

Abstract
Filamentous fungi are the predominant source of lignocellulolytic enzymes used in industry for the transformation of plant biomass into high-value molecules and biofuels. The rapidity with which new fungal genomic and post-genomic data are being produced is vastly outpacing functional studies. This underscores the critical need for developing platforms dedicated to the recombinant expression of enzymes lacking confident functional annotation, a prerequisite to their functional and structural study. In the last decade, the yeast Pichia pastoris has become increasingly popular as a host for the production of fungal biomass-degrading enzymes, and particularly carbohydrate-active enzymes (CAZymes). This study aimed at setting-up a platform to easily and quickly screen the extracellular expression of biomass-degrading enzymes in P. pastoris. We first used three fungal glycoside hydrolases (GHs) that we previously expressed using the protocol devised by Invitrogen to try different modifications of the original protocol. Considering the gain in time and convenience provided by the new protocol, we used it as basis to set-up the facility and produce a suite of fungal CAZymes (GHs, carbohydrate esterases and auxiliary activity enzyme families) out of which more than 70% were successfully expressed. The platform tasks range from gene cloning to automated protein purifications and activity tests, and is open to the CAZyme users’ community.
Keywords: recombinant proteins, expression platform, CAZymes, Pichia pastoris, factorial approach, automation, glycoside hydrolase
Introduction
Lignocellulosic biomass is recognized as a sustainable source of mixed sugars for producing second generation biofuels by fermentation, and for the synthesis of biomaterials (Lynd et al., 2008). In industry, saccharification of lignocellulose is performed enzymatically using fungal enzyme cocktails (Himmel et al., 2007). Due to the recalcitrant nature of lignocellulosic biomass, the development of more efficient fungal enzyme preparations is an active research field with the sequencing of hundreds of fungal genomes at the US Department of Energy Joint Genome Institute 1,000 fungal genome project, (Grigoriev et al., 2014). This wealth of genomic information opens new horizons and opportunities to improve the enzymatic conversion of biomass.
However, due to throughput differences the production of recombinant fungal proteins lags far behind that of data generated by –omic approaches, which creates a bottleneck in the process of unraveling the function of novel CAZymes. Although historically filamentous fungi (e.g., Trichoderma reesei, Aspergillus niger) and other yeast (e.g., Saccharomyces cerevisiae, Yarrowia lipolytica) have been used for expressing native or recombinant fungal enzymes (Dashtban et al., 2009), the yeast Pichia pastoris has become the premier example of yeast species used for the production of recombinant proteins (Cregg et al., 2000; Damasceno et al., 2012). Advantages of this yeast include the use of efficient and tightly regulated promoters, higher expression levels for lower cost than insect and mammalian cells. Compared to prokaryotic expression, it provides better protein folding, allows post-translational modifications (N- and O-glycosylation, methylation, and acylation), and is able to secrete high levels of recombinant proteins in the culture medium. In addition, it secretes very low levels of endogenous proteins none of which has been reported to be active on lignocellulosic biomass, which simplifies protein purification.
Finally, because filamentous fungi and yeast are phylogenetically close, P. pastoris is the organism of choice for expressing different classes of active fungal CAZymes, such as GHs (Bauer et al., 2006; Couturier et al., 2011a,b), CE (Topakas et al., 2010, 2012) and AA enzymes [i.e., lytic polysaccharide mono-oxygenases (Kittl et al., 2012; Bey et al., 2013)]. Having these many advantages over other expression systems makes P. pastoris an organism of choice for the production of industrial enzymes (Rabert et al., 2013).
Despite obvious advantages, we have found that heterologous expression of fungal enzymes in P. pastoris remained time-consuming, and reasoned that protocols for transformation, screening of transformants, cultures, purification of recombinant proteins could benefit from streamlining to try to increase protein production-throughput. In this study, we have used in-house fungal genes encoding CAZymes [GH5, GH11, GH45 (Couturier et al., 2011a,b)] that we previously successfully expressed in P. pastoris using pPICZα inducible vector, to establish a simpler standard protocol. In this paper, we have analyzed, and modified whenever possible, different steps of the protocol set-up by Invitrogen for expressing proteins in P. pastoris from the design of expression constructs to the assay of purified proteins, with the ultimate goal of setting-up a lab-scale recombinant protein production facility. This platform now makes the link between upstream expert annotation of CAZymes (www.cazy.org) (Lombard et al., 2014) and downstream automated assay of enzymatic activities (Lafond et al., 2012) and crystallogenesis (Sulzenbacher et al., 2002).
Materials and Methods
Enzymes
Taq (screening purpose) and Platinum pfx (cloning purpose) DNA polymerases (Invitrogen) were used in PCR experiments.
Expression Constructs
Coding sequences to express were PCR amplified, and then inserted into Invitrogen vectors pPICZαA (Couturier et al., 2011a,b), pGAPZαA and pBGP1 (Sasagawa et al., 2011) (this study) inframe with N-terminal yeast α-secretion factor and C-terminal His6 tag encoding sequences. Plasmid pBGP1 was obtained by removing the Gateway cassette from pBGP1-DEST (Sasagawa et al., 2011) by XhoI/XbaI restriction digestion. The primers used were: GGGGCTAAAGAACTCGAGAAAAGAGAGGCTGAAGCTCTCCCCCAAGCACAAGGTG and TTTTCTAGACCCGCCGGGAGAGCATTGATAG for Podospora anserine GH5 (GenBank: HM357135.1), GGGGCTAAAGAACTCGAGAAAAGAGAGGCTGAAGCTGCCCCCGGTGAGCTTCCT and TTTTCTAGACCGTGTGTCTGGACATAAATGTC for P. anserine GH11 (GenBank: HM357137.2), and GGGGCTAAAGAACTCGAGAAAAGAGAGGCTGAAGCTGATGTCCCACTTTGGGGCCAATG and TCTAGACCTTCGTCCGTACGAGCACA for P. pastoris GH45 (GenBank: CAY71902.1). Except pBGP1, resulting recombinant expression plasmids were linearized with PmeI (pPICZαA) or AvrII (pGAPZαA) restriction enzymes.
Preparation of Frozen Electrocompetent Cells and Transformation
Cells were grown as described by Invitrogen (easyselect_man.pdf) with the following modification: when OD600 was about 1.4, cells recovered from 250 ml culture were resuspended in 100 ml YPD supplemented with 20 ml 1 M HEPES pH8 and 2.5 ml 1 M DTT, and stored at 30°C for 15’. After washing as described in easyselect_man.pdf, they were eventually resuspended in 0.5 ml of cold 1 M sorbitol and 60 μl aliquots were frozen at -80°C.
Electroporation was performed as described in easyselect_man.pdf using 60 μl of thawed electrocompetent cells.
Liquid Phase Selection and Expression Screening
General Strategy
Both were performed in 24-wells deepwell plates. Indicated volumes are for one well. An aliquot (200 μl) of sorbitol cell suspension was used to inoculate 2 ml YPD supplemented with 100 μg/ml Zeocin, which was then incubated for 3 days at 30°C under 225 rpm rotary shaking. This step has two aims: it selects transformed cells as do Zeocin-containing agar plates, and serves as a pre-culture for the following recombinant protein expression screening. The latter was performed by inoculating 5 ml Zeocin-free growth medium with 50 μl pre-culture and then incubating at 30°C under shaking. At the end of the expression period, culture supernatants were recovered by centrifugation for 5’ at 4000 g and their protein content analyzed by SDS-PAGE and/or enzymatic activity assay.
Constitutive Expression (pGAPZαA)
Protein expression screening was performed by inoculating 5 ml YPD with 50 μl pre-culture and then incubating for 4 days at 30°C under shaking.
Inducible expression (pPICZαA)
Protein expression screening was performed by inoculating 5 ml BMGY (easyselect_man.pdf) with 50 μl pre-culture, and then incubating overnight at 30°C under shaking. The plate was then spun as above and the cell pellet was resuspended in 1 ml BMMY (easyselect_man.pdf). The culture medium was daily supplemented with 3% methanol for 4 days.
Automated Purification of Recombinant Proteins
Non-Automated Pre-Purification Steps
(i) At the end of the expression period, 24-wells deepwell plates were spun for 5’ at 4000 g to pellet the cells. Supernatants were transferred well to well to another 24-wells deepwell plate, and each well was supplemented with 1/10 supernatant volume of 250 mM Tris pH8. (ii) Two hundred microliters of a 50% suspension of Ni SepharoseTM High Performance (GE Healthcare 17-5268-02) were loaded into each well of a 96-wells deepwell filter plate (AcroPrepTM Advance 1 ml, 1 μm Glass Fiber, PALL PN 8131). (iii) 24-wells deepwell plates containing the supernatants were put on a TECAN Freedom Evo robot, and the 96-wells deepwell filter plate on the first manifold of the same robot (Navarro et al., 2010).
Automated Steps
Indicated volumes are for one well. The automate removed ethanol from Sepharose beads by vacuum aspiration for 30″ at -50 millibars (mb), and then added 200 μl buffer A (10 mm Tris pH7.8, 150 mM NaCl and 10 mM imidazole). After 2′, buffer A was eliminated by vacuum aspiration for 30″ at -50 mb. This step was repeated once. The robot transferred 800 μl of each culture supernatant from the 24-wells deepwell plate to each well of the 96-wells deepwell filter plate. After 3′ incubation, the supernatant was eliminated by vacuum aspiration for 60″ at -50 mb. If more than 1 ml supernatant was to be processed, this step was repeated the required number of times. At the end of the binding step, the robot washed the beads twice with buffer A as above, and then dried them by an additional 5″ -700 mb vacuum aspiration. The robot transferred the filter plate on top of a 96-wells microplate that had been put on the second manifold of the robot beforehand, and added 200 μl buffer B (10 mm Tris pH7.8, 150 mM NaCl and 500 mM imidazole) to each well of the filter plate. After 5′ incubation, the robot transferred the elution volume from each well of the filter plate to the corresponding well of the 96-wells microplate underneath by two successive vacuum aspirations for 20″ at -50 mb. Complete recovery of elution volume was achieved by performing a second vacuum aspiration for 5″ at -700 mb. The filter plate was then transferred back to the first manifold where beads were regenerated by performing the following operations. (i) Buffer B (200 μl) was added and after 2′ incubation removed by vacuum aspiration for 30″ at -50 mb. This step was repeated once. (ii) Buffer A (200 μl) was added and after 2′ incubation removed by vacuum aspiration for 30″ at -50 mb. (iii) Finally, beads were supplemented in 400 μl of 20% ethanol. The filter plate was sandwiched between two adhesive aluminum sheets (Dutscher reference 106524) and stored at 4°C.
Enzymatic Activity Assays
Indicated volumes are for one well. GH5, GH11, and GH45 enzymatic activities were assayed by measuring the hydrolysis of 1% azo-glucomannan, azo-xylan, and azo-CM-cellulose (Megazyme), respectively. The assay is based on GH-dependent hydrolysis of a dyed substrate that produces low-molecular weight dyed fragments which remain in solution on addition of a precipitant solution. High-molecular weight material is removed by centrifugation and the color of the supernatant is measured. In practice, culture supernatants or purified enzymes (10 μl) were added to 50 μl of azo-substrate solution in 50 mM sodium acetate pH4.8 in 96-well plates. The latter were heat-sealed with aluminum sheet using the robot PlateLoc sealer, and incubated at 40°C for 2 h under 800 rpm rotary shaking. Ethanol (150 μl) was added to precipitate non-hydrolysed substrate. After 10′ at room temperature, plates were centrifuged at 4000 g for 5′. Supernatants were collected and their optical density measured at 590 nm. Activities were expressed as percentage of azo-substrate used. The 100% value was the OD590 of a well to which no ethanol and no enzyme had been added.
Results and Discussion
In a previous work, three GH (GH5, GH11, and GH45) were successfully expressed in P. pastoris by strictly following the protocol proposed by Invitrogen (easyselect_man.pdf). Analysis of the original protocol resulted in a list of different steps that could be subject of optimization in terms of time, cost, or convenience. The list was made of: (1) using frozen competent cells instead of extemporaneously preparing competent cells; (2) using less plasmid for electroporation; (3) skipping the selection of recombinant clones on plate; (4) replacing tubes and flasks with deep-well plates in the screening process. In addition to simplifying the original protocol, the purification of secreted His-tagged proteins was automated to increase throughput.
Expression Constructs
In a first trial, GH5, GH11, and GH45 coding sequences were sub-cloned by restriction/ligation (R/L) into episomal pBGP1 (Sasagawa et al., 2011) and integrative pGAPZαA and pPICZαA expression vectors as described in Section “Materials and Methods,” and recombinant expression was evaluated by SDS-PAGE in the supernatant of culture media. Using integrative vectors was expected to result in genetically stable P. pastoris strains as they are integrated into the yeast chromosome at the cost of more laborious screening, whereas an episomal vector could be isolated by a simple plasmid preparation procedure. In addition, episomal vectors have higher transformation efficiencies and more than one copy per cell. It was therefore of interest to assess whether these features were correlated to recombinant protein expression level. Results are reported in Figures 1A–C. Expression of GH5 and GH45 was plasmid-dependant: GH5 was better expressed by pPICZαA than by pGAPZαA whereas it was the opposite for GH45. No GH11 was visible in the culture medium of any plasmid condition. To check whether GH11 was expressed, it was purified by affinity chromatography on Ni-NTA as described in Section “Materials and Methods” and analyzed by SDS-PAGE (Figure 1D). GH11 was slightly better expressed by pPICZαA than by pGAPZαA, although at levels definitely lower than those of GH5 and GH45. On the basis of above results, integrative vectors pGAPZαA and pPICZ were chosen for expressing fungal CAZymes. Two attempts to make DNA constructs easier resulted in negative expression results: the use of Gateway cloning instead of R/L (Additional File 1), and the use of PCR to both generate and linearize integrative expression constructs in a single experiment (Liu et al., 2008; Yu et al., 2012) (Additional File 2). In conclusion, the classical R/L technique followed by linearization by restriction of expression integrative constructs before transformation was used in the rest of this study, as proposed by Invitrogen (easyselect_man.pdf).
FIGURE 1.
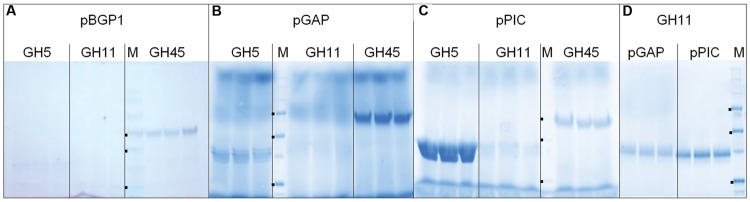
Expression results of different DNA constructs. (A–C), 25 μl of culture supernatant using the indicated expression plasmid were directly analyzed by SDS-PAGE. (D) GH11 from (B) and (C) was purified by affinity chromatography on Ni-NTA, and 25 μl of elution fraction were analyzed by SDS-PAGE. M, molecular weight markers. (A) 180, 130, 100, 70 (■), 55 (■), 40, 35, 25 (■), 15, 10 kDa. (B–D) 250, 150, 100, 75 (■), 50 (■), 37, 25 (■), 20, 10 kDa.
Transformation
Frozen Electrocompetent Cells
Invitrogen advices not using frozen electrocompetent cells because freeze/thaw cycles reduce cell transformation efficiency. However, extemporaneously preparing electrocompetent cells is time consuming, which proportionally increases the cost price of recombinant proteins. The protocol used throughout this study and described in Section “Materials and Methods” was kindly provided by one of our collaborators (EIPL) and proved successful with all our expression constructs. No attempt was made to substitute electroporation with chemical transformation because the transformation efficiency was considered too low for the liquid phase transformation described below, although a recently launched chemical compound (BG2) seems promising in this respect (Filyak et al., 2013).
Amount of DNA for Transformation
As mentioned above, highest transformation efficiencies are not required in a recombinant protein expression facility because few clones are sufficient for expressing a recombinant protein. Since the number of individual colonies on agar plates is proportional to the amount of DNA used for transformation for a given transformation efficiency, we assessed the possibility to reduce the amount of DNA used for transformation. Invitrogen protocol suggests using 10 μg of linear expression construct per transformation. However, we found that as low as 0.5 μg of linear expression construct was enough for obtaining the necessary number of transformants for expressing recombinant proteins. Incidentally, using less DNA also allows using plasmid DNA mini-prep and not midi-prep kits, which is both faster and cheaper. An additional gain of time could be to skip the expression construct linearization by restriction, but we found that electroporating yeast cells with 0.5 μg of non-linearized integrative expression construct failed to give rise to colonies on agar plates (not illustrated). In conclusion, 0.5 μg of linear expression construct were decided to be, respectively, the default value and DNA state used on the expression platform.
Plating-Free Transformation
Invitrogen’s protocol implies plating cells on Zeocin agar plates after electroporation for selecting transformants. We reasoned that there were no reason why transformants could not be selected in liquid rather than in solid phase (i.e., on agar) provided the selection reagent Zeocin was included in the culture medium. Thereafter, we refer to selection of transformants by plating on Zeocin-containing agar plates as “solid phase” selection, and to selection of transformants by growing in YPD-Zeocin as “liquid phase” selection. The only weakness of the liquid phase option is that transformants are screened as a pool and not as individual clones. As a consequence, the expression screen result is the average of all clones that have been selected by the antibiotic, which can lead to suboptimal expression results.
We compared both approaches to assess whether having individual clones was critical for our project. X33 cells were transformed with linearized pGAPZαA constructs bearing coding sequences for GH5, GH11, or GH45. After the sorbitol step, 200 μl of cell suspension in sorbitol were plated and then incubated at 30°C for 3 days as usual, and another 200 μl of the same cell suspension in sorbitol were used for liquid phase selection as described in Section “Materials and Methods”. When clones began to appear on agar, they were individually used to inoculate 5 ml YPD-Zeocin and then allowed to grow and express the recombinant protein for 4 days. Expression of recombinant proteins of all solid and liquid phase experimental points was assessed by assaying GH5, GH11, and GH45 enzymatic activities. Results (Figure 2) definitely show that, at least for our three reference proteins, there was no difference between either selection mean. In addition to using less Zeocin than solid phase selection [2 μl in 2 ml YPD instead of 25 μl in 25 ml YPD containing 1.2% agar (YPDA; i.e., the volume poured per plate)], liquid phase selection does not preclude individual clones to be selected on plate if necessary since it uses only 200 out of 1000 μl of the sorbitol suspension of transformed cells and so an additional 200 μl aliquot can be plated and grown in parallel with the liquid phase selection. In conclusion, the liquid phase selection was chosen as default option for our facility because it allows a yes/no answer to be obtained more quickly than solid state screening. In case of negative or poor response, plated clones can be used as a rescue.
FIGURE 2.
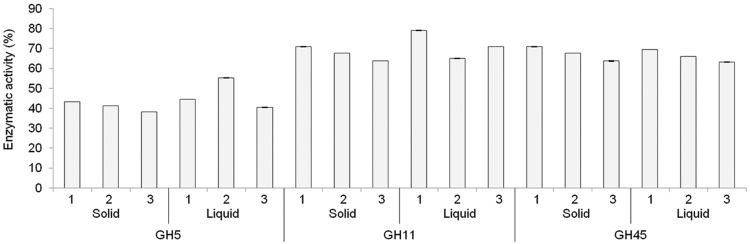
Plating is dispensable for expressing recombinant GH in Pichia pastoris. Liquid and solid phase selections were performed as described in the results section. For the solid phase selection, three individual clones (numbered 1, 2, 3 on the x axis) were grown each in triplicate in individual wells of a 24-wells deep-well plate. For the liquid phase selection, three individual wells (numbered 1, 2, 3 on the x axis) of a 24-wells deep-well were seeded each with 200 out of 1000 μl of the cell suspension in sorbitol solution after electroporation and assayed in triplicate. Results show the mean enzymatic activity assayed in culture supernatants, with standard deviations of the triplicate assay.
Culture of Transformants
Culture Dish
Based on our experience in recombinant expression screening in Escherichia coli (Noguere et al., 2012) and in filamentous fungi (Alberto et al., 2009), the ANSI/SLAS (American National Standards Institute/Society for Laboratory Automation and Screening) format was preferred to classical tubes and flasks for growing cells. This format has two advantages. First, dishes stand alone and do not require additional equipment such as tube or flask holders, which is convenient when many samples have to be processed in parallel as it is often the case on a dedicated platform. Second, it was devised for use with automates, which allows for a direct transfer from culture incubators to robot benchwork.
Although 96-wells deepwell plates have been successfully used for growing and co-expressing recombinant proteins in P. pastoris (Geier et al., 2013), 24-wells deepwell plates better fitted with the requirements of this type of platform. By accommodating larger culture volumes than 96-wells deepwell plates (5 instead of 1 ml per well), 24-wells deepwell plates act as a magnifying tool of recombinant protein expression for the following reason. Since recombinant proteins secreted by P. pastoris in the culture medium are C-terminally His6-tagged, they can be concentrated up to about 25 times by collecting them by affinity chromatography on Ni-NTA beads if elution from the affinity column is performed using 200 μl elution buffer (see further). Hence, even tiny levels of expression can be detected by SDS-PAGE analysis and Coomassie blue staining. By contrast, each well of a 96-wells deepwell plates provides five times less signal on gel for the same expression level because it can accommodate only 1 ml of culture medium. In addition, since oxygen transfer in 24-wells deepwell plates is higher than in 96-wells deepwell plates (Duetz, 2007), one may expect higher expression levels in the former than in the latter. Baffled 16-wells deepwell plates used for growing filamentous fungi (Alberto et al., 2009) were eventually not considered for use on the platform because the number of wells was too small for the intended screening throughput and their geometry unfitted for the automate six needle pipetting arm. In conclusion, 24-wells deepwell plates were chosen as default culture dish.
Managing Zeocin Consumption
Yeast expression plasmids of pGAPZ and pPICZ series confer Zeocin resistance to transformed cells, which allows for easy selection of the latter after plating on Zeocin-containing agar plates. However, Zeocin has two failings: it is expensive and degrades rapidly with time. In particular, it is light sensitive. Therefore, Zeocin-containing agar plates cannot be stored for long periods of time before use and must be protected from light throughout the process ranging from pouring to plating, which makes their handling inconvenient.
We addressed the issues of cost and degradability by testing the possibility of directly adding Zeocin to suspensions of electroporated cells in sorbitol solution rather than to agar plates.
In practice, X33 cells were electroporated with episomal Gateway plasmid pBGP1-DEST bearing GH5 coding sequence. After the sorbitol step, 100 out of 1000 μl cell suspension were spread on either a Zeocin plate made of 25 ml of YPDA supplemented with 25 μl of 100 mg/ml Zeocin solution, or on agar plates made of 25 ml of Zeocin-free YPDA. In the latter case, cell suspensions were supplemented with 0, 1, 5, 10, or 25 μl of 100 mg/ml Zeocin solution immediately before spreading on Zeocin-free plates. Plates were then incubated at 30°C for 4 days, and the number of colonies was compared. Results (Figure 3) indicate that adding 1 μl of Zeocin solution directly to the cell suspension just before plating provided roughly the same result as plating the same volume of the same cell suspension on an agar plate supplemented with 25 μl of Zeocin solution.
FIGURE 3.

Managing Zeocin consumption. The experiment was performed as described in the results section. After 4 days at 30°C, plates were photographed. In the upper row, Zeocin was added directly to the cell suspension before plating on Zeocin deficient plates. Numbers on top indicated the volume of Zeocin mother solution added to 150 μl of cell suspension. The lonely plate below the upper row is the reference plate. In that case, the cell suspension was not supplemented with Zeocin, but was plated on Zeocin-containing plate. This reference plate was located between plates “0” and “1” of the upper row because it roughly contained an intermediate number of clones between these two plates.
Adding Zeocin to the cell suspension immediately before plating rather than in the agar plate allows (i) dividing Zeocin consumption by about 50 for the same selection efficiency, (ii) preparing YPDA plates in advance of use and then storing them without particular caution except asepsis. In conclusion, if the liquid phase selection described in a previous paragraph cannot be performed because isolating clones is mandatory for some reason, this can be done at lower cost and more conveniently by adding Zeocin to the cell suspension just before plating rather than in the YPDA plate.
Prevention of Bacterial Contamination
Because integrative plasmids are generally used for transforming P. pastoris cells for which maintaining antibiotic selection pressure after transformants have been selected is unnecessary, and because the antibiotic used to select transformants (Zeocin) is expensive, no antibiotic is used for growing clones thereafter. Sometimes, however, the absence of antibiotic in rich culture media such as YPD allows bacterial contaminations to occur with the mandatory loss of the contaminated batch. Penicillin and Streptomycin are routinely used to prevent bacterial contaminations in eukaryotic cell line cultures. Being eukaryotes, yeasts should not be sensitive to antibiotics commonly used by microbiologists (ampicillin, chloramphenicol, kanamycin, tetracycline), which could thus be added to the culture medium without deleterious effect while preventing such contaminations.
To check that antibiotics had really no effect on yeast growth, a clone of P. pastoris cells expressing GH5 from pGAPZαA was grown for 4 days in YPD in the presence or absence of the antibiotics listed above at concentrations used in laboratories to select resistant E. coli. Biomass was assessed by measuring the optical density of the culture at 600 nm at the end of that period, which definitely demonstrated the total innocuousness of this series of antibiotics (Figure 4A). The amount of recombinant protein secreted in the culture medium was next compared. To that end, His6-tagged GH5 was concentrated by affinity chromatography on Ni-NTA beads under different conditions that are detailed in the next paragraph. Results (Figure 4B) showed no difference between protein expression in the presence or absence of antibiotic, suggesting that antibiotics have no negative effect on the over expression of a recombinant protein in P. pastoris. In conclusion, if bacterial contaminations are an issue commonly used antibiotics can provide good protection at much lower cost than Zeocin, and without effect on P. pastoris cell growth or protein expression.
FIGURE 4.

Antibiotics have no effect on P. pastoris growth nor on protein expression. (A) Effect of antibiotics on cell growth. The y axis is the optical density of a 1/300 dilution of the culture. None, no antibiotic; Amp, 100 μg/ml ampicillin; Chlo, 34 μg/ml chloramphenicol; Kana, 50 μg/ml kanamycin; Tetra, 15 μg/ml tetracycline; Zeo, 100 μg/ml Zeocin. (B) Recombinant proteins purified by affinity chromatography on Ni-NTA beads from 4 ml culture. From 100 μl of elution volume, 15 μl were analyzed by SDS-PAGE along with 15 μl of non-purified supernatant (lanes S). M, molecular weight markers; from top to bottom: 116, 66.2, 45, 35, 25, 18.4, 14.4 kDa. Numbers 50, 100, 200 in lanes Chlo and Tetra indicate the volume in μl of 50% Ni-NTA beads suspension used. See results for details.
Protein Purification
Recombinant proteins were expressed as fusion proteins with an N-terminal signal peptide and a C-terminal His6-tag. This combination has two consequences. First, recombinant proteins are secreted in the culture medium as are biomass-degrading enzymes by filamentous fungi in their native environment. Second, this diluted protein solution can be concentrated up to about 25 times (compare, for example, lanes S to any other lane in Figure 4B) by collecting tagged proteins by affinity chromatography on Ni-NTA beads. The latter has two requirements: high specific binding is favored by slightly alkaline pH (about 8), and low non-specific binding requires high salt concentrations. Since YPD is devoid of any added salt and its pH ≈6, the addition (Figure 4B) of 0.3 M NaCl with (lanes N8) or without (lanes N) 100 mM Tris pH8 was compared to no addition (lanes 0). Results indicate that such additions were not required. However, when the culture medium pH is willingly maintained acidic (pH3) as in the fractional factorial approach described in the next paragraph, buffering using a final concentration of 100 mM Tris pH8 is mandatory before binding. The amount of Ni-NTA beads proved not limiting since increasing them from 50 to 200 μl of a 50% beads suspension did not increase the recovery yield (Figure 4B, lanes Chlo and Tetra).
Since deepwell plates can be directly transferred from a shaking incubator to a robot benchwork, the recovery of His6-tagged recombinant proteins by affinity chromatography on Ni-NTA was automated. To that end, Ni-NTA beads were used in deepwell filter plates as described in Section “Materials and Methods.” Note that 100 μl of beads per well were used because using 50 μl provided less reproducible results (not illustrated). The flowchart of the automated process is illustrated by Figure 5, and a thorough description of the robot can be found in (Navarro et al., 2010). After elution, recombinant His6-tagged proteins were analyzed by gel electrophoresis using pre-cast gels, and by assaying their enzymatic activity (Figure 6).
FIGURE 5.
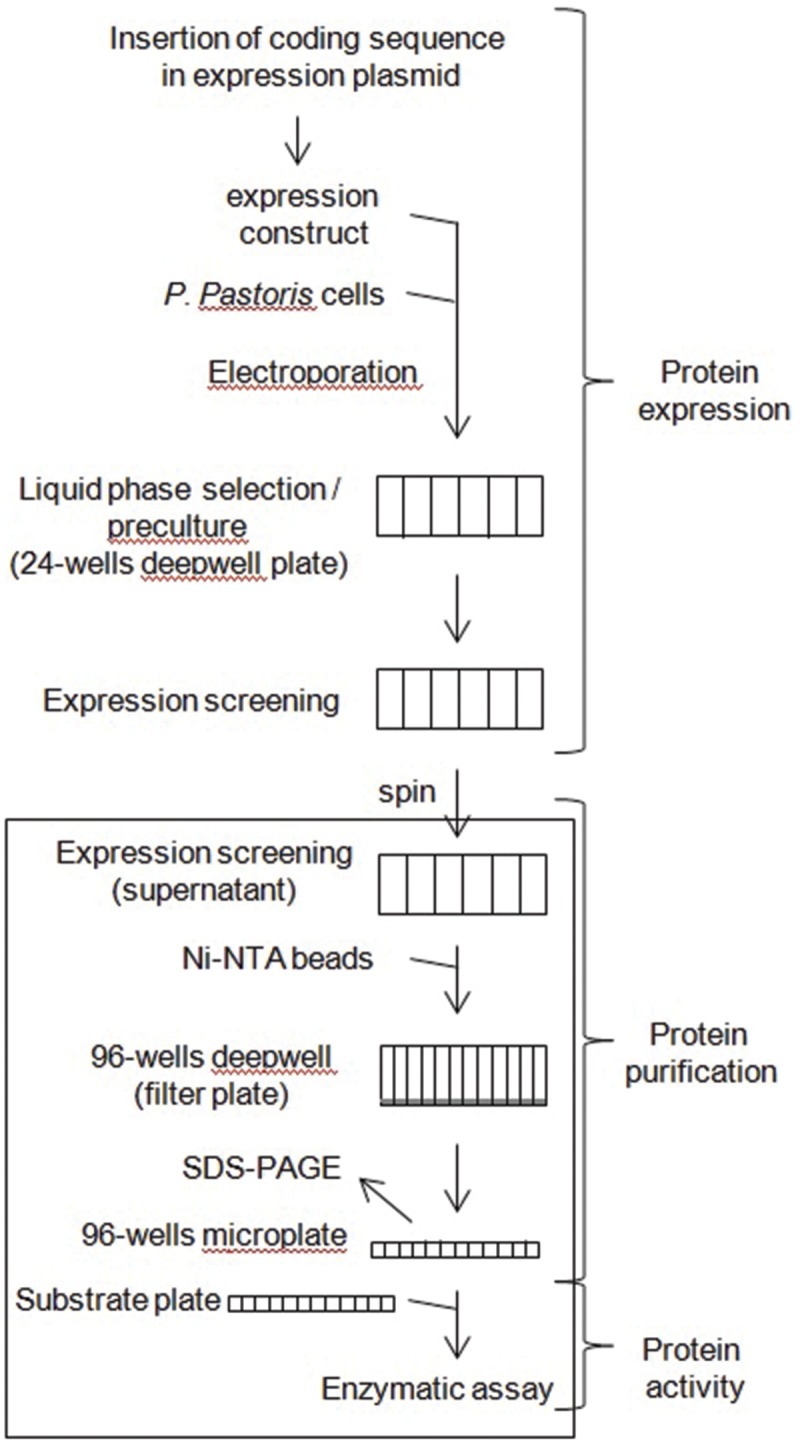
Pichia pastoris recombinant protein expression platform. Automated purification by affinity chromatography on Ni-NTA and enzymatic activities assays of His6-tagged recombinant proteins are boxed. See Section “Materials and Methods” for details.
FIGURE 6.

Constitutive and inducible expression of a recombinant protein in P. pastoris using fractional factorial approaches. Recombinant GH11 was purified by affinity chromatography on Ni-NTA as described in Section “Materials and Methods” and in Figure 5, and analyzed by SDS-PAGE and enzymatic assay. (A) Constitutive expression using pGAPZαA. (B) Inducible expression using pPICZαA. Parameter value combinations of the fractional factorial approach are indicated on top of each experimental point. Glu, Glucose; Gly, Glycerol; Sac, Saccharose; Sor, Sorbitol. M0.1, M1, M3, M10 stand for 0.1, 1, 3, 10% methanol concentration, respectively. M, molecular weight markers (same as Figure 1A; GH11 migrates as the 40 kDa band). See Additional File 3 for details.
Expression Conditions
It is our experience that modifying expression parameters in E. coli by trial and error is less effective than testing the same parameters in a factorial approach, mainly because parameter interaction is lost in the former case (Benoit et al., 2007; Noguere et al., 2012). Therefore, GH11 expression was tested in two different fractional factorial approaches using pGAPZαA and pPICZαA vectors, respectively. A detailed description of the method and of the results is in Additional File 3. In short, both approaches converged to the conclusion that expression was proportional to the temperature, and that an alkaline pH was more favorable than an acidic one (Figure 6).
Conclusion
The recombinant protein production facility using P. pastoris should avoid using episomal and/or Gateway vectors because the former uselessly request maintaining expensive Zeocin selection throughout the expression process, and the latter failed at efficiently expressing three reference proteins. We have observed that expressing three reference CAZymes was plasmid-dependent. However, since testing both constitutive and inducible expression in parallel might prove impractical for large projects, a serial approach is more realistic. In case of negative result, swapping plasmids is made easy by the identity of cloning regions and reading frames of both vectors. Expression constructs should better be made using classical R/L followed by linearization by restriction. However, large projects may require using synthetic genes with yeast-optimized codons. After electroporation, transformants should be selected with Zeocin in liquid phase as a pool and not as individual colonies on agar plates. However, since electroporated cells can be stored for weeks at 4°C as sorbitol suspensions and only 200 μl out of 1000 are used for liquid phase selection, another 200 μl aliquot should be plated in parallel to later screen for higher producers (i.e., bearing gene multi-copies) if required. In addition, in case of bacterial contamination it is possible to start a new culture from liquid phase selection pre-culture (i.e., without the need for another electroporation) in the presence of one or more of the anti-microbial antibiotics tested in the present study. For large projects, basic expression conditions (standard culture media, 27°C) are preferred in first intention. By contrast, fractional factorial approaches may be performed in case of single protein project low expression yields and for high-added value proteins. In the ANSI/SLAS format, 24-wells deepwell plates are used for growing yeasts, liquid phase selection of transformants and expression screening. Recombinant proteins secreted in the culture medium are purified and concentrated by automated affinity chromatography on NiNTA beads.
A comparative flowchart of the classical protocol set-up by Invitrogen (pgapz_man.pdf) and of the simplified protocol described here for expressing biomass-degrading fungal enzymes is provided in Figure 7. Overall, the simplified protocol allows dividing by two the time required to go from transformation to expression screening, and was used as a basis for setting-up a recombinant protein production facility for the heterologous expression of fungal CAZymes in P. pastoris (Figure 5). Table 1 summarizes all the results obtained using P. pastoris as host for expressing fungal biomass-degrading enzymes in replacement of native or recombinant expression in other fungi such a A. niger and T. reesei (Levasseur et al., 2004; Poidevin et al., 2009). Switching from the original Invitrogen protocol to the simplified protocol described in this report and now routinely used on the platform has resulted in an increased use of recombinant expression in yeast from a few coding sequences in 2011 to a total of more than 40 today, out of which more than 50% were expressed at high level (>100 mg/L). The fungal CAZymes functionally produced originate from several filamentous fungi and belong to different CAZy families within the GH, CE, and AA classes. The platform is accessible to the community of CAZyme users upon request.
FIGURE 7.

Comparative flowchart of Invitrogen (left) and our (right) protocol for expressing recombinant proteins in P. pastoris. In this example, constitutive expression was chosen. Numbers on the left and on the right are the estimated number of days required by the considered step, with their sum (total) at the bottom.
Table 1.
The different enzymes expressed on the platform are reported.
| Fungal strain | CAZy family | CBM | Predicted activity | Genbank ID | cDNA (bp) | Produced | Yield | Reference |
|---|---|---|---|---|---|---|---|---|
| Ustilago maydis | GH3 | – | β-glucosidase | 3634215 | 2619 | No | – | – |
| Podospora anserina | GH5 | – | β-1,4 mannanase | 6194921 | 1236 | Yes | >100 | Couturier et al., 2011a |
| P. anserina | GH5 | – | β-1,4 mannanase | 6187561 | 1388 | No | – | – |
| P. anserina | GH6 | 1 | Cellobiohydrolase | 6187194 | 1544 | Yes | >500 | Poidevin et al., 2013 |
| P. anserina | GH6 | – | Endoglucanase | 6187311 | 1319 | Yes | >500 | Poidevin et al., 2013 |
| P. anserina | GH6 | 1 | Cellobiohydrolase | 6188027 | 1258 | Yes | >500 | Poidevin et al., 2013 |
| P. anserina | GH10 | 1 | β-1,4 xylanase | 6188644 | 1387 | No | – | – |
| P. anserina | GH11 | – | β-1,4 xylanase | 6197075 | 742 | No | – | – |
| P. anserina | GH11 | 1 | β-1,4 xylanase | 6187330 | 1060 | Yes | <100 | Couturier et al., 2011a |
| P. anserina | GH26 | 35 | β-1,4 mannanase | 6188179 | 1466 | Yes | >100 | Couturier et al., 2011a |
| P. anserina | GH27 | – | α-galactosidase | 6195125 | 1370 | No | – | – |
| U. maydis | GH27 | 35 | α-galactosidase | 3632217 | 1719 | No | – | – |
| P. anserina | GH43 | – | β-xylosidase | 6187343 | 1629 | No | – | – |
| Pichia pastoris | GH45 | 1 | β-1,4 glucanase | 8201351 | 1845 | Yes | >500 | Couturier et al., 2011b |
| P. anserina | GH51 | – | α-L-arabinofuranosidase | 6188578 | 2251 | Yes | >100 | Couturier et al., 2011a |
| U. maydis | GH51 | – | α-L-arabinofuranosidase | 3628887 | 2016 | Yes | >100 | unpublished |
| U. maydis | GH62 | – | α-L-arabinofuranosidase | 3632410 | 996 | Yes | >100 | Siguier et al., 2014 |
| P. anserina | GH62 | – | α-L-arabinofuranosidase | 6188704 | 1068 | Yes | >100 | Couturier et al., 2011a; Siguier et al., 2014 |
| P. anserina | GH67 | – | α-glucuronidase | 6189855 | 2591 | No | – | – |
| P. anserina | GH93 | – | arabinanase | 6188440 | 1274 | Yes | >100 | unpublished |
| P. anserina | GH131 | 1 | β-1,4 glucanase | 6187659 | 1093 | Yes | >500 | Lafond et al., 2012 |
| Talaromyces funiculosus | CE1 | 1 | Cinnamoyl esterase | AJ291496 | 1059 | Yes | n.d. | unpublished |
| Penicillium purpurogenum | CE1 | 1 | Acetyl xylan esterase | AF529173 | 1146 | Yes | n.d. | unpublished |
| Volvariella volvacea | CE1 | 1 | Acetyl xylan esterase | ABI63599 | 1047 | Yes | n.d. | unpublished |
| Pycnoporus cinnabarinus | CE1 | 1 | Carbohydrate esterase (CE) | CDO72503 | 1128 | Yes | n.d. | unpublished |
| P. anserina | CE15 | 1 | Methylglucuronic esterase | 6187208 | 1446 | Yes | <100 | Katsimpouras et al., 2014 |
| P. anserina | CE15 | – | Methylglucuronic esterase | 6189833 | 1188 | Yes | <100 | unpublished |
| P. anserina | CE16 | 1 | Acetyl esterase | 6189799 | 1329 | Yes | <100 | unpublished |
| U. maydis | AA3_2 | – | GMC oxidoreductase | 3632124 | 1911 | Yes | >500 | unpublished |
| U. maydis | AA3_2 | – | Glucose oxidase | 3631645 | 1905 | Yes | >100 | unpublished |
| P. anserina | AA3_2 | – | Cellobiose dehydrogenase | 6188032 | 2376 | Yes | >500 | Bennati-Granier et al., 2015 |
| Pycnoporus cinnabarinus | AA3_2 | – | Cellobiose dehydrogenase | AF081574 | 4500 | Yes | >500 | Bey et al., 2011 |
| U. maydis | AA5_1 | – | Glyoxal oxidase | 3629211 | 1959 | No | – | – |
| U. maydis | AA5_1 | – | Glyoxal oxidase | 3630482 | 2589 | No | – | – |
| P. anserina | AA9 | 1 | Lytic polysaccharide monooxygenase | 6195934 | 1056 | Yes | >100 | Bey et al., 2013 |
| P. anserina | AA9 | 1 | Lytic polysaccharide monooxygenase | 6192283 | 1043 | Yes | >500 | Bey et al., 2013 |
| P. anserina | AA9 | – | Lytic polysaccharide monooxygenase | 6192256 | 783 | No | – | – |
| P. anserina | AA9 | – | lytic polysaccharide monooxygenase | 6190208 | 1038 | Yes | >100 | Bennati-Granier et al., 2015 |
| P. anserina | AA9 | 1 | Lytic polysaccharide monooxygenase | 6191260 | 934 | Yes | <100 | Bennati-Granier et al., 2015 |
| P. anserina | AA9 | – | Lytic polysaccharide monooxygenase | 6195000 | 902 | Yes | <100 | Bennati-Granier et al., 2015 |
| P. anserina | AA9 | – | Lytic polysaccharide monooxygenase | 6196037 | 778 | Yes | <100 | Bennati-Granier et al., 2015 |
| P. anserina | AA9 | 1 | Lytic polysaccharide monooxygenase | 6187700 | 1113 | Yes | >100 | Bennati-Granier et al., 2015 |
Yield, amount of secreted recombinant protein (mg/L).
Author Contributions
MH performed most of the experiments.
SG set up and optimized NiNTA protein purification protocols in collaboration with DN who was more involved in the automation thereof.
AG was in charge of factorial approaches.
J-GB had the idea of setting-up a recombinant protein expression facility using P. pastoris and proposed that CB be in charge of its set-up. He also provided several guidelines and participated in the writing of the manuscript.
CB preliminarily tested different experimental options developed in this study, coordinated the effort of above people and wrote the paper.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr Frederic Carrière (Enzymology at Interfaces and Physiology of Lipolysis, EIPL, Marseille, France) for the protocol for preparing frozen electro competent cells and for allowing one of us to perform preliminary experiments in his lab where we greatly appreciated the valuable assistance of Justine Baignol. We thank Pr Manabu Arioka at the Laboratory of Microbiology, Department of Biotechnology, the University of Tokyo for providing pBGP1-DEST. This study was funded by the French National Research Agency (ANR FUNLOCK ANR-13-BIME-0002-01).
Abbreviations
- AA
auxiliary activities
- CAZy
carbohydrate-active enzymes
- CE
carbohydrate esterase
- GH
glycoside hydrolase
- kDa
kiloDalton
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2015.01002
References
- Alberto F., Navarro D., de Vries R. P., Asther M., Record E. (2009). Technical advance in fungal biotechnology: development of a miniaturized culture method and an automated high-throughput screening. Lett. Appl. Microbiol. 49 278–282. 10.1111/j.1472-765X.2009.02655.x [DOI] [PubMed] [Google Scholar]
- Bauer S., Vasu P., Persson S., Mort A. J., Somerville C. R. (2006). Development and application of a suite of polysaccharide-degrading enzymes for analyzing plant cell walls. Proc. Natl. Acad. Sci. U.S.A. 103 11417–11422. 10.1073/pnas.0604632103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennati-Granier C., Garajova S., Champion C., Grisel S., Haon M., Zhou S., et al. (2015). Substrate specificity and regioselectivity of fungal AA9 lytic polysaccharide monooxygenases secreted by Podospora anserina. Biotechnol. Biofuels 8 90 10.1186/s13068-015-0274-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit I., Coutard B., Oubelaid R., Asther M., Bignon C. (2007). Expression in Escherichia coli, refolding and crystallization of Aspergillus niger feruloyl esterase A using a serial factorial approach. Protein Expr. Purif. 55 166–174. 10.1016/j.pep.2007.04.001 [DOI] [PubMed] [Google Scholar]
- Bey M., Berrin J. G., Poidevin L., Sigoillot J. C. (2011). Heterologous expression of Pycnoporus cinnabarinus cellobiose dehydrogenase in Pichia pastoris and involvement in saccharification processes. Microb. Cell Fact. 10 113 10.1186/1475-2859-10-113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bey M., Zhou S., Poidevin L., Henrissat B., Coutinho P. M., Berrin J. G., et al. (2013). Cello-oligosaccharide oxidation reveals differences between two lytic polysaccharide monooxygenases (family GH61) from Podospora anserina. Appl. Environ. Microbiol. 79 488–496. 10.1128/AEM.02942-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier M., Feliu J., Haon M., Navarro D., Lesage-Meessen L., Coutinho P. M., et al. (2011a). A thermostable GH45 endoglucanase from yeast: impact of its atypical multimodularity on activity. Microb. Cell Fact. 10 103 10.1186/1475-2859-10-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couturier M., Haon M., Coutinho P. M., Henrissat B., Lesage-Meessen L., Berrin J. G. (2011b). Podospora anserina hemicellulases potentiate the Trichoderma reesei secretome for saccharification of lignocellulosic biomass. Appl. Environ. Microbiol. 77 237–246. 10.1128/AEM.01761-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg J. M., Cereghino J. L., Shi J., Higgins D. R. (2000). Recombinant protein expression in Pichia pastoris. Mol. Biotechnol. 16 23–52. 10.1385/MB:16:1:23 [DOI] [PubMed] [Google Scholar]
- Damasceno L. M., Huang C. J., Batt C. A. (2012). Protein secretion in Pichia pastoris and advances in protein production. Appl. Microbiol. Biotechnol. 93 31–39. 10.1007/s00253-011-3654-z [DOI] [PubMed] [Google Scholar]
- Dashtban M., Schraft H., Qin W. (2009). Fungal bioconversion of lignocellulosic residues; opportunities and perspectives. Int. J. Biol. Sci. 5 578–595. 10.7150/ijbs.5.578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duetz W. A. (2007). Microtiter plates as mini-bioreactors: miniaturization of fermentation methods. Trends Microbiol. 15 469–475. 10.1016/j.tim.2007.09.004 [DOI] [PubMed] [Google Scholar]
- Filyak Y., Finiuk N., Mitina N., Bilyk O., Titorenko V., Hrydzhuk O., et al. (2013). A novel method for genetic transformation of yeast cells using oligoelectrolyte polymeric nanoscale carriers. Biotechniques 54 35–43. 10.2144/000113980 [DOI] [PubMed] [Google Scholar]
- Geier M., Braun A., Fladischer P., Stepniak P., Rudroff F., Hametner C., et al. (2013). Double site saturation mutagenesis of the human cytochrome P450 2D6 results in regioselective steroid hydroxylation. FEBS J. 280 3094–3108. 10.1111/febs.12270 [DOI] [PubMed] [Google Scholar]
- Grigoriev I. V., Nikitin R., Haridas S., Kuo A., Ohm R., Otillar R., et al. (2014). MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res. 42 D699–D704. 10.1093/nar/gkt1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., et al. (2007). Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315 804–807. 10.1126/science.1137016 [DOI] [PubMed] [Google Scholar]
- Katsimpouras C., Bénarouche A., Navarro D., Karpusas M., Dimarogona M., Berrin J. G., et al. (2014). Enzymatic synthesis of model substrates recognized by glucuronoyl esterases from Podospora anserina and Myceliophthora thermophila. Appl. Microbiol. Biotechnol. 98 5507–5516. 10.1007/s00253-014-5542-9 [DOI] [PubMed] [Google Scholar]
- Kittl R., Kracher D., Burgstaller D., Haltrich D., Ludwig R. (2012). Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5 79 10.1186/1754-6834-5-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafond M., Navarro D., Haon M., Couturier M., Berrin J. G. (2012). Characterization of a broad-specificity β-glucanase acting on β-(1,3)-, β-(1,4)-, and β-(1,6)-glucans that defines a new glycoside hydrolase family. Appl. Environ. Microbiol. 78 8540–8546. 10.1128/AEM.02572-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levasseur A., Benoit I., Asther M., Asther M., Record E. (2004). Homologous expression of the feruloyl esterase B gene from Aspergillus niger and characterization of the recombinant enzyme. Protein Expr. Purif. 37 126–133. 10.1016/j.pep.2004.05.019 [DOI] [PubMed] [Google Scholar]
- Liu Z., Pscheidt B., Avi M., Gaisberger R., Hartner F. S., Schuster C., et al. (2008). Laboratory evolved biocatalysts for stereoselective syntheses of substituted benzaldehyde cyanohydrins. Chembiochem 9 58–61. 10.1002/cbic.200700514 [DOI] [PubMed] [Google Scholar]
- Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42 D490–D495. 10.1093/nar/gkt1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynd L., Laser M. S., Bransby D., Dale B. E., Davison B., Hamilton R., et al. (2008). How biotech can transform biofuels. Nat. Biotechnol. 26 169–172. 10.1038/nbt0208-169 [DOI] [PubMed] [Google Scholar]
- Navarro D., Couturier M., Damasceno da Silva G. G., Berrin J. G., Rouau X., Asther M., et al. (2010). Automated assay for screening the enzymaticrelease of reducing sugars from micronized biomass. Microb. Cell Fact. 9 58 10.1186/1475-2859-9-58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguere C., Larsson A. M., Guyot J. C., Bignon C. (2012). Fractional factorial approach combining 4 Escherichia coli strains, 3 culture media, 3 expression temperatures and 5 N-terminal fusion tags for screening the soluble expression of recombinant proteins. Protein Expr. Purif. 84 204–213. 10.1016/j.pep.2012.05.011 [DOI] [PubMed] [Google Scholar]
- Poidevin L., Feliu J., Doan A., Berrin J. G., Bey M., Coutinho P. M., et al. (2013). Insights into exo- and endoglucanase activities of family 6 glycoside hydrolases from Podospora anserina. Appl. Environ. Microbiol. 79 4220–4229. 10.1128/AEM.00327-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poidevin L., Levasseur A., Paës G., Navarro D., Heiss-Blanquet S., Asther M., et al. (2009). Heterologous production of the Piromyces equi cinnamoyl esterase in Trichoderma reesei for biotechnological applications. Lett. Appl. Microbiol. 49 673–678. 10.1111/j.1472-765X.2009.02734.x [DOI] [PubMed] [Google Scholar]
- Rabert C., Weinacker D., Pessoa A., Jr., Farías J. G. (2013). Recombinants proteins for industrial uses: utilization of Pichia pastoris expression system. Braz. J. Microbiol. 44 351–356. 10.1590/S1517-83822013005000041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa T., Matsui M., Kobayashi Y., Otagiri M., Moriya S., Sakamoto Y., et al. (2011). High-throughput recombinant gene expression systems in Pichia pastoris using newly developed plasmid vectors. Plasmid 65 65–69. 10.1016/j.plasmid.2010.08.004 [DOI] [PubMed] [Google Scholar]
- Siguier B., Haon M., Nahoum V., Marcellin M., Burlet-Schiltz O., Coutinho P. M., et al. (2014). First structural insights into αα-L-arabinofuranosidases from the two GH62 glycoside hydrolase subfamilies. J. Biol. Chem. 289 5261–5273. 10.1074/jbc.M113.528133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzenbacher G., Gruez A., Roig-Zamboni V., Spinelli S., Valencia C., Pagot F., et al. (2002). A medium-throughput crystallization approach. Acta Crystallogr. D Biol. Crystallogr. 58 2109–2115. 10.1107/S0907444902013938 [DOI] [PubMed] [Google Scholar]
- Topakas E., Moukouli M., Dimarogona M., Vafiadi C., Christakopoulos P. (2010). Functional expression of a thermophilic glucuronyl esterase from Sporotrichum thermophile: identification of the nucleophilic serine. Appl. Microbiol. Biotechnol. 87 1765–1772. 10.1007/s00253-010-2655-7 [DOI] [PubMed] [Google Scholar]
- Topakas E., Moukouli M., Dimarogona M., Vafiadi C., Christakopoulos P. (2012). Expression, characterization and structural modelling of a feruloyl esterase from the thermophilic fungus Myceliophthora thermophila. Appl. Microbiol. Biotechnol. 94 399–411. 10.1007/s00253-011-3612-9 [DOI] [PubMed] [Google Scholar]
- Yu X. W., Wang R., Zhang M., Xu Y., Xiao R. (2012). Enhanced thermostability of a Rhizopus chinensis lipase by in vivo recombination in Pichia pastoris. Microb. Cell Fact. 11 102 10.1186/1475-2859-11-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.