

Abstract
The LRRK2 G2019S mutation is found at higher frequency among Parkinson disease (PD) patients of Ashkenazi Jewish (AJ) ancestry. This study was designed to test whether an internet-based approach could be an effective approach to screen and identify mutation carriers. Individuals with and without PD of AJ ancestry were recruited and consented through an internet-based study website. An algorithm was applied to a series of screening questions to identify individuals at increased risk to carry the LRRK2 G2019S mutation. About 1000 individuals completed the initial screening. Around 741 qualified for mutation testing and 650 were tested. Seventy-two individuals carried at least one LRRK2 G2019S mutation; 38 with PD (12.5%) and 34 without (10.1%). Among the AJ PD participants, each affected first-degree relative increased the likelihood the individual was LRRK2+ [OR = 4.7; 95% confidence interval = (2.4–9.0)]. The same was not observed among the unaffected AJ subjects (P = 0.11). An internet-based approach successfully screened large numbers of individuals to identify those with risk factors increasing the likelihood that they carried a LRRK2 G2019S mutation. A similar approach could be implemented in other disorders to identify individuals for clinical trials, biomarker analyses and other types of research studies.
Keywords: Parkinson disease, LRRK2, genetic testing
Introduction
Parkinson disease (PD) is the second most frequent neurodegenerative disorder among the elderly. Familial forms of PD comprise only 10% of cases (Elbaz et al. 1999) but have yielded important pathophysiological insights through the identification of novel genes contributing to PD susceptibility. Mutations in five genes result in Mendelian forms of PD. Two genes, leucine-rich repeat kinase 2 (LRRK2) and synuclein, alpha (non-A4 component of amyloid precursor) (SNCA), are mutated in autosomal dominant forms of PD. Three genes, parkin RBR E3 ubiquitin protein ligase (PARK2), DJ1 (PARK7) and PTEN-induced putative kinase 1 (PINK1) are mutated in autosomal recessive forms of PD.
The LRRK2 G2019S mutation is the most common, single cause of PD that has been identified to date. This mutation is found at higher frequency in populations from Northern Africa as well as individuals of Ashkenazi (Eastern European) ancestry. Among PD patients of Ashkenazi Jewish (AJ) ancestry, screening studies have found the frequency of the LRRK2 G2019S mutation to be as high as 15–20% (Orr-Urtreger et al. 2007; Ozelius et al. 2006). The penetrance of this mutation has been reported to be reduced (Goldwurm et al. 2007; Healy et al. 2008; Hentati et al. 2014; Hulihan et al. 2008; Latourelle et al. 2008; Troiano et al. 2010), with the most recent estimates suggesting that the penetrance may be as low as 30% at age 80 years (Marder et al. 2014).
The Parkinson’s Progression Markers Initiative (PPMI) is an observational clinical study using advanced imaging, biologic sampling, and clinical and behavioral assessments to identify biomarkers of PD progression. The focus of PPMI is to identify individuals at greatest risk of developing PD in order to detect biomarkers of phenoconversion and early disease progression. One of the challenges to identify these biomarkers is the identification of individuals at increased risk of PD in whom biomarkers could be monitored. One approach to identify individuals at increased risk of PD is to enrich for subjects more likely to have genetic risk factors, such as the LRRK2 G2019S mutation. Even with the availability of this risk factor, it is still financially costly and time consuming to screen large numbers of individuals to identify those that carry this mutation.
The PPMI study has developed an innovative approach to screen large numbers of individuals from enriched populations. In this study, we focused on the recruitment of individuals of AJ ancestry and then further enriched the sample by asking about a family history of PD. To reduce costs, all individuals completed their screening and consenting process online and provided a saliva sample through the mail for genetic testing. As we describe in this report, our strategy has proven extremely successful and led to the identification of a large number of individuals, both with and without a diagnosis of PD, who carry a LRRK2 G2019S mutation. These individuals have now been invited to participate in the biomarker aspect of the PPMI study.
Methods
Outreach and participant screening
The Michael J. Fox Foundation prepared recruitment material that highlighted the higher frequency of the LRRK2 G2019S mutation in the AJ population. The recruitment materials were widely distributed through print, e-mail and in-person campaigns to individuals of AJ ancestry. In addition, recruitment materials were also distributed broadly by the Michael J. Fox Foundation to individuals interested in PD research. Interested individuals were directed to the Michael J. Fox Foundation PPMI website for an initial screening to determine if they had risk factors that indicated an increased risk of a LRRK2 mutation (Fig.1). Individuals who met the initial criteria were then provided a link which directed them to a website at Indiana University.
Figure 1.
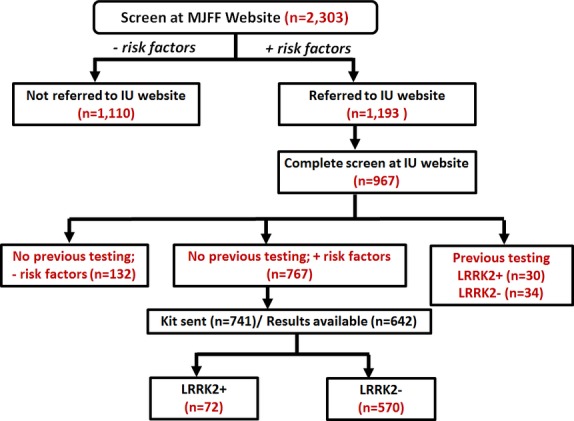
Study overview.
The website at Indiana University presented a fact sheet about LRRK2 and PD. Individuals could then choose to continue to the online Informed Consent document. The Informed Consent document described the study, provided additional information about LRRK2 and PD, and provided the risks and benefits of study participation. Individuals were then asked if they had any questions about the study. If they indicated that they had questions, they were asked to provide their name and contact information so that study personnel could contact them to answer their questions. Individuals who indicated that they had questions were not given the option to agree to the electronic consent or continue with the screening process until after they had been contacted by study personnel. If the individual did not have any questions, they indicated that they agreed to the terms of the Informed Consent form by providing their name. They were then asked to provide their contact information (mailing address, e-mail address and telephone number).
After providing their contact information, the participant was directed to an online study case report form (Fig.1). Participants were asked a series of questions to determine whether they had risk factors that would increase the likelihood that they carried a LRRK2 G2019S or R1441G mutation (Table1). Questions included whether or not they had PD, if they were of AJ ancestry, and whether or not they had a first-degree relative with PD. An algorithm based on the participant’s responses determined the individual’s eligibility to participate in the next phase of the study (Fig.2). Individuals with a diagnosis of PD had to report AJ ancestry and/or a first-degree relative with PD to qualify to receive genetic testing. To qualify for genetic testing, participants who did not have a diagnosis of PD were required to be of AJ ancestry and to also have a first-degree relative with PD. Any individual, regardless of PD status, who reported a first-degree relative with a LRRK2 mutation also qualified for genetic testing. A map of the US PPMI sites was displayed and participants were reminded that participation in the full PPMI study would require that they visit a PPMI site several times over the upcoming years. They were then asked to select the PPMI site that would be most convenient for them to go to for study visits. At the end of the screening questions and site selection, the individual received a message indicating whether they were eligible to participate in the genetic screening phase of the study.
Table 1.
Results for the screening questions for 642 individuals with results available for LRRK2 G2019S genetic testing
| Question | Response | Number of responses (%) | No. LRRK2 G2019S+ (%) (n = 72) | No. LRRK2 G2019S− (%) (n = 570) |
|---|---|---|---|---|
| 1. Do you have Parkinson disease? | Yes | 305 (47.5) | 38 (52.8) | 267 (46.8) |
| No | 337 (52.5) | 34 (47.2) | 303 (53.2) | |
| 2. Have you previously been tested for a possible LRRK2 gene mutation? (no report found to be available) | Yes | 13 (2.0) | 5 (6.9) | 8 (1.4) |
| No | 582 (90.7) | 60 (83.3) | 522 (91.6) | |
| Do not know | 47 (7.3) | 7 (9.7) | 40 (7.0) | |
| 3. Are you of Eastern European (Ashkenazi) Jewish descent? | Yes | 579 (90.2) | 67 (93.1) | 512 (89.8) |
| No | 43 (6.7) | 3 (4.2) | 40 (7.0) | |
| Do not know | 20 (3.1) | 2 (2.3) | 18 (3.2) | |
| 4. Are you of Basque (Northern Spain) descent? (newer question, 356 responding) | Yes | 3 (0.8) | 0 (0.0) | 3 (0.5) |
| No | 335 (94.1) | 35 (48.6) | 300 (52.6) | |
| Do not know | 18 (5.1) | 4 (5.6) | 14 (2.5) | |
| 5. Do any of the following also have PD? | Father | 222 (34.6) | 26 (36.1) | 196 (34.4) |
| Mother | 161 (25.1) | 25 (34.7) | 136 (23.9) | |
| Brother | 50 (7.8) | 5 (6.9) | 45 (7.9) | |
| Sister | 35 (5.5) | 5 (6.9) | 30 (5.3) | |
| Children | 9 (1.4) | 1 (1.4) | 8 (1.4) | |
| 6. Do you have a first-degree relative (father, mother, full sibling, child) with a positive LRRK2 gene test? | Yes | 57 (8.9) | 12 (16.7) | 44 (7.7) |
| No | 143 (22.3) | 14 (19.4) | 129 (22.6) | |
| Do not know | 443 (69.0) | 46 (63.9) | 397 (69.6) |
Figure 2.

Screening algorithm.
Individuals who responded that they had already undergone genetic testing were informed that they would be contacted and asked to send a copy of their test results to the study coordinator for verification. If a positive test result was verified, the individual was offered genetic counseling and then referred directly to a PPMI site to learn more about the PPMI study.
Sample collection and genetic testing
After completing the online screening, each participant was contacted to review their responses and confirm that they wished to receive a saliva collection kit from which DNA would be obtained. Once confirmed, an Oragene DISCOVER (OGR-500) kit (DNA Genotek Inc., Kanata, Ontario, Canada) was shipped to them by mail along with directions for saliva collection. The sample was then shipped back to Indiana University. Samples received at Indiana University were sent for genetic testing at Massachusetts General Hospital. Samples were tested for the LRRK2 G2019S or LRRK2 R144G mutation based on the participant’s response to the screening questions.
Genomic DNA was extracted from patients’ peripheral leukocytes (blood) or buccal epithelial cells (saliva) using QIAcube DNA Purification System by QIAGEN (Valencia, CA). The standard protocols provided by the manufacturer were followed. Patient DNA was PCR amplified using appropriate primer pairs depending on the specific mutation screened (LRRK2 exon 31 c.44321C>A, p.Arg1441Gly; LRRK2 exon 41 c.6055G>A, p.Gly2019Ser). The primers were manufactured by Invitrogen (Life Technologies, Grand Island, NY) and each primer pair was used to amplify its corresponding exon and the adjacent intronic sequences. Positive mutations were identified by comparison of bidirectional sequence data against reference sequence.
Genetic testing results were sent by Massachusetts General Hospital to Indiana University. An appointment was made for the genetic counselor to review the genetic testing results with the participant by telephone. Following counseling, the participant was provided with a written summary of the genetic testing results and counseling session.
Statistical analysis
Logistic regression was performed to identify those variables that predicted whether an individual was LRRK2 positive (LRRK2+) or LRRK2 negative (LRRK2−). Due to the differential screening criteria for affected and unaffected individuals, analyses were performed in each group separately. Analyses performed in the two subject groups of AJ descent (PD and unaffected) included as potential predictors age, gender and the number of first-degree relatives with PD. Secondary analyses were performed testing for the effect of each type of relative with PD (father, mother, brother, sister, offspring).
Results
The Michael J. Fox Foundation and Indiana University websites were activated on February 25, 2014. All results are reported for individuals who completed their consent at the Indiana University site by September 1, 2014. Missing data or kits are truncated as of October 20, 2014. A total of 2303 individuals completed the initial survey at the Michael J. Fox Foundation website and 1193 individuals qualified to be directed to the Indiana University website for further screening (see Fig.1).
A total of 967 individuals completed the consent process at the Indiana University website. Responses to the screening questions are shown in Table1. Eighty-one individuals reported having previous genetic testing which they believed indicated they carried a LRRK2 mutation. Sixty-four of these individuals were able to provide a copy of their testing report to Indiana University for review. Upon review of the genetic testing results, slightly more than half of this group (53.1%) did not carry a LRRK2 G2019S mutation. The majority of these results were from testing performed by 23andMe. Of the remaining 17 individuals who indicated previous testing, 4 could not be reached by the study coordinator and 13 could not produce a copy of their results. For those 13 unable to provide their test results, responses to the screening questions were used to determine eligibility for a genetic testing.
Table2 summarizes basic demographic information for the 899 individuals who completed the screening questions at the Indiana University website to determine eligibility for genetic testing. Of these 899 individuals, 395 reported a diagnosis of PD and 504 did not. The study coordinator attempted to contact all individuals to verify their screening responses. Of note, 14 individuals who had reported AJ ancestry during the survey reported that they were not actually of AJ descent during these calls, and eight individuals who had reported a relative with a known LRRK2 mutation could not confirm that such testing had been performed.
Table 2.
Summary and demographic information for individuals who completed initial screening at the Indiana University website by January 9, 2014
| Number | % | % M | % PD | |
|---|---|---|---|---|
| Consented through WRI site, no previous testing | 899 | 100 | 42 | 44 |
| Qualified for genetic testing | 767 | 85 | 45 | 47 |
| Qualified and confirmed through WRI | 741 | 82 | 45 | 47 |
| Did not qualify through WRI | 132 | 15 | 26 | 29 |
| Unable to contact to confirm further participation | 15 | 2 | 20 | 47 |
| Declined further participation in study | 11 | 1 | 46 | 27 |
| Kits sent | 7411 | 100 | 45 | 47 |
| Kits not returned | 91 | 12 | 33 | 45 |
| Kits with LRRK2 G2019S results from MGH | 642 | 87 | 47 | 48 |
| LRRK2 G2019S | 642 | 100 | 47 | 48 |
| LRRK2 G2019S- | 570 | 89 | 47 | 47 |
| LRRK2 G2019S+ | 72 | 11 | 44 | 53 |
| Consented through WRI site, with previous testing report available | 64 | 100 | 41 | 66 |
| LRRK2 G2019S- | 34 | 53 | 35 | 79 |
| LRRK2 G2019S+ | 30 | 47 | 47 | 50 |
Genetic testing results have not been received for 5 individuals, and there were 3 sample failures.
An algorithm was applied to determine eligibility for genetic testing (Fig.2). Individuals with a diagnosis of PD had to report at least one of the following to qualify: (1) AJ ancestry (n = 299); (2) first-degree relative with PD (n = 58); or (3) first-degree relative with a LRRK2 mutation (n = 1). A total of 358 individuals with PD met these requirements for a saliva kit. Individuals who did not have a diagnosis of PD were required to meet one of the two following criteria: (1) have a first-degree relative with a LRRK2 mutation (n = 18); or (2) be of AJ ancestry and have a first-degree relative with PD (n = 391). About 409 unaffected individuals met these requirements for a saliva kit used for genetic testing.
Only a small number (n = 26) who qualified to receive a saliva kit elected not to do so or could not be reached by the study coordinator to confirm participation. Among the 741 individuals sent a saliva kit, 650 (87.7%) returned the filled kit to Indiana University. The vast majority of individuals were screened for the LRRK2 G2019S mutation (99.9%). Only five individuals qualified to be screened for the LRRK2 R1441G mutation in addition to the LRRK2 G2019S, and one for the LRRK2 R1441G mutation only. None of the subjects were positive for the LRRK2 R1441G mutation.
LRRK2 G2019S testing results were available for a total of 642 individuals (Table1). Results for 5 individuals were still pending and 3 samples failed when tested. This group of 642 individuals included 305 with a diagnosis of PD (47.5%) and 337 who did not. The total number of individuals carrying a LRRK2 G2019S mutation was 72 (11.2%). Among those with PD, the number carrying a LRRK2 G2019S mutation was 38 (12.5%) and among those without a diagnosis of PD, the number was 34 (10.1%). There were two individuals who were homozygous for the LRRK2 G2019S mutation. Both had been previously diagnosed with PD; however, only one had a family history of PD.
The family history question on the screening survey was a strong predictor of LRRK2 mutation status. A majority (76.4%) of individuals found to have a LRRK2 G2019S mutation had at least one relative with PD, and 9.7% reported more than one relative with PD. Among those carrying the LRRK2 mutation, the most frequently reported relative with PD was a parent; 36.1% reported their father with PD, and 34.7% reported an affected mother. Two LRRK2 G2019S heterozygotes (2.8%) reported both their parents had PD.
Of the individuals with LRRK2 G2019S testing results available, there were 254 AJ individuals with PD. Although the majority did not carry a LRRK2 mutation (n = 219, 86.2%), there were 35 individuals who did (13.8%). Neither age nor gender were significant predictors of LRRK2 mutation status (P > 0.94). However, the number of affected relatives was a significant predictor of mutation status (Wald χ2 = 21.0, df = 1, P < 4.9 × 10−6). For each additional affected first-degree relative, the odds that the study participant had a LRRK2 mutation increased by 4.7 [95% confidence interval (CI) = (2.4–9.0)]. Secondary analyses revealed that having an affected father significantly increased the odds the participant carried a LRRK2 mutation by 3.7 [P = 0.01, 95% CI = (1.4–9.8)]. If the participant’s mother was affected, the odds the participant was LRRK2+ more than doubled, with an odds ratio of 8.1 [P = 3.9 × 10−5, 95% CI = (3.0–22.1)].
Of the individuals with results available, there were 325 AJ individuals who did not report a diagnosis of PD. Of those, 293 were LRRK2− (90.1%), and 32 were LRRK2+ (9.9%). None of the tested variables was a significant predictor of LRRK2 status (gender P = 0.83, age at interview P = 0.08, number of relatives P = 0.11).
Discussion
The identification of individuals who have inherited a LRRK2 mutation is challenging. In this study, we designed an efficient approach that allowed us to screen nearly 1000 individuals to determine if they carried risk factors that increased the likelihood that they carried a LRRK2 mutation. With this strategy, we newly identified 72 individuals who carry a LRRK2 G2019S mutation and also confirmed the result for 30 individuals who had received previous genetic testing. Thus, in only 6 months, we identified over 100 individuals who qualified to participate in the PPMI biomarker study focused on longitudinal evaluation of individuals carrying a LRRK2 G2019S mutation.
Previous studies have reported that approximately 15–20% of PD patients of AJ ancestry carry the LRRK2 G2019S mutation. In this study, we found a rate of 12.5%. This is slightly lower than the previously reported rate, but still supports the higher frequency of this mutation in this population. Prior to initiating this study, we predicted that unaffected individuals of AJ ancestry who had a first-degree relative with PD would also have an elevated frequency of the G2019S mutation. We anticipated that this rate would be half that of the PD population. In this study, we found the rate to be 10.2%, slightly higher than we would have anticipated. This may be in part due to some unaffected individuals being referred to the study by a family member who learned of their LRRK2 carrier status.
From this study, we confirmed the importance of collecting family history information prior to genetic testing for the LRRK2 G2019S mutation. Individuals with PD were not required to have another family member with PD to qualify for genetic testing. However, the presence of each additional affected family member with a mutation increased the likelihood that an AJ individual with PD carried the LRRK2 mutation by 4.7%. Individuals who were unaffected were not eligible for genetic testing unless they had a first-degree relative with PD. We found that among this group, the number of additional affected family members was not a significant predictor of LRRK2 status.
We did find that individuals with an affected parent were more likely to carry a LRRK2 mutation as compared with those who did not have an affected parent. Furthermore, in our study, having an affected mother was a stronger predictor of LRRK2+ status as compared with an affected father. These results appear to be consistent with an earlier study by Alcalay et al. (2013) which found that the penetrance of LRRK2 G2019S may be higher in women than men. If female carriers of the LRRK2 mutation are more likely to be affected, we would expect to find more affected mothers than affected fathers in our sample. This finding requires further evaluation in additional samples.
Potential study participants appeared to be unsure of previous genetic testing in family members. On the initial screen, 92 individuals reported that a family member had been previously tested and found to carry a LRRK2 mutation. When the subject was contacted by the study coordinator to confirm this information, the majority was unable to confirm that a genetic test had been performed and was positive. In these cases, if the subject still met criteria for genetic testing, they were sent a saliva kit. If they did not qualify for a genetic testing, they did not participate further in the study.
The cost of recruiting subjects through an internet-based strategy as used in this study is difficult to quantify. Study personnel developed the website and databases required for this study. A full time coordinator was available to answer questions and also spoke by telephone to each individual who qualified for genetic testing prior to the shipping of the saliva kit. This personal interaction likely increased the commitment of the study participants. Surprisingly, few reminders to return a saliva kit were required in this subject group. Anecdotally, subjects found it convenient to participate in the study without leaving their home. Thus, by coupling a convenient online participation with a telephone call with study staff, we likely ensured that those subjects who received the saliva kit were likely to also return the kit and receive study results.
The approach used in this study could be applied quite easily for the testing of other PD susceptibility genes or to other disorders. We have just initiated recruitment for another risk factor, glucocerebrosidase (GBA), using the same study design. There is an ongoing study that has implemented an internet-based program for AJ carrier screening for multiple genetic disorders (Grinzaid et al. 2014). There is growing interest to utilize precision genomics, which will target disease treatment based on the patient’s underlying genetic risk profile. It will be essential that trials can be initiated that recruit participants with particular risk factors or mutations. Approaches such as ours could dramatically increase the rate at which such focused patient populations could be efficiently identified.
One weakness of the study was a targeted recruitment, focused largely on individuals of AJ ancestry. Therefore, the conclusions drawn about the participation rates and the frequency of LRRK2 G2019S mutation are not applicable to individuals of other ancestry. However, results suggest that this recruitment strategy is successful in identifying an at-risk population. This study had several other strengths. The population being recruited was highly engaged, and for the most part had access to the internet. Only a small number of individuals (<20) required the screening questionnaire to be mailed to them because they did not have internet access. As a group, this subject population was motivated to participate fully in the study. Among those who qualified for genetic testing based on their screening questions, only 11 (1.4%) did not wish to receive a saliva kit and 15 (2.0%) did not respond to the coordinator’s telephone call to confirm their continued participation. Upon receiving a saliva kit, 87.7% returned the filled saliva kit. A total of 91 individuals (12.3%) did not return their saliva kit. Given that this is a study conducted largely by mail and the internet, the return rate for the saliva kits is very high.
In summary, we used an internet-based approach to screen large numbers of individuals to identify those with risk factors increasing the likelihood that they carried a mutation contributing to PD susceptibility. This was a highly efficient approach that in only 6 months yielded over 100 individuals who carried a LRRK2 mutation. We believe that a similar approach could be implemented in other disorders to identify individuals for clinical trials, biomarker analyses, and other types of research studies.
Acknowledgments
We thank the participants and their families for their support of this research study.
Appendix
Steering committee
Kenneth Marek, MD1 (Principal Investigator); Danna Jennings, MD1 (Site Investigator, Olfactory Core, PI); Shirley Lasch, MBA1; Caroline Tanner, MD, PhD9 (Site Investigator);Tanya Simuni, MD3 (Site Investigator); Christopher Coffey, PhD4 (Statistics Core, PI); Karl Kieburtz, MD, MPH5 (Clinical Core, PI); Renee Wilson, MA5; Werner Poewe, MD7 (Site Investigator); Brit Mollenhauer, MD8 (Site Investigator); Douglas Galasko, MD15; Tatiana Foroud, PhD 16 (Genetics Coordination Core, BioRepository PI); Todd Sherer, PhD6; Sohini Chowdhury6; Mark Frasier, PhD6; Catherine Kopil, PhD6; Vanessa Arnedo6.
Study cores
Clinical Coordination Core: Cynthia Casaceli, MBA5; Imaging Core: John Seibyl, MD 1; Susan Mendick, MPH1; Norbert Schuff, PhD9; Statistics Core: Christopher Coffey, PhD4; Chelsea Caspell 4; Liz Uribe4; Eric Foster 4; Katherine Gloer PhD4; Jon Yankey MS4; Bioinformatics Core: Arthur Toga, PhD10 Principal Investigator), Karen Crawford, MLIS10; BioRepository: Danielle Elise Smith16; Paola Casalin12; Giulia Malferrari12; Bioanalytics Core: John Trojanowski, MD, PhD13 (Principal Investigator), Les Shaw, PhD13 (Co-Principal Investigator); Genetics Core: Andrew Singleton, PhD14, (Principal Investigator); Genetics Coordination Core: Cheryl Halter16.
Site investigators
David Russell, MD, PhD1 Site; Stewart Factor, DO17; Penelope Hogarth, MD18; David Standaert, MD, PhD19; Robert Hauser, MD, MBA20; Joseph Jankovic, MD21; Matthew Stern, MD13; Lama Chahine, MD13; Shu-Ching HU, MD PhD22; Samuel Frank, MD23; Claudia Trenkwalder, MD8,; Wolfgang Oertel MD34; Irene Richard, MD24; Klaus Seppi, MD7; Eva Reiter, MD7; Holly Shill, MD 25; Hubert Fernandez, MD 26; Anwar Ahmed, MD26; Daniela Berg, MD 27; Isabel Wurster MD27; Zoltan Mari, MD28; David Brooks, MD29; Nicola Pavese, MD29; Paolo Barone, MD, PhD30; Stuart Isaacson, MD31; Alberto Espay, MD, MSc 32; Dominic Rowe, MD, PhD33; Melanie Brandabur MD2; James Tetrud MD2; Grace Liang MD10Karen Marder35; Jean-Christophe Corvol36; Jose Felix Martí Masso37; Eduardo Tolosa38; Jan O. Aasly39; Nir Giladi40; Leonidas Stefanis41.
Coordinators
Laura Leary1; Cheryl Riordan1; Linda Rees, MPH1; Barbara Sommerfeld, RN, MSN17; Cathy Wood-Siverio, MS17; Alicia Portillo18; Art Lenahan18; Karen Williams3; Stephanie Guthrie, MSN19; Ashlee Rawlins19; Sherry Harlan20; Christine Hunter, RN21; Baochan Tran13; Abigail Darin13; Carly Linder13; Gretchen Todd22; Cathi-Ann Thomas, RN, MS23; Raymond James, RN23; Cheryl Deeley, MSN24; Courtney Bishop BS24; Fabienne Sprenger, MD7; Diana Willeke8; Sanja Obradov25; Jennifer Mule26; Nancy Monahan26; Katharina Gauss27; Kathleen Comyns9 Deborah Fontaine, BSN, MS, RN, GNP, MS 15; Christina Gigliotti15; Arita McCoy28; Becky Dunlop28; Bina Shah, BSc29; Susan Ainscough30; Angela James32; Rebecca Silverstein31; Kristy Espay32; Madelaine Ranola33; Helen M. Santana35; Nelly Ngono36; Elisabet Rezola37 (; Delores Vilas Rolan38; Bjorg Waro39; Anat Mirlman40; Maria Stamelou41.
Industry Scientific Advisory Board
Thomas Comery, PhD42; Spyros Papapetropoulos, MD, PhD42; Bernard Ravina, MD, MSCE43; Igor D. Grachev, MD, PhD44; Jordan S. Dubow, MD45; Michael Ahlijanian, PhD46; Holly Soares, PhD46; Suzanne Ostrowizki, MD, PhD47; Paulo Fontoura, MD, PhD47; Alison Chalker, PhD48; David L. Hewitt, MD48; Marcel van der Brug, PhD49; Alastair D. Reith, PhD50; Peggy Taylor, ScD51; Jan Egebjerg, PhD52; Mark Minton, MD53; Andrew Siderowf, MD, MSCE53; Pierandrea Muglia, PhD54; Robert Umek, PhD55; Ana Catafau, MD, PhD56.
1Institute for Neurodegenerative Disorders, New Haven, CT; 2The Parkinson’s Institute, Sunnyvale, CA; 3Northwestern University, Chicago, IL; 4University of Iowa, Iowa City, IA; 5Clinical Trials Coordination Center, University of Rochester, Rochester, NY; 6The Michael J. Fox Foundation for Parkinson’s Research, New York, NY; 7Innsbruck Medical University, Innsbruck, Austria; 8Paracelsus-Elena Klinik, Kassel, Germany; 9University of California, San Francisco, CA; 10Laboratory of Neuroimaging (LONI), University of Southern California; 11Coriell Institute for Medical Research, Camden, NJ; 12BioRep, Milan, Italy; 13University of Pennsylvania, Philadelphia, PA; 14National Institute on Aging, NIH, Bethesda, MD; 15University of California, San Diego, CA; 16Indiana University, Indianapolis, IN; 17Emory University of Medicine, Atlanta, GA; 18Oregon Health and Science University, Portland, OR; 19University of Alabama at Birmingham, Birmingham, AL; 20University of South Florida, Tampa, FL; 21Baylor College of Medicine, Houston, TX; 22University of Washington, Seattle, WA; 23Boston University, Boston, MA; 24University of Rochester, Rochester, NY; 25Banner Research Institute, Sun City, AZ; 26Cleveland Clinic, Cleveland, OH; 27University of Tuebingen, Tuebingen, Germany; 28Johns Hopkins University, Baltimore, MD; 29Imperial College of London, London, UK; 30University of Salerno, Salerno, Italy; 31Parkinson’s Disease and Movement Disorders Center, Boca Raton, FL; 32University of Cincinnati, Cincinnati, OH; 33Macquarie University, Sydney Australia; 34Philipps University Marburg, Germany; 35Columbia Medical, New York, NY; 36Pitié-Salpêtrière Hospital, Paris France; 37University of Donostia-Service of Neurology Hospital, San Sebastian, Spain; 38University of Barcelona-Hospital Clinic of Barcelona, Barcelona, Spain; 39Norwegian University of Science and Technology, Trondheim, Norway; 40Tel Aviv Sourasky Medical Center, Tel Aviv, Isreal; 41Foundation for Biomedical research of the Academy of Athens, Athens, Greece; 42Pfizer, Inc., Groton, CT; 43Biogen Idec, Cambridge, MA; 44GE Healthcare, Princeton, NJ; 45AbbVie, Abbot Park, IL; 46Bristol-Myers Squibb Company; 47F.Hoffmann La-Roche, Basel, Switzerland; 48Merck & Co., North Wales, PA; 49Genentech, Inc., South San Francisco, CA; 50GlaxoSmithKline, Stevenage, United Kingdom; 51Covance, Dedham, MA; 52H. Lundbeck A/S; 53Avid Radiopharmaceuticals, Philadelphia, PA; 54UCB Pharma S.A., Brussels, Belgium; 55Meso Scale Discovery; 56Piramal Life Sciences, Berlin, Germany.
Conflict of Interest
Kenneth Marek has ownership in Molecular NeuroImaging, LLC and is a consultant for Pfizer, GE Healthcare, Merck, Lilly, BMS, Piramal, Prothena, Neurophage, LTI, and Roche.
References
- Alcalay RN, Mirelman A, Saunders-Pullman R, Tang MX, Mejia Santana H, Raymond D, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord. 2013;28:1966–1971. doi: 10.1002/mds.25647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbaz A, Grigoletto F, Baldereschi M, Breteler MM, Manubens-Bertran JM, Lopez-Pousa S, et al. Familial aggregation of Parkinson’s disease: a population-based case-control study in Europe. EUROPARKINSON Study Group. Neurology. 1999;52:1876–1882. doi: 10.1212/wnl.52.9.1876. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Zini M, Mariani L, Tesei S, Miceli R, Sironi F, et al. Evaluation of LRRK2 G2019S penetrance: relevance for genetic counseling in Parkinson disease. Neurology. 2007;68:1141–1143. doi: 10.1212/01.wnl.0000254483.19854.ef. [DOI] [PubMed] [Google Scholar]
- Grinzaid KA, Page PZ, Denton JJ, Ginsberg J. Creation of a National, At-home Model for Ashkenazi Jewish Carrier Screening. J Genet Couns. 2014 doi: 10.1007/s10897-014-9800-x. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentati F, Trinh J, Thompson C, Nosova E, Farrer MJ. Aasly JO. LRRK2 parkinsonism in Tunisia and Norway: a comparative analysis of disease penetrance. Neurology. 2014;83:568–569. doi: 10.1212/WNL.0000000000000675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, et al. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet Neurol. 2008;7:591–594. doi: 10.1016/S1474-4422(08)70116-9. [DOI] [PubMed] [Google Scholar]
- Latourelle JC, Sun M, Lew MF, Suchowersky O, Klein C, Golbe LI, et al. The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med. 2008;6:32. doi: 10.1186/1741-7015-6-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder K, Tang M, Alcalay RN, Meija-Santana H, Raymond D, Mirelman A, et al. Age specific penetrance of the LRRK2 G2019S mutation in the Michael J Fox Ashkenazi Jewish (AJ) LRRK2 consortium. Neurology. 2014;82:S17.002. doi: 10.1212/WNL.0000000000001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Shifrin C, Rozovski U, Rosner S, Bercovich D, Gurevich T, et al. The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: is there a gender effect? Neurology. 2007;69:1595–1602. doi: 10.1212/01.wnl.0000277637.33328.d8. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, et al. LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- Troiano AR, Elbaz A, Lohmann E, Belarbi S, Vidailhet M, Bonnet AM, et al. Low disease risk in relatives of North African lrrk2 Parkinson disease patients. Neurology. 2010;75:1118–1119. doi: 10.1212/WNL.0b013e3181f39a2e. [DOI] [PubMed] [Google Scholar]