

Abstract
Under pathophysiological conditions, infiltration of leukocyte plays a key role in the progression of the neuroinflammatory reaction in the CNS. Prostaglandin E2 (PGE2) is known to accumulate at lesion sites of the post-ischemic brain. Although post-ischemic treatments with cyclooxygenase-2 inhibitors reduce blood-brain barrier (BBB) leukocyte infiltration, the direct effect of PGE2 on BBB has not been fully implemented. Therefore, the direct effect of increasing PGE2 infusion on translocation of labeled albumin into the brain was assessed. Under anesthesia rats were drilled stereo-taxicaly a burr hole in the right forebrain and PGE2 was infused into the forebrain and the hole was occluded. The animals were then injected with fluorescent labeled albumin (FA), via internal right jugular vein and decapitated at different infusion time points. The forebrain was removed and each forebrain hemisphere was homogenized and fluorescence intensities were measured in the supernatant. The fluorescence intensities measured in the right and left forebrain hemispheres of the control group (0.0 μg PGE2) were almost identical. Four hours after infusion of PGE2 at doses higher than 250 μg, fluorescence intensity increased in the right forebrain supernatant, even if it was not statistically significant. The fluorescence intensity was detectable in the brain supernatant 4 h after infusion of PGE2 in doses higher than 250 μg PGE2. The highest fluorescence intensity was 16 h after infusion of 500 μg PGE2, which returned to near control values after 48 h. Increased fluorescence intensity in the brain following PGE2 infusion is concluded to be associated with disruption of the BBB.
Keywords: Blood-brain barrier, Ischemia, Neuroinflammatory, Prostaglandin
INTRODUCTION
Inflammation is one of the mechanisms known to participate in the progression of brain injury (1,2). A large number of studies indicate that blockade of the neuroin-flammatory process considerably reduces ischemic brain injury (3,4,5). Prostaglandin E2 (PG E2), one of the most likely candidates for the manifestation of the inflammation, is known to be accumulated at the lesion sites of the post-ischemic brain (1,6,7). These observations have been confirmed by biochemical studies that reported a significant increase in the expression of the PGE2 synthesizing enzyme, cyclooxygenase (COX-2), following ischemia (8,9,10). However, there is a debate on the specific role of PGE2 in cerebral ischemia (1). A recent study has found beneficial effects (11), another report claimed harmful (12), and an article reported PG E2 did not affect ischemic brain injury (13). However, several lines of evidence indicated that after the onset of ischemia, there is a significant disruption of the blood-brain barrier (BBB), followed by a massive infiltration of leukocytes (3,14,15). These reports suggested that disruption of BBB might be the main cause of neuroinflammatory process in the brain. While it is becoming clear that increased expression of COX-2 is associated with ischemic brain, it is by no means clear that a cause-and-effect relationship exists between the increased PGE2 and BBB permeability directly. This study examines the time course and direct effects of increasing PG E2 infusion on translocation of labeled albumin into the brain.
MATERIALS AND METHODS
Chemicals
PG E2 (Prostin E2, 10 mg/ml) was purchased from Upjohn (Puurs Belgium), fluorescein isothiocyante isomer 1 (FITC), Sephadex G-25 and rat serum albumin were procured from Sigma (Poole, Dorset, UK). Unless stated, all reagents were of the highest grade and made up in double glass-distilled water.
Fluorescein-labeled rat serum albumin
Preparation of fluorescein-labeled rat serum albumin was carried out by the method of Nargessi and coworkers (16). Equal volumes of FITC solution (1 mg/ml) and rat serum albumin solution (0.25 mg protein/ml) were mixed and stirred overnight at 4 °C. A Sephadex G-25 column (1.2 × 20 cm) was then loaded by the FITC/serum albumin mixture.
The entire fluorescein labeled albumin (FALB) band was identified by its fluorescence under the UV light in a dark room. The FALB band eluted with 2 ml bicarbonate buffer (pH 9) and stored at -20 °C. Unconjugated FITC remained in the column. The pH of the labeled was adjusted to 7.4 and fluorescence intensity of FALB solution was measured using a Parkin-Elmer (Norwalk, CT) LSE spectrophotometer-fluorimeter with the excitation wavelength of 495 nm and an emission wavelength of 540 nm.
Animals and preparations
Eight-week-old male Wistar rats weighing 200-220 g were purchased from the Pasteur Institute (Tehran, Iran). The animals were maintained with respect to the animal welfare regulation in animal house (6 rats in each group) in a regulated environment (25 ± 1 °C; 50 to 55% relative humidity; 12 h light/dark cycle), with free access to food (Pars Dam Co., Iran) and water. All procedures were approved by the Ethical Committee of the Isfahan University of Medical Sciences, and conducted in accordance with the ‘Principles of Laboratory Animal Care’ (National Institutes of Health publication no. 86-23, revised 1985).
The animal surgery was performed between 8-8:30 AM. Briefly rat was anesthetized by intramuscular injection of ketamine (100 mg/kg) and xylazine (10 mg/kg) solution. The animals were then placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA). The scalp and underlying soft tissues were removed. The animal was drilled stereotaxicaly a burr hole in the right midline 3 mm anterior to bregma and 40 μl PG E2 solutions (62.5, 125, 250, 500 μg PG E2 diluted in normal saline) was infused slowly into the right forebrain by a fixed mycrosyrange for over 10 minutes, while only normal saline was infused into the right forebrain of the control group (sham treatment group). The hole was occluded and the animal was closely monitored for 60 min. The animal was then injected with 1 ml fluorescein labeled albumin (100 μg albumin/ml), via internal right jugular vein. The rats were killed by decapitation at different infusion time points: 4, 8, 16, 24 and 48 h. The forebrain was then rapidly removed and each forebrain hemisphere was weighed up and homogenized in 5 ml of ice-cold saline Triton X-100. The homogenate was centrifuged for 10 min at 16000 g and fluorescence intensity was measured in supernatant at the excitation wavelength of 495 nm and emission wavelength of 540 nm. Results were expressed relative to an arbitrary scale of fluorescence intensity.
Statistical analysis
Statistical analysis was performed using the Statistical Package of Social Science (SPSS) version 17. Data are presented as means ± standard deviation (SD). Experimental data from the right forebrain hemisphere are compared to their own left forebrain hemisphere values by pairing Student's t-test. The level of significance was set at P<0.05. The corresponding parameters were also compared using analysis of variance (ANOVA).
RESULTS
Changes in BBB permeability to fluorescein-labeled albumin were measured at right and left forebrain hemispheres after infusion of the range concentrations of PGE2 at different infusion time points. The results are summarized in Table 1. The fluorescence intensities measured in the right and left forebrain hemispheres of the control group (0.0 μg PG E2) were almost identical. Four hours after infusion of PG E2 at doses higher than 250 μg (see method section) fluorescence intensity increased in the right forebrain supernatant, even if it was not statistically significant. However, the fluorescence intensities in the right forebrain suprnatant markedly increased 16 h after infusion of 500 μg PGE2, which returned to near control values after 48 h. The fluorescence intensities in left forebrain suprnatant increased compared to the sham operation group.
Table 1.
The effect of PGE2 infusion on the translocation of fluorescein-labeled albumin into blood brain barrier.
Fig. 1 shows the time course of translocation of FALB after 500 μg PGE2 infusion. The values of fluorescence intensities increased in the treatment groups 16 and 24 h after infusion compared to corresponding sham values (P<0.05). Four hours after perfusion, the levels of tissue fluorescence intensities increased greatly in right forebrain compared with the left forebrain supernatant.
Fig. 1.
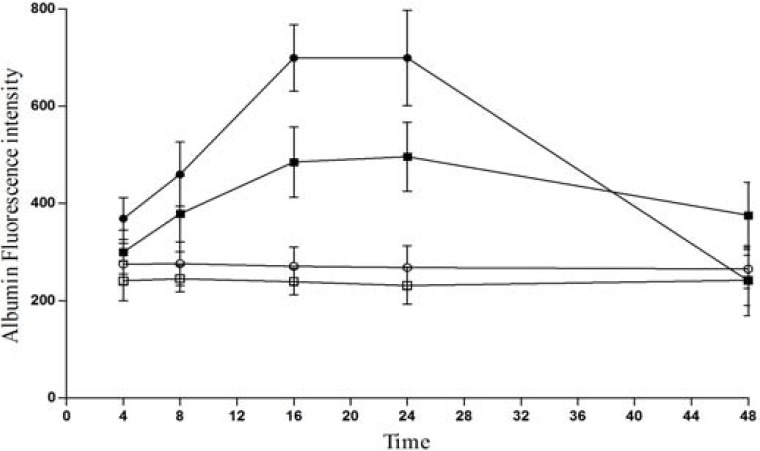
The time course of translocation of fluorescent labeled albumin. Animals were injected with 1 ml fluorescein labeled albumin (100 μg albumin/ml), via internal right jugular vein and PGE2 at doses of 500 μg was infused into the rat right brain hemisphere (see Method section), The fluorescence intensities were measured at infusion time points of 4, 8, 16, 24, and 48 h after infusion in right (-●-) and left (-■-) forebrain supenatant of treatment groups and right (-○-) and left (-□-) sham groups.
Fig. 2 compares the fluorescence intensities measured in the suprnatants prepared from right and left brain hemispheres 16 h after infusion of different doses of PGE2 (62.5, 125, 250 and 500 μg). As it can be seen the fluorescence intensities are significantly higher in the suprnatants prepared from right forebrain (P<0.05). The results indicated that translocation of FALB is dose dependent with respect to infeusion of PGE2, noticeably FALB would be augmented with doses of PGE2 higher than 125 μg.
Fig. 2.
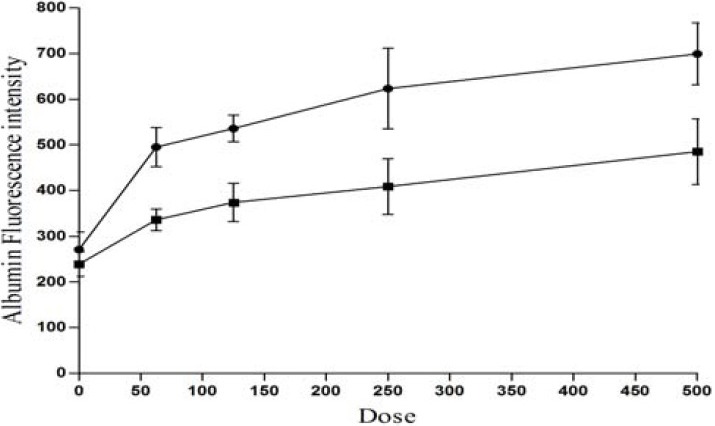
Dose response curve of PGE2 with respect to the fluorescence intensities. Animals were injected with 1 ml fluorescein labeled albumin (100 μg albumin/ml), via their internal right jugular vein and PGE2 at dose ranges from 0 to 500 μg were infused into the rat right brain hemisphere (see Method section), and the fluorescence intensities were measured 16 h after infusion at right (-●-) and left (-■-) brain supenatant.
DISCUSSION
The present study assessed for the first time the contribution of PGE2 accumulation on BBB damage and infiltration of serum albumin in brain in an in vivo model. Furthermore, to the best of our knowledge, a detailed time course and dosages of PGE2 function, and its relation to the development of BBB disruption had not been previously investigated. Post-ischemic inflammation has recently emerged as an important factor responsible for the development of the ischemic brain injury. In this regard, the present findings indicate that increasing PGE2 concentrations in right forebrain hemispheres resulted in BBB disruption and infiltration of the labeled albumin. The translocation of labeled albumin 24 h after infusion of PGE2 is consistent with the observation of Krizanac-Bengez and coworkers who showed that the COX2 inhibitor entirely protected BBB permeability in an in vitro BBB model using rat brain microvascular endothelial cells (17).
Our findings, however, is not in agreement with the suggestion made by McCullough and colleagues that the disruption of the PGE2 receptors aggravated neuronal death after transient forebrain ischemia, proposing that PGE2 has a neuroprotective effect on postischemic injury (18). In addition, the effect of PGE2 in the in vitro studies has been controversial, with results showing both toxic and protective effects on neuronal survival (11). However, Interpretation of these results, is difficult in part due to the complexity and heterogeneity of the brain regions with multiple actions of PGE2 receptors and treatment protocols. A potential link between PGE2 expression and PGE2 synthesis, which are involved in BBB damage, should be also considered as the main role of PGE2 -mediated mechanism involved in brain cell inflammatory conditions (19,21).
It is suggested that the restoration of cerebral blood flow after ischemia may provoke damage to the BBB, which may cause infiltration and aggravate brain edema (3). These observations shed more light into the specific role of the PGE2 function in ischemic brain injury, and might have important implications for the potential use of COX inhibitors or agents modulating PGE2 synthesis in different clinical stages of cerebral ischemia.
The model presented in this study demonstrated that PGE2 accumulation in the brain is responsible for the expansion of BBB disruption frankly, which is well-known factor for brain damage after cerebral ischemia (21,22). This is interpreted as being consistent with the COX-2 activation after ischemic brain injury (23,24).
CONCLUSION
It is concluded that the cellular and microvascular response to PG E2 action is mediated through changes in the ultrastructure of the brain accompanied by an increase in BBB permeability.
ACKNOWLEDGMENTS
This work was supported by the Research Council of Khorasgan Branch, Islamic Azad University, Isfahan, Iran. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
REFERENCES
- 1.Dirnagl U. Inflammation in stroke: the good, the bad, and the unknown. Ernst Schering Res Found Workshop. 2004;47:87–99. doi: 10.1007/978-3-662-05426-0_5. [DOI] [PubMed] [Google Scholar]
- 2.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 3.Batteur-Parmentier S, Margaill I, Plotkine M. Modulation by nitric oxide of cerebral neutrophil accumulation after transient focal ischemia in rats. J Cereb Blood Flow Metab. 2000;20:812–819. doi: 10.1097/00004647-200005000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Wide therapeutic time window for nimesulide neuroprotection in a model of transient focal cerebral ischemia in the rat. Brain Res. 2004;1007:98–108. doi: 10.1016/j.brainres.2004.01.078. [DOI] [PubMed] [Google Scholar]
- 5.Candelario-Jalil E, Mhadu NH, Gonzalez-Falcon A, Garcia-Cabrera M, Munoz E, Leon OS, et al. Effects of the cyclooxygenase-2 inhibitor nimesulide on cerebral infarction and neurological deficits induced by permanent middle cerebral artery occlusion in the rat. J Neuroinflammation. 2005;2:3. doi: 10.1186/1742-2094-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Davis S, Lees K, Donnan G. Treating the acute stroke patient as an emergency: current practices and future opportunities. Int J Clin Pract. 2006;60:399–407. doi: 10.1111/j.1368-5031.2006.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pepicelli O, Fedele E, Berardi M, Raiteri M, Levi G, Greco A, et al. Cyclo-oxygenase-1 and -2 differently contribute to prostaglandin E2 synthesis and lipid peroxidation after in vivo activation of N-methyl-D-aspartate receptors in rat hippocampus. J Neurochem. 2005;93:1561–1567. doi: 10.1111/j.1471-4159.2005.03150.x. [DOI] [PubMed] [Google Scholar]
- 9.Sasaki T, Kitagawa K, Yamagata K, Takemiya T, Tanaka S, Omura-Matsuoka E, et al. Amelioration of hippocampal neuronal damage after transient forebrain ischemia in cyclooxygenase-2-deficient mice. J Cereb Blood Flow Metab. 2004;24:107–113. doi: 10.1097/01.WCB.0000100065.36077.4A. [DOI] [PubMed] [Google Scholar]
- 10.Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikeda-Matsuo Y, Ota A, Fukada T, Uematsu S, Akira S, Sasaki Y. Microsomal prostaglandin E synthase-1 is a critical factor of stroke-reperfusion injury. Proc Natl Acad Sci. 2006;103:11790–11795. doi: 10.1073/pnas.0604400103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iadecola C, Sugimoto K, Niwa K, Kazama K, Ross ME. Increased susceptibility to ischemic brain injury in cyclooxygenase-1-deficient mice. J Cereb Blood Flow Metab. 2001;21:1436–1441. doi: 10.1097/00004647-200112000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Cheung RT, Pei Z, Feng ZH, Zou LY. Cyclooxygenase-1 gene knockout does not alter middle cerebral artery occlusion in a mouse stroke model. Neuroscience Lett. 2002;330:57–60. doi: 10.1016/s0304-3940(02)00738-3. [DOI] [PubMed] [Google Scholar]
- 14.Martín A, Rojas S, Chamorro Á, Falcón C, Bargalló N, Planas AM. Why does acute hyperglycemia worsen the outcome of transient focal cerebral ischemia? Role of corticosteroids, inflammation, and protein O-glycosylation. Stroke. 2006;37:1288–1295. doi: 10.1161/01.STR.0000217389.55009.f8. [DOI] [PubMed] [Google Scholar]
- 15.Stanimirovic D, Satoh K. Inflammatory mediators of cerebral endothelium: a role in ischemic brain inflammation. Brain Pathol. 2000;10:113–126. doi: 10.1111/j.1750-3639.2000.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nargessi RD, Landon J, Smith DS. Non-separation fluoroimmunoassay of human albumin in biological fluids. Clin. Chim. 1978;89(3):461–7. doi: 10.1016/0009-8981(78)90410-2. [DOI] [PubMed] [Google Scholar]
- 17.Krizanac-Bengez L, Mayberg MR, Cunningham E, Hossain M, Ponnampalam S, Parkinson FE, et al. Loss of shear stress induces leukocyte-mediated cytokine release and blood-brain barrier failure in dynamic in vitro blood-brain barrier model. J Cell Physiol. 2006;206:68–77. doi: 10.1002/jcp.20429. [DOI] [PubMed] [Google Scholar]
- 18.McCullough L, Wu L, Haughey N, Liang X, Hand T, Wang Q, et al. Neuroprotective function of the PGE 2 EP2 receptor in cerebral ischemia. J Neurosci. 2004;24:257–268. doi: 10.1523/JNEUROSCI.4485-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cipollone F, Fazia ML, Iezzi A, Cuccurullo C, De Cesare D, Ucchino S, et al. Association between prostaglandin E receptor subtype EP4 overexpression and unstable phenotype in atherosclerotic plaques in human. Arterioscler Thromb Vasc Biol. 2005;25:1925–1931. doi: 10.1161/01.ATV.0000177814.41505.41. [DOI] [PubMed] [Google Scholar]
- 20.Khan KM, Howe LR, Falcone DJ. Extracellular matrix-induced cyclooxygenase-2 regulates macrophage proteinase expression. J Biol Chem. 2004;279:22039–22046. doi: 10.1074/jbc.M312735200. [DOI] [PubMed] [Google Scholar]
- 21.Pavlovic S, Du B, Sakamoto K, Khan KM, Natarajan C, Breyer RM, et al. Targeting prostaglandin E2 receptors as an alternative strategy to block cyclooxygenase-2-dependent extracellular matrix-induced matrix metalloproteinase-9 expression by macrophages. J Biol Chem. 2006;281:3321–3328. doi: 10.1074/jbc.M506846200. [DOI] [PubMed] [Google Scholar]
- 22.Abbott NJ. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol. 2000;20:131–147. doi: 10.1023/A:1007074420772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Couturier JY, Ding-Zhou L, Croci N, Plotkine M, Margaill I. 3-Aminobenzamide reduces brain infarction and neutrophil infiltration after transient focal cerebral ischemia in mice. Exp Neurol. 2003;184:973–980. doi: 10.1016/S0014-4886(03)00367-4. [DOI] [PubMed] [Google Scholar]
- 24.Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, et al. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]