

Abstract
Purpose
To characterize in detail the phenotype and genotype of patients with pericentral retinal degeneration (PRD).
Methods
Patients were screened for an annular ring scotoma ranging from 3° to 40° (n = 28, ages 24–71) with kinetic perimetry. All patients had pigmentary retinopathy in the region of the dysfunction. Further studies included cross-sectional and en face imaging, static chromatic perimetry, and electroretinography. Molecular screening was performed.
Results
Genotypes of 14 of 28 PRD patients were identified: There were mutations in eight different genes previously associated with autosomal dominant or autosomal recessive RDs. Kinetic fields monitored in some patients over years to more than a decade could be stable or show increased extent of the scotoma. Electroretinograms were recordable but with different severities of dysfunction. Patterns of photoreceptor outer nuclear layer (ONL) loss corresponded to the distribution of visual dysfunction. Outer nuclear layer thickness topography and en face imaging indicated that the greatest disease expression was in the area of known highest rod photoreceptor density.
Conclusions
Molecular heterogeneity was a feature of the PRD phenotype. Many of the molecular causes were also associated with other phenotypes, such as maculopathies, typical retinitis pigmentosa (RP) and cone–rod dystrophy. The pericentral pattern of retinal degeneration is thus confirmed to be an uncommon phenotype of many different genotypes rather than a distinct disease entity.
Keywords: cone, optical coherence tomography, perimetry, rod
Pericentral retinal degeneration patients were studied for visual function and retinal structure. Molecular diagnoses were identified in one-half the patients. This pericentral pattern is thus confirmed to be an uncommon phenotype of many different genotypes rather than a distinct disease entity.
Distinct patterns of visual field loss in forms of retinitis pigmentosa (RP) and Usher syndrome have been previously studied and classified.1 The more common visual field patterns include an altitudinal distribution of disease and a midperipheral ring scotoma, the latter being the classical description in RP.2 Less common is a pattern of dysfunction that affects the retinal region surrounding the central few degrees, variably described as pericentral, perimacular, or paramacular retinal degeneration.1,3–12 It has been debated whether the retinal degenerations included under the diagnosis of pericentral retinal degeneration (PRD) are a distinct entity.4,13–15 Attempts have been made to identify the genetic cause of individuals with presumed autosomal recessive (ar) PRD but these have not been revealing.11,16 Some autosomal dominant (ad) Norwegian families with affected members showing a pericentral pattern of disease had missense mutations in the RHO (rhodopsin) or TOPORS (topoisomerase I-binding arginine/serine rich) genes.12,17
We identified a cohort of RD patients with a pericentral pattern of disease and then studied their phenotype by measures of visual function and retinal imaging. Gene screening was performed and one-half of the patients were found to have mutations in genes previously associated with ad or ar retinal degenerations.
Methods
Subjects
In a population of 1074 patients diagnosed as nonsyndromic RD of all genetic types, there were 28 patients given the clinical diagnosis of PRD (ages 24–71 at first visit; Table) based on the presence of an annular scotoma ranging from 3° to 40° in their kinetic visual field; all patients had pigmentary retinopathy within and beyond the vascular arcades. Twelve patients were evaluated on multiple visits, while the remainder had single visits only. In our population of 129 patients with ABCA4-RD,18–24 we identified a group with fovea-sparing (n = 10; ages 22–63) and compared their function and structural findings with those of the PRD patients (Supplementary Material). Eight patients with the diagnosis of RP or Usher syndrome but without a pericentral disease expression (ages 9–58) were also included for comparison of optical coherence tomography (OCT) results. Procedures followed the Declaration of Helsinki and the study was approved by the institutional review board (IRB). Informed consent, assent and parental permission were obtained and the work was Health Insurance Portability and Accountability Act (HIPAA)–compliant.
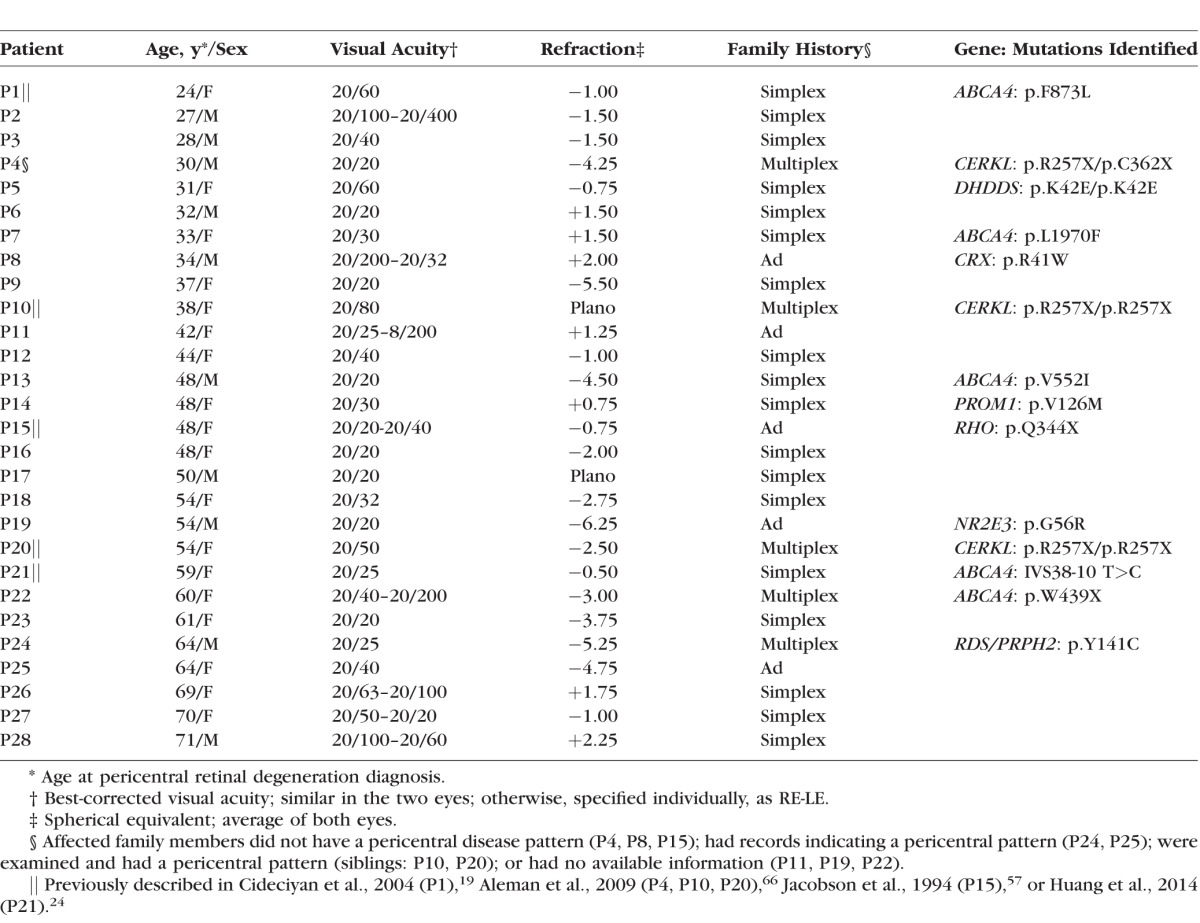
Table.
Clinical and Molecular Characteristics of the Pericentral Retinal Degeneration Patients
Visual Function
Patients underwent a complete eye examination including best-corrected visual acuity, electroretinography (ERG), and Goldman kinetic visual fields (using V-4e and I-4e test targets). Full-field ERGs were performed using International Society for Clinical Electrophysiology of Vision (ISCEV) standard stimuli.25 Dark- and light-adapted chromatic static threshold perimetry (200-ms duration, 650- and 500-nm stimuli in dark and 600-nm in light, 1.7° diameter target) was also performed. Sensitivity was measured at 2° intervals along the horizontal meridian spanning 60°, and with a full-field test of 72 loci on a 12° grid.26,27 The difference between dark-adapted sensitivities to 500- and 650-nm stimuli was used to determine if rods or cones or both mediated vision. The 72 loci set was split into two parts for separate analysis of the peripheral visual field (51 loci with eccentricities ≥ 30°). For the peripheral field, location-specific rod and cone sensitivity losses were averaged to produce an aggregate metric used for ranking the patients and to correlate rod and cone function (coefficient of determination, r2). Other details of the techniques, data analyses, and normal results have been described.26,27
Imaging Studies
Retinal cross-sections along horizontal and vertical meridians crossing the fovea were obtained with OCT. The principles of the method and our recording and analysis techniques have been published.28–30 Briefly, overlapping OCT scans that were 4.5 or 9 mm in length were used to cover the horizontal and vertical meridians up to 9 mm eccentricity from the fovea. At least three OCTs were obtained at each retinal location. Post-acquisition processing of OCT data was performed with custom programs (MATLAB 7.5; MathWorks, Natick, MA, USA). Longitudinal reflectivity profiles (LRPs) making up the OCT scans were aligned by straightening the major RPE reflection. Nuclear layers were defined as previously published.30 Outer nuclear layer (ONL) thickness in each patient was quantified, plotted as a function of eccentricity, and compared with the normal range (mean ±2 SD; n = 15; age range, 8–62 years).
En face imaging was performed in a subset of patients with a confocal scanning laser ophthalmoscope (Spectralis; Heidelberg Engineering GmbH, Heidelberg, Germany) as previously published.21,31,32 Retinal and subretinal features were imaged with 820-nm near-infrared (NIR) light in the reflectance (REF) mode. Health of the RPE was estimated with a reduced-illuminance autofluorescence imaging (RAFI) method using 790-nm NIR excitation and 488-nm short-wavelength (SW) excitation. Near-infrared–RAFI and SW-RAFI images were obtained with a fixed sensitivity setting of 105%. Near-infrared–REF, NIR-RAFI, and SW-RAFI were performed in the ‘high-speed' mode where retinal regions (30° × 30° square or 55° diameter circular) were sampled onto a 768 × 768 pixel image. Automatic real-time (ART) averaging feature of the manufacturer's software was used to average 21 frames. For SW-RAFI, macular images were obtained; for NIR-REF and NIR-RAFI overlapping neighboring regions were obtained. Wide-angle composite images were generated post acquisition by digitally stitching individual images using manually specified corresponding retinal landmark pairs.
Molecular Screening Strategy
Twenty-three of 28 patients with pericentral dysfunction were the only known affected individuals in their families (simplex; n = 18) or they had affected siblings (multiplex; n = 5). These patients were first screened for mutations in arRP-associated genes using APEX microarrays (490, 518, 594 variants arrays; Asper Ophthalmics, Tartu, Estonia) or through the Carver Nonprofit Genetic Testing Laboratory (Iowa City, IA, USA). Five patients from ad pedigrees were screened for mutations in adRP-associated genes through the Carver Laboratory or with APEX microarrays (385 or 414 variants arrays; Asper Ophthalmics). Because the adRP arrays included mutations in RHO and TOPORS, genes previously reported in PRD patients,12,17 and in RDS, which was causative in one pericentral patient,33 simplex and multiplex patients who were negative on arRP panels were also screened on the adRP arrays. All patients without a molecular diagnosis were screened for mutations in ABCA4 with APEX microarrays (496, 519, or 577 variants arrays; Asper Ophthalmics),34 by Carver Laboratory or by Casey Eye Institute Molecular Testing Laboratory (Portland, OR, USA). ABCA4 molecular testing methods in the 10 patients with fovea-sparing ABCA4–RD (Supplementary Material) have been reported.19–24
Results
Clinical and Molecular Characteristics of the Pericentral RD Cohort
The PRD patients ranged in age from 24 to 71 years (average, 47; SD, 14 years) when first diagnosed. Best-corrected visual acuities at first visit were 20/20 to 20/100 in the best-seeing eye. There was no common refractive error (spherical equivalent range, +2.25 to −6.25; Table). Longitudinal data for visual acuity were available in 11 patients and follow-up intervals ranged from 2 to 18 years (average, 7 years). Visual acuities remained the same in 7/11 (64%) patients; the intervals for these patients were 2 to 9 years (average, 4 years). The remaining four patients showed substantial reductions in acuity over time (e.g., P21 from 20/25 to HM over 11 years; P11 from 20/25 to 20/200 over 17 years; P26 from 20/63 to HM in 4 years; P27 from 20/20 to CF in 14 years); the intervals in this group were longer on average (11 years). The acuity changes were not explained by increasing cataract. On ophthalmoscopic examination, all patients had some vessel attenuation and a waxy appearance to the optic nerve head. Pigmentary abnormalities such as granularity, depigmentation, and some atrophic changes were present within the vessel arcades with some changes extending to greater eccentricities. In general, the peripheral fundus tended to be more normal in appearance than the pericentral region.
Genetic testing revealed underlying molecular causes of the retinal degeneration in 14 patients of this PRD cohort (Table). Four patients had homozygous or compound heterozygous mutations in ar retinal degeneration-associated genes: DHDDS (P5) and CERKL (P4, P10, P20). Four patients had mutations known to cause ad retinal degenerations: CRX (P8), RHO (P15), NR2E3 (P19), and RDS (P24). A novel PROM1 variant that is predicted to be damaging through PolyPhen-2 was identified in P14. Five patients (P1, P7, P13, P21, P22) had single ABCA4 mutant alleles, which are considered to be disease-causing (p.L1970F35; p.W439X36; p.F873L37; IVS38-10 T>C and p.V552I38). The PRD patients who were heterozygous for ABCA4 mutations did not show fundus features that would be considered typical of Stargardt disease such as yellow-white irregular flecks in the posterior pole.19–24 The patients with single mutant ABCA4 alleles were also screened for mutations in genes previously reported as disease-causing in pericentral disease, RHO, RDS, and TOPORS, as well as PROM1; these results were negative. A comparison of the PRD patients to a subset of fovea-sparing ABCA4-RD patients with two mutant alleles indicated the central ring of dysfunction was much smaller in the latter group (see Supplementary Materials).
Pericentral Pattern of Visual Dysfunction
Representative kinetic perimetry results from nine patients with pericentral patterns of retinal dysfunction are shown (Fig. 1A). Most patients had a complete annular relative and/or absolute scotoma surrounding fixation, but proximity of the inner edge to the fovea and extent of the outer edge into the midperiphery varied. Some patients had pericentral scotomas that were incomplete (Fig. 1A: P13, P17, P18, P14). The peripheral visual field extent to the V-4e target was mainly within normal limits (26/28; 92.8%); abnormalities in peripheral field detection of the I-4e target, however, were present in many patients (16/28; 57.1%) and are illustrated by the fields of P24, P16, and P28 (Fig. 1A, bottom row). We analyzed quantitatively the horizontal and vertical extent of the central island of vision and the pericentral scotoma in all the patients (Figs. 1B, 1C). The mean horizontal and vertical extents of the central island of vision were 6.5° (SD, 4.0°) and 5.5° (SD, 2.9°), respectively. The mean horizontal extent of the pericentral scotoma was 19.5° (SD, 7.1°) toward the nasal field side and 26.6° (SD, 12.3°) toward the temporal field. The mean vertical extent of the pericentral scotoma was 21.3° (SD, 8.2°) toward the inferior field and 18.4° (SD, 8.3°) superiorly.
Figure 1.

Kinetic visuals fields in patients with pericentral RD. (A) Kinetic visual fields using V- and I-4e test targets in one eye of nine representative PRD patients. P28 did not detect the I-4e target whereas the other patients shown had measurable fields with both target sizes. Gray areas: relative scotomas; black areas: absolute scotomas. (B) Horizontal (nasal and temporal field) extent of the pericentral scotomas (gray) and of the central island of vision (white) within the scotoma for the entire cohort of pericentral RP patients. Patient numbers (Table) are along the left vertical axis. (C) Vertical extent (superior and inferior field) of the pericentral scotomas (gray) and of the central island (white) within the scotoma for these patients. Patient numbers are along the lower horizontal axis of the graph.
This study wanted to determine if the cohort of RD patients remained stable during the period of observation. Longitudinal data in a subset of patients are shown (Figs. 2A–E). P1, over a 9-year interval, showed no remarkable changes in extent of the pericentral scotoma or in the peripheral field (Fig. 2A). P25, over a 4-year period, progressed from an incomplete relative and absolute pericentral deficit to a complete relative pericentral scotoma with an increase in the peripheral extent of the scotoma and a reduction in the central island. P21, during a 12-year follow-up, had an apparent increase in the peripheral extent of the pericentral scotoma, and the small central island was no longer detectable (and acuity became reduced). P26, during a 5-year follow-up period, progressed from having an extensive absolute pericentral deficit that spared a small central island to loss of perception with this island (and reduced acuity). P27 had multiple visits over a 12-year time period. At 70 years of age, the patient had a normal extent of peripheral visual field and a relative pericentral scotoma. There was progression over approximately 2 years to a larger relative scotoma and reduced peripheral field to the I-4e target; at age 78, there was an absolute pericentral scotoma with no detection of I-4e across the field. There was an absolute central scotoma by age 82 with no detection of a central island of vision; visual acuity was much reduced at that age (Fig. 2E).
Figure 2.

Serial kinetic fields in pericentral RD. (A–E) Longitudinal kinetic field data in five pericentral RD patients showing responses to V-4e and I-4e targets. Gray areas: relative scotomas; black areas: absolute scotomas.
Rod and Cone Function in Pericentral RD
Twenty-two of the patients had ERGs recorded, and all had detectable signals. Representative waveforms for rod b-wave, mixed cone–rod, and cone flicker ERGs in three patients with different severities of dysfunction are illustrated (Fig. 3A). P11 has ERGs that fall within the normal limits for the four parameters measured (Fig. 3B). P9 has amplitudes reduced to approximately one-third of normal mean for all waveforms. P19 has lower amplitudes for all responses than P9 with rod and mixed responses approximately 15% of normal mean and cone flicker 24% of normal. Of the 22 patients, 5 (23%) had normal ERGs to all stimuli; one patient (4%) retained normal cone flicker only. The remaining 16 patients (73%) had abnormal ERGs for all parameters: rod b-wave amplitudes ranged from 32 to 133 μV, mean 79 μV (normal mean = 299 μV; −2 SD = 195 μV); mixed cone–rod a-waves ranged from 43 to 168 μV, mean 99 μV (normal mean = 297 μV; −2 SD = 167 μV); mixed b-waves from 72 to 254 μV, mean 168 μV (normal mean = 497 μV; −2 SD = 275 μV); and cone flicker amplitudes from 23 to 101 μV, mean 59 μV (normal mean = 172 μV; −2 SD = 102 μV; Fig. 3B).
Figure 3.

Rod and cone function in pericentral RD. (A) Representative ERG waveforms from three PRD patients with different degrees of retinal dysfunction. (B) Four measured ERG parameters in the 22 patients with recordings. The three gray symbols to the left correspond to patients whose waveforms are shown in (A). In the center of each parameter column are all patient data (circles) and a box defining ±2 SD and the mean normal. To the right in each column is a boxplot summarizing the data for the patient group, indicating interquartile range (gray box), 10/90th percentiles (error bars), 5/95th percentiles (symbols above and below), median (thicker line within box) and mean (thin line). (C) Dark-adapted (500 nm, top) and light-adapted (600 nm, bottom) horizontal sensitivity profiles in four pericentral RP patients (square filled symbols connected by lines) compared with normal data (shaded area, ±2 SD from mean). For dark-adapted sensitivities, photoreceptor mediation based on two-color (500 nm, 650 nm) testing, is shown above the results: R, rod-mediated; M, mixed rod- and cone-mediated; C, cone-mediated. Hatched bar: physiologic blind spot. (D) Maps of rod sensitivity loss (500 nm dark-adapted, top) and cone sensitivity loss (600 nm light-adapted, bottom) in the same four patients. Gray scale has 16 levels, representing 0- to 40-dB losses for rods and 0- to 30-dB losses for cones. Black square: physiologic blind spot represented at 12° in the temporal field. N, nasal; T, temporal; I, inferior; S, superior visual field.
Static threshold perimetry was performed in the light- and dark-adapted states in all 28 patients. Representative horizontal sensitivity profiles in four patients are shown (Fig. 3C). P13 has a central island with normal rod and cone function, a pericentral area of rod and cone dysfunction and, at greater eccentricities, a return to near normal rod function and normal cone sensitivity. The central island in P19 is cone-mediated in the dark-adapted state and normal in function in the light-adapted state. The pericentral dysfunction in P19 is greater for rods than in P13 but the light-adapted cone profile is similar to that in P13. Eccentric to the pericentral deficit, the profiles of the two patients are similar. P16, like P19, has a cone-mediated central locus but a wider pericentral deficit for rods and cones. There is mainly a residual cone central island in P7 with an extensive pericentral region of dysfunction of rod and cone function.
Maps of rod and cone sensitivity loss across the visual field are shown for the same four patients (Fig. 3D). The pericentral rod dysfunction in P13 extends to greater eccentricities than does the cone dysfunction; more peripheral rod and cone function is within normal limits for the most part. The rod dysfunction noted centrally in the horizontal profile of P19 continues into the periphery; cone function, however, is abnormal at far peripheral loci but otherwise mainly within normal limits. P16 has a large central rod scotoma and a pericentral cone scotoma with the preserved central cone island evident in the sensitivity profile. Both rod and cone function in the periphery show abnormalities. P7 has a rod central scotoma which exceeds that in P16. The pericentral cone dysfunction is also more extensive than that in P16 and there is greater sensitivity loss for rods and cones in the peripheral field.
In summary, there was a range of severities of peripheral dysfunction in the pericentral patients. A group of PRD patients had peripheral rod sensitivity losses that were no greater than 1 log unit on average (nine patients, 32%), as exemplified by P13. A second group with sensitivity losses ranging from 1 to 3 log units included 14 patients (50%), exemplified by P19 and P16. The remaining five patients (18%), exemplified by P7, had rod sensitivity losses in excess of 3 log units on average. Cone sensitivity losses in the periphery were relatively well correlated with rod sensitivity losses (r2 = 0.77). There was no strong correlation between the extent of the pericentral scotoma (as measured by kinetic perimetry; Figs. 1B, 1C) and the severity of peripheral visual sensitivity loss for both rods and cones (r2 = 0.18 and 0.2, respectively).
Retinal Structural Basis of Pericentral Dysfunction
We studied the microanatomical basis of the pericentral functional abnormalities using OCT across the central 60° of retina. Typical disease expressions of RP at relatively early stages can show preserved ONL thickness within a wide extent of central retina and, in the mildest expressions, can follow a near normal distribution of ONL (Fig. 4A). We have previously reported greater preservation of ONL in the superior than inferior retina in vertical profiles crossing the fovea in different forms of RP.29,39–41 Horizontal profiles of ONL thickness are relatively symmetrical around the fovea but details of any nasal-temporal asymmetry tend to be obscured by the optic nerve in the nasal retina. Eight patients with the diagnosis of RP or Usher syndrome (ages 9–58) and at different stages of disease illustrate the patterns of structural abnormality typically noted (Fig. 4A). Vertically, there is greater extent of ONL thickness in the superior than inferior retina; horizontally, there is relatively symmetrical nasal and temporal extent.
Figure 4.

Retinal laminar architecture in patients with pericentral RD. (A–C) Cross-sectional OCT images along the vertical and horizontal meridians through the fovea in a 58-year-old patient with RP simplex (A) compared with P19 (B) and P1 (C), two patients with PRD. Outer nuclear layer is highlighted in blue in the scans (top). Vertical (middle) and horizontal (bottom) thickness profiles of ONL in eight patients with RP or Usher syndrome (A) and 20 PRD patients (B, C) are shown. Gray areas: normal limits (±2 SD from mean). (D) Serial ONL thickness profiles along the vertical and horizontal meridians in P27 and P11, two PRD patients. Gray areas: normal limits (±2 SD from mean). (E) Topographic map of ONL thickness in a 45-year-old woman with normal vision (top, left). For reference, topography of rod photoreceptor density in the human retina (reprinted and modified from Curcio CA, Sloan KR, Kaline RE, Hendrickson AE. Human photoreceptor topography. J Comp Neurol. 1990; 292:497–523; top, right). Lighter intensities correspond to higher spatial densities and darker intensities to lower densities (intensity bar is in rod cells X 1000/mm2). Black oval: optic nerve (ON) head. Outer nuclear layer thickness topography in two PRD patients, P19 and P23 (bottom left and right, respectively). Traces of major blood vessels and location of optic nerve head are overlaid on each map (depicted as right eyes).
Patterns of ONL loss in two groups of patients with pericentral dysfunction are shown (Figs. 4B, 4C). Both groups could have normal or abnormal foveal ONL thickness surrounded by a decrease in ONL thickness to nearly nondetectable levels within a few degrees of the fovea.
Outer nuclear layer eccentric to the abnormal pericentral region can increase and attain normal thickness or remain reduced. The first group of 10 patients represents those with an average scotoma extent less than the median value from kinetic perimetry (considering both horizontal and vertical extents; Fig. 1) and includes P4, P8 to P10, P12, P19, P23, and P27 (Fig. 4B). The second group had greater than the median scotoma extent and includes P1, P2, P6, P11, P15, P16, P18, P20, P21, and P24 (Fig. 4C). The vertical and horizontal extent of ONL thickness loss in the pericentral region differs between the two groups. The first group, as predicted from the scotoma extents, has a smaller pericentral region around fixation with diminished ONL, and beyond the ONL loss there can be a return to normal or near normal thickness (Fig. 4B). In the vertical cross-sections there appears to be a wider zone of loss superiorly than inferiorly; in the horizontal cross-sections there appears to be a greater extent of the pericentral deficit toward the nasal retina than temporally. The second group has more extensive pericentral ONL loss with some patient data not returning to normal ONL within the scan extent (Fig. 4C).
Two PRD patients, P27 and P11, were followed with OCT over a span of 12 and 9 years, respectively (Fig. 4D). P27 shows a progressive pericentral ONL deficit with superior retinal loss of ONL eventually exceeding that of the inferior retina. The ONL thickness profiles in P11 are apparently more advanced and do not change as dramatically over the interval examined. The main change is a decrease in foveal ONL at the later age. Outer nuclear layer thickness topography across an expanse of central retina in two PRD patients (P19, P23; Fig. 4E, bottom left and right) is shown compared with an average normal ONL map (Fig. 4E, top left) and rod photoreceptor topography in a comparable region (Fig. 4E, top right; modified from Refs. 29 and 42). Normal ONL thickness topography shows a peak at the fovea and there is a decline with distance from the fovea. There is increased thickness in the superior versus inferior retina; and thickness into the nasal retina is greater than temporally. The superior ONL thickness corresponds with the rod hotspot identified in photoreceptor quantitation studies42 and rod photoreceptor density is greater nasally than temporally. Both patients show a pericentral ONL deficit that tends to follow the rod density pattern and mirrors the areas of greater ONL thickness in normal retina.
With en face imaging, PRD patients showed an annular region displaying greater visibility of choroidal features (Fig. 5, arrowheads). Near-infrared–RAFI intensity in this annular region could range from very low (Fig. 5C) to being nearly the same intensity as surrounding regions without choroidal features (Fig. 5F). Corresponding regions on NIR-REF imaging tended to show locally increased signal (lower insets in Figs. 5C, 5D, 5F). The combination of choroidal visibility and increased reflectivity has been shown to likely correspond to RPE depigmentation or atrophy.32 The central retinal area within the pericentral annulus of RPE disturbance could consist of an elliptical region surrounded by a hyperautofluorescent ring with (Figs. 5C, 5D) or without (Figs. 5B, 5E) a buffer zone of homogeneous signal. Some patients showed smaller regions of macular atrophy (Figs. 5C, 5F). The boundary to the peripheral region of the pericentral annulus was beyond the eccentricity of the optic nerve, and could show a hyperfluorescent ring (Figs. 5C, 5E) or display a smoother transition (Figs. 5B, 5F).
Figure 5.

Digitally-stitched wide-field near-infrared (NIR) reduced-illuminance autofluorescence imaging (RAFI) results of a representative healthy subject compared with five patients with pericentral RD. Upper insets: short-wavelength (SW) RAFI; lower insets: NIR reflectance (REF) images in the same eyes. (A) Near-infrared–RAFI of the 16-year-old normal subject demonstrates higher signal near the fovea smoothly transitioning to a lower signal in the pericentral and midperipheral regions. Blood vessels and the optic nerve appear dark. Short-wavelength–RAFI shows central depression corresponding to the macular pigment absorption. (B–F) Near-infrared–RAFI of pericentral RD patients P6, P12, P16, P19, and P11 demonstrate an annular region (arrowheads) of greater visibility of choroidal pattern of blood vessels implying depigmentation of the RPE and greater penetration of excitation light to choroidal layers. This region is bounded centrally by a relatively preserved macular region with or without regions of atrophy, and a relatively preserved peripheral region beyond the eccentricity of the optic nerve head. Near-infrared–REF images (lower insets) show locally increased reflectivity corresponding to the regions of greater choroidal visibility. Macular SW-RAFI images (upper insets) demonstrate that some of the pericentral demelanization corresponds to RPE atrophy, whereas others retain RPE lipofuscin signal. All images are shown as equivalent right eyes and contrast stretched for visibility of features. Pair of calibrations shown in (F) apply to all panels; both upper and lower insets are displayed at the same magnification.
Discussion
Comparison With Other Cohorts of Pericentral RD
Among previous reports of RD patients with pericentral patterns of disease, there are three descriptions of cohorts with at least 10 patients.11,12,17 Common to the three studies are data for visual acuities and ERGs. One study was mainly of simplex RP patients with a pericentral distribution of disease.11 In this group with pericentral RP, six patients had molecular screening for known mutations in GNAT1, CNGB1, GCAP, CRX, CNGB3, RPE65, CRALBP, RDH5, RGR, and RGS9 and the results were negative; one patient was negative for USH2A mutations.11 Visual acuities in the cohort were 20/20 to 20/40 at first visit. Electroretinograms were recordable but abnormally reduced. Longitudinal data on visual acuity indicated that 4 of 18 patients eventually had reductions of more than two lines. Some ERGs became reduced while others did not. It was concluded that, compared with other studies of groups of RP patients not specified to have a pericentral distribution, there was slower progression of disease in simplex or multiplex pericentral RP.11
There are two reports of ad pericentral disease, both from Norwegian pedigrees. One of the studies was the first to identify a molecular basis of the pericentral phenotype and two missense mutations in the RHO gene were found.17 Visual acuities were 20/20 to 20/25 in at least one eye of 16 of 22 patients but were reduced in all others. Electroretinograms were listed in four patients and these were abnormal; most were recordable. Longitudinal visual acuity data for six of the patients, representing at least 2 decades of follow-up, showed a remarkable decline in two patients but stability in the others. Another ad Norwegian pedigree showed pericentral disease in most of the affected family members and a TOPORS mutation was found to cosegregate with the disease.12 Visual acuities ranged from normal to severely abnormal; ERGs were abnormal except in one patient who had a normal ERG. Both of the ad pericentral studies described variability of expression from mild to severe.
In our PRD cohort, visual acuity was 20/20 to 20/40 in at least one eye of 21/28 (75%) patients at first visit. Electroretinograms showed a range of results from normal to subnormal, but all signals were recordable. Available longitudinal data in a subset of our cohort showed that acuities could remain the same or progress, as reported in previous studies. The dimensions of the pericentral scotoma could also extend both away from and toward the fovea, the latter change not surprisingly accompanied by loss of visual acuity (Fig. 2). Static perimetry indicated that there was both rod and cone dysfunction contributing to the pericentral disease and to the peripheral dysfunction in some patients. The OCT measures of photoreceptor layer thickness provide a morphologic correlate to the psychophysical findings. There is only one histopathologic report of a postmortem donor patient presumed to have pericentral RD.5 Our finding of normal central islands of ONL in some of the patients (Figs. 4B, 4C) would relate to the normal foveal architecture in that donor retina; the reduced rod function but normal cone function centrally in some of our patients (e.g., P16, Fig. 3C) could represent the observation of reduced rods at and beyond the foveal slope; the return of ONL thickness to normal or near normal levels in many of the patients eccentric to the pericentral scotoma would be the equivalent of areas of preserved rods and cones in the far periphery of the donor retina.5
Molecular Heterogeneity in Pericentral RP
Of the 28 patients in the current PRD cohort, 14 (50%) were identified to have a molecular abnormality that was previously associated with inherited retinal degeneration. Four genes and the specific mutations had previously been considered causative for ad degenerations. NR2E3, recognized initially as the cause of an ar disease of retinal development with accompanying degeneration,43–45 was subsequently determined to be a cause of adRP (RP37).46,47 The G56R mutation identified in P19 is a relatively common single mutation cause of adRP.48 The phenotype literature on this form of adRP does not provide sufficient information to decide if a pericentral pattern has been noted previously; most patients are considered severely affected with early-onset night blindness and constricted visual fields with nondetectable rod ERGs46–48; OCTs, however, for two such patients suggest a pericentral loss of ONL,49 and thereby resemble the phenotype in P19. Of interest, the G56R mutation is in the DNA-binding domain of NR2E3 and has been shown to have interactions with CRX,50 the disease gene identified in P8. CRX, and specifically Arg41 mutations, has been associated with cone–rod dystrophy,51 macular dystrophy, or autosomal dominant RP.52,53 The depictions of phenotype for the Arg41Trp mutation in the literature do not clearly represent a pericentral pattern of disease, although a frameshift mutation (Cys202Sfs*17) does show this pattern by OCT.53
Two patients had mutations in RDS/PRPH2 or RHO, photoreceptor-specific genes associated with ad retinal degenerations. P24 had the Y141C RDS mutation, which has been reported to exhibit phenotypic variability, with some patients showing maculopathy only and others having widespread retinal degeneration (reviewed in Ref. 54). The Y141C knockin mouse model indicated outer segment disc abnormalities in rod and cone photoreceptors.55 P15 had the Q344ter RHO mutation, which has been studied in adRP families and in mouse models.56–59 The mechanism of disease is thought to result from abnormal trafficking and localization of mutant rhodopsin to the rod outer segment.58,59 Of interest, P15, when first examined at age 24,56 showed a full kinetic field to both V-4e and I-4e targets and did not show pericentral dysfunction, but a pericentral phenotype was documented when the patient was reexamined in the current study at age 48. Mutations in PROM1 are described as causing RP, macular dystrophy, and cone–rod degeneration and both ad and ar inheritance patterns are reported.60–62 PROM1 has been suggested to have a role in photoreceptor disk morphogenesis.60
The remaining three genes identified in our PRD cohort have been associated only with ar disease. The gene encoding DHDDS has been determined to be a cause of arRP in Ashkenazi Jewish patients.63,64 DHDDS plays a role in the N-glycosylation of many proteins, such as rhodopsin. Descriptions of phenotype in the patients reported to date do not specifically mention pericentral RD64,65 but the sequence of changes in chromatic perimetric profiles in one such patient over 14 years passes through a stage that shows a pericentral distribution (Fig. 3D, P463), although the eventual severity of disease is greater than in the current pericentral cohort. The homozygous K42E DHDDS mutation in pericentral patient P5 is the same as the common mutation previously published. Three patients had mutations in the CERKL gene.66 CERKL has been considered as protective against apoptosis from oxidative stress.67 ABCA4, the common cause of Stargardt disease and ar cone–rod dystrophy,68,69 was detected as single mutant alleles in five patients. A comparison of fovea-sparing ABCA4-RD patients with the PRD patients is in the Supplementary Material.
The present results indicate there is no single molecular cause of the pericentral phenotype. Unexpected was the finding that some of the genotypes associated with PRD in our cohort have been reported to cause multiple clinical expressions. For example, RDS, CRX, PROM1, and ABCA4 have all been associated with RP, macular dystrophy and cone–rod dystrophy.53,54,61,62,69,70 RHO is associated with different patterns of disease, from early retina-wide rod loss to initially delimited altitudinal or sectoral defects.29,71 A pericentral disease distribution has been reported in a patient with a RHO T58R mutation (Fig. 4C in Ref. 29). CERKL has been described as RP, cone–rod dystrophy and as a phenotype of RP with macular atrophy.72 These observations lead to the hypothesis that the pericentral distribution of disease is not only molecularly heterogeneous but may often be associated with genes that are known to lead to phenotypic heterogeneity. Why individuals within and between families with the same genotype manifest different disease phenotypes is unclear.
Rod Cell Density and Pericentral Retinal Disease
The regional retinal distribution of the most pronounced cell loss and dysfunction in PRD corresponds to the retinal region in humans with the highest density of rod photoreceptors.42 Based on the results of the current study, there are multiple genes and molecular mechanisms now associated with the disease pattern and in categories, such as transcription factors, photoreceptor outer segment membrane proteins, an oxidative stress protectant, and a key enzyme in dolichol biosynthesis. Do the genetic causes share any mechanism that could explain the pericentral distribution? Common to all of these genes are known or potential roles played by the mutant gene products on rod photoreceptor outer segment development and maintenance. However, the fact that these genotypes are also associated with other phenotypes makes any theory of how the most rod–cell dense retinal region is specifically more vulnerable than other regions unconvincing. Among the possibilities is a hypothetical interaction of the retinal stress caused by the RD genotype with the molecular gradients involved in photoreceptor patterning73; natural variations in the latter may produce the pericentral photoreceptor cell death rather than other patterns. Evaluation of such a hypothesis is currently not possible since the molecular basis of photoreceptor patterning remains mostly unknown.
Supplementary Material
Acknowledgments
Supported by grants from Hope for Vision (Aventura, FL, USA) Foundation Fighting Blindness (Columbia, MD, USA), Macula Vision Research Foundation (West Conshohocken, PA, USA), the Chatlos Foundation (Longwood, FL, USA), and National Eye Institute/National Institutes Health (EY 013203; Bethesda, MD, USA).
Disclosure: R. Matsui, None; A.V. Cideciyan, None; S.B. Schwartz, None; A. Sumaroka, None; A.J. Roman, None; M. Swider, None; W.C. Huang, None; R. Sheplock, None; S.G. Jacobson, None
References
- 1. Grover S,, Fishman GA,, Brown J,, Jr. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology. 1998; 105: 1069–1075. [DOI] [PubMed] [Google Scholar]
- 2. Gonin J. Le scotome annulaire dans la dégénérescence pigmentaire de la rétine. Ann d'Ocul. 1901; 125: 101–130. [Google Scholar]
- 3. Francois J,, De Rouck A,, Cambie E,, De Laey JJ. Visual functions in pericentral and central pigmentary retinopathy. Ophthalmologica. 1972; 165: 38–61. [DOI] [PubMed] [Google Scholar]
- 4. Krill AE. Central and Pericentral Retinitis Pigmentosa. Krill's Hereditary Retinal and Choroidal Diseases Volume 2. Hagerstown: Harper & Row Publishing Co.; 1977. [Google Scholar]
- 5. Szamier RB,, Berson EL. Histopathologic study of an unusual form of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1982; 22: 559–570. [PubMed] [Google Scholar]
- 6. Hayasaka S,, Fukuda K,, Tsuchiya M,, Mizuno K. Pericentral pigmentary retinal degeneration. Jpn J Ophthalmol. 1985; 29: 161–169. [PubMed] [Google Scholar]
- 7. Grøndahl J. Pericentral retinal dystrophy. Acta Ophthalmol (Copenh). 1987; 65: 344–351. [DOI] [PubMed] [Google Scholar]
- 8. Traboulsi EI,, O'Neill JF,, Maumenee IH. Autosomal recessive pericentral pigmentary retinopathy. Am J Ophthalmol. 1988; 106: 551–556. [DOI] [PubMed] [Google Scholar]
- 9. Noble KG. Peripapillary (pericentral) pigmentary retinal degeneration. Am J Ophthalmol. 1989; 108: 686–690. [DOI] [PubMed] [Google Scholar]
- 10. Durlu YK,, Burumcek E,, Devranoglu K,, Mudun AB,, Karacorlu S,, Arslan MO. Associated ocular findings in pericentral pigmentary retinopathy. Acta Ophthalmol Scand. 1997; 75: 101–103. [DOI] [PubMed] [Google Scholar]
- 11. Sandberg MA,, Gaudio AR,, Berson EL. Disease course of patients with pericentral retinitis pigmentosa. Am J Ophthalmol. 2005; 140: 100–106. [DOI] [PubMed] [Google Scholar]
- 12. Selmer KK,, Grøndahl J,, Riise R,, et al. Autosomal dominant pericentral retinal dystrophy caused by a novel missense mutation in TOPORS gene. Acta Ophthalmol. 2010; 88: 323–328. [DOI] [PubMed] [Google Scholar]
- 13. Franceschetti A,, François J,, Babel J. Chorioretinal Hérédodegenerations. Springfield: Charles C. Thomas; 1974. [Google Scholar]
- 14. Duke-Elder S,, Dobree JH. Diseases of the retina. : Duke-Elder S, System of Ophthalmology, Volume 10. St. Louis, MO: The C.V. Mosby Co.; 1967: 591–593. [Google Scholar]
- 15. Deutman F. Rod-cone dystrophy: primary, hereditary, pigmentary, retinopathy, retinitis pigmentosa. : Krill A, Krill's Hereditary Retinal and Choroidal Diseases, Volume 2. New York, NY: Harper & Row Publishing Co.; 1977: 479–556. [Google Scholar]
- 16. Bass SJ,, Noble KG. Autosomal dominant pericentral retinochoroidal atrophy. Retina. 2006; 26: 717–719. [DOI] [PubMed] [Google Scholar]
- 17. Grøndahl J,, Riise R,, Heiberg A,, Leren T,, Christoffersen T,, Bragadottir R. Autosomal dominant retinitis pigmentosa in Norway: a 20-year clinical follow-up study with molecular genetic analysis. Two novel rhodopsin mutations: 1003delG and I179F. Acta Ophthalmol Scand. 2007; 85: 287–297. [DOI] [PubMed] [Google Scholar]
- 18. Cideciyan AV,, Aleman TS,, Swider M,, et al. Mutations in ABCA4 result in accumulation of lipofuscin before slowing of the retinoid cycle: a reappraisal of the human disease sequence. Hum Mol Genet. 2004; 13: 525–534. [DOI] [PubMed] [Google Scholar]
- 19. Cideciyan AV,, Swider M,, Aleman TS,, et al. ABCA4-associated retinal degenerations spare structure and function of the human papillary retina. Invest Ophthalmol Vis Sci. 2005; 46: 4739–4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aleman TS,, Cideciyan AV,, Windsor EA,, et al. Macular pigment and lutein supplementation in ABCA4-associated retinal degenerations. Invest Ophthalmol Vis Sci. 2007; 48: 1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cideciyan AV,, Swider M,, Aleman TS,, et al. Reduced-illuminance autofluorescence imaging in ABCA4-associated retinal degenerations. J Opt Soc Am A Opt Image Sci Vis. 2007; 24: 1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cideciyan AV,, Swider M,, Aleman TS,, et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet. 2009; 18: 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cideciyan AV,, Swider M,, Aleman TS,, et al. Macular function in macular degenerations: repeatability of microperimetry as a potential outcome measure for ABCA4-associated retinopathy trials. Invest Ophthalmol Vis Sci. 2012; 53: 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang WC,, Cideciyan AV,, Roman AJ,, et al. Inner and outer retinal changes in retinal degenerations associated with ABCA4 mutations. Invest Ophthalmol Vis Sci. 2014; 55: 1810–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aleman TS,, Lam BL,, Cideciyan AV,, et al. Genetic heterogeneity in autosomal dominant retinitis pigmentosa with low-frequency damped electroretinographic wavelets. Eye (Lond). 2009; 23: 230–233. [DOI] [PubMed] [Google Scholar]
- 26. Jacobson SG,, Voigt WJ,, Parel JM,, et al. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology. 1986; 93: 1604–1611. [DOI] [PubMed] [Google Scholar]
- 27. Roman AJ,, Schwartz SB,, Aleman TS. Quantifying rod photoreceptor-mediated vision in retinal degenerations: dark-adapted thresholds as outcome measures. Exp Eye Res. 2005; 80: 259–272. [DOI] [PubMed] [Google Scholar]
- 28. Huang Y,, Cideciyan AV,, Papastergiou GI,, et al. Relation of optical coherence tomography to microanatomy in normal and rd chickens. Invest Ophthalmol Vis Sci. 1998; 39: 2405–2416. [PubMed] [Google Scholar]
- 29. Aleman TS,, Cideciyan AV,, Sumaroka A,, et al. Retinal laminar architecture in human retinitis pigmentosa caused by rhodopsin gene mutations. Invest Ophthalmol Vis Sci. 2008; 49: 1580–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jacobson SG,, Aleman TS,, Cideciyan AV,, et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc Natl Acad Sci U S A. 2005; 102: 6177–6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jacobson SG,, Cideciyan AV,, Huang WC,, et al. TULP1 mutations causing early-onset retinal degeneration: preserved but insensitive macular cones. Invest Ophthalmol Vis Sci. 2014; 55: 5354–5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cideciyan AV,, Swider M,, Jacobson SG. Autofluorescence imaging with near-infrared excitation: normalization by reflectance to reduce signal from choroidal fluorophores. Invest Ophthalmol Vis Sci. 2015; 56: 3393–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Manes G,, Guillaumie T,, Vos WL,, et al. High prevalence of PRPH2 in autosomal dominant retinitis pigmentosa in France and characterization of biochemical and clinical features. Am J Ophthalmol. 2015; 159: 302–314. [DOI] [PubMed] [Google Scholar]
- 34. Jaakson K,, Zernant J,, Külm M,, et al. Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat. 2003; 22: 395–403. [DOI] [PubMed] [Google Scholar]
- 35. Rozet JM,, Gerber S,, Souied E,, et al. Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies. Eur J Hum Genet. 1998; 6: 291–295. [DOI] [PubMed] [Google Scholar]
- 36. Fujinami K,, Lois N,, Davidson AE,, et al. A longitudinal study of Stargardt disease: clinical and electrophysiologic assessment, progression, and genotype correlations. Am J Ophthalmol. 2013; 155: 1075–1088. [DOI] [PubMed] [Google Scholar]
- 37. Webster AR,, Héon E,, Lotery AJ,, et al. An analysis of allelic variation in the ABCA4 gene. Invest Ophthalmol Vis Sci. 2001; 42: 1179–1189. [PubMed] [Google Scholar]
- 38. Michaelides M,, Chen LL,, Brantley MA,, Jr,, et al. ABCA4 mutations and discordant ABCA4 alleles in patients and siblings with bull's-eye maculopathy. Br J Ophthalmol. 2007; 91: 1650–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Herrera W,, Aleman TS,, Cideciyan AV,, et al. Retinal disease in Usher syndrome III caused by mutations in the clarin-1 gene. Invest Ophthalmol Vis Sci. 2008; 49: 2651–2660. [DOI] [PubMed] [Google Scholar]
- 40. Williams DS,, Aleman TS,, Lillo C,, et al. Harmonin in the murine retina and the retinal phenotypes of Ush1c-mutant mice and human USH1C. Invest Ophthalmol Vis Sci. 2009; 50: 3881–3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jacobson SG,, Cideciyan AV,, Gibbs D,, et al. Retinal disease course in Usher syndrome 1B due to MYO7A mutations. Invest Ophthalmol Vis Sci. 2011; 52: 7924–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Curcio CA,, Sloan KR,, Kaline RE,, Hendrickson AE. Human photoreceptor topography. J Comp Neurol. 1990; 292: 497–523. [DOI] [PubMed] [Google Scholar]
- 43. Jacobson SG,, Marmor MF,, Kemp CM,, Knighton RW. SWS, (blue) cone hypersensitivity in a newly identified retinal degeneration. Invest Ophthalmol Vis Sci. 1990; 31: 827–838. [PubMed] [Google Scholar]
- 44. Haider NB,, Jacobson SG,, Cideciyan AV. Mutation of a nuclear receptor gene NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet. 2000; 24: 127–131. [DOI] [PubMed] [Google Scholar]
- 45. Wright AF,, Reddick AC,, Schwartz SB. Mutation analysis of NR2E3 and NRL genes in enhanced S cone syndrome. Hum Mutat. 2004; 24: 439. [DOI] [PubMed] [Google Scholar]
- 46. Coppieters F,, Leroy BP,, Beysen D,, et al. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet. 2007; 81: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Escher P,, Gouras P,, Roduit R,, et al. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum Mutat. 2009; 30: 342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gire AI,, Sullivan LS,, Bowne SJ,, et al. The Gly56Arg mutation in NR2E3 accounts for 1–2% of autosomal dominant retinitis pigmentosa. Mol Vis. 2007; 13: 1970–1975. [PubMed] [Google Scholar]
- 49. Escher P,, Tran HV,, Vaclavik V,, Borruat FX,, Schorderet DF,, Munier FL. Double concentric autofluorescence ring in NR2E3-p.G56R-linked autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2012; 53: 4754–4764. [DOI] [PubMed] [Google Scholar]
- 50. Roduit R,, Escher P,, Schorderet DF. Mutations in the DNA-binding domain of NR2E3 affect in vivo dimerization and interaction with CRX. PLoS One. 2009; 4: e7379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Swain PK,, Chen S,, Wang QL,, et al. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron. 1997; 19: 1329–1336. [DOI] [PubMed] [Google Scholar]
- 52. Sohocki MM,, Sullivan LS,, Mintz-Hittner HA,, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998; 63: 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hull S,, Arno G,, Plagnol V. The phenotypic variability of retinal dystrophies associated with mutations in CRX with report of a novel macular dystrophy phenotype. Invest Ophthalmol Vis Sci. 2014; 55: 6934–6944. [DOI] [PubMed] [Google Scholar]
- 54. Boon CJ,, den Hollander AI,, Hoyng CB,, Cremers FP,, Klevering BJ,, Keunen JE. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008; 27: 213–235. [DOI] [PubMed] [Google Scholar]
- 55. Stuck MW,, Conley SM,, Naash MI. The Y141C knockin mutation in RDS leads to complex phenotypes in the mouse. Hum Mol Genet. 2014; 23: 6260–6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jacobson SG,, Kemp CM,, Sung CH,, Nathans J. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am J Ophthalmol. 1991; 112: 256–271. [DOI] [PubMed] [Google Scholar]
- 57. Jacobson SG,, Kemp CM,, Cideciyan AV,, Macke JP,, Sung CH,, Nathans J. Phenotypes of stop codon and splice site rhodopsin mutations causing retinitis pigmentosa. Invest Ophthalmol Vis Sci. 1994; 35: 2521–2534. [PubMed] [Google Scholar]
- 58. Sung CH,, Makino C,, Baylor D,, Nathans J. A rhodopsin gene mutation responsible for autosomal dominant retinitis pigmentosa results in a protein that is defective in localization to the photoreceptor outer segment. J Neurosci. 1994; 14: 5818–5833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Concepcion F,, Chen J. Q344ter mutation causes mislocalization of rhodopsin molecules that are catalytically active: a mouse model of Q344ter-induced retinal degeneration. PLoS One. 2010; 5: e10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang Z,, Chen Y,, Lilo C,, et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest. 2008; 118: 2908–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Michaelides M,, Gaillard MC,, Escher P,, et al. the PROM1 mutation p.R373C causes an autosomal dominant bull's eye maculopathy associated with rod, rod-cone and macular dystrophy. Invest Ophthalmol Vis Sci. 2010; 51: 4771–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Littink KW,, Koenekoop RK,, van den Born LI,, et al. Homozygosity mapping in patients with cone-rod dystrophy: novel mutations and clinical characterizations. Invest Ophthalmol Vis Sci. 2010; 51: 5943–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zelinger L,, Banin E,, Obolensky A,, et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet. 2011; 88: 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Züchner S,, Dallman J,, Wen R,, et al. Whole exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am J Hum Genet. 2011; 88: 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Venturini G,, Koskiniemi-Kuendig H,, Harper S,, Berson EL,, Rivolta C. Two specific mutations are prevalent causes of recessive retinitis pigmentosa in North American patients of Jewish ancestry. Genet Med. 2015; 17: 285–290. [DOI] [PubMed] [Google Scholar]
- 66. Aleman TS,, Soumittra N,, Cideciyan AV,, et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest Ophthalmol Vis Sci. 2009; 50: 5944–5954. [DOI] [PubMed] [Google Scholar]
- 67. Garanto A,, Vicente-Tejedor J,, Riera M,, et al. Targeted knockdown of Cerkl, a retinal dystrophy gene, causes mild affectation of the retinal ganglion cell layer. Biochim Biophys Acta. 2012; 1822: 1258–1269. [DOI] [PubMed] [Google Scholar]
- 68. Fishman GA,, Stone EM,, Grover S,, Derlacki DJ,, Haines HL,, Hockey RR. Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene. Arch Ophthalmol. 1999; 117: 504–510. [DOI] [PubMed] [Google Scholar]
- 69. Fishman GA,, Stone EM,, Eliason DA,, Taylor CM,, Lindeman M,, Derlacki DJ. ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch Ophthalmol. 2003; 121: 851–855. [DOI] [PubMed] [Google Scholar]
- 70. Burke TR,, Tsand SH. Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet. 2011; 32: 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cideciyan AV,, Hood DC,, Huang Y,, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A. 1998; 95: 7103–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Khan AO,, Abu-Safieh L. Rod-cone dystrophy with initially preserved visual acuity despite early macular involvement suggests recessive CERKL mutations [published online ahead of print February 19 2014]. Ophthalmic Genet. doi:http://dx.doi.org/10.3109/13816810.2014.889168. [DOI] [PubMed]
- 73. Schulte D,, Bumsted-O'Brien KM. Molecular mechanisms of vertebrate retina development: implications for ganglion cell and photoreceptor patterning. Brain Res. 2008; 1192: 151–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.