doi: 10.1152/physrev.1983.63.4.1420. [DOI] [PubMed] [Google Scholar]
- 26.Bieber LL. Carnitine. Annu Rev Biochem. 1988;57:261–283. doi: 10.1146/annurev.bi.57.070188.001401. [DOI] [PubMed] [Google Scholar]
- 27.Chang B, Nishikawa M, Nishiguchi S, Inoue M. L-carnitine inhibits hepatocarcinogenesis via protection of mitochondria. Int J Cancer. 2005;113:719–729. doi: 10.1002/ijc.20636. [DOI] [PubMed] [Google Scholar]
- 28.Reznick AZ, Kagan VE, Ramsey R, Tsuchiya M, Khwaja S, Serbinova EA, et al. Antiradical effects in L-propionyl carnitine protection of the heart against ischemia-reperfusion injury: the possible role of iron chelation. Arch Biochem Biophys. 1992;296:394–401. doi: 10.1016/0003-9861(92)90589-o. [DOI] [PubMed] [Google Scholar]
- 29.Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 30.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 31.Vendemiale G, Grattagliano I, Altomare E, Turturro N, Guerrieri F. Effect of acetaminophen administration on hepatic glutathione compartmentation and mitochondrial energy metabolism in the rat. Biochem Pharmacol. 1996;52:1147–1154. doi: 10.1016/0006-2952(96)00414-5. [DOI] [PubMed] [Google Scholar]
- 32.Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F. Mitochondria and cell death. Mechanistic aspects and methodological issues. Europ J Biochem. 1999;264:687–701. doi: 10.1046/j.1432-1327.1999.00725.x. [DOI] [PubMed] [Google Scholar]
- 33.Pessayre D, Mansouri A, Haouzi D, Fromenty B. Hepatotoxicity due to mitochondrial dysfunction. Cell Biol Toxicol. 1999;15:367–373. doi: 10.1023/a:1007649815992. [DOI] [PubMed] [Google Scholar]
- 34.Sullivan PG, Thompson MB, Scheff SW. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp Neurol. 1999;160:226–234. doi: 10.1006/exnr.1999.7197. [DOI] [PubMed] [Google Scholar]