

Abstract
Glycosaminoglycans (GAGs) such as chondroitin are ubiquitous disaccharide carbohydrate chains that contribute to the formation and function of proteoglycans at the cell membrane and in the extracellular matrix. Although GAG-modifying enzymes are required for diverse cellular functions, the role of these proteins in human development and disease is less well understood. Here, we describe two sisters out of seven siblings affected by congenital limb malformation and malignant lymphoproliferative disease. Using Whole-Genome Sequencing (WGS), we identified in the proband deletion of a 55 kb region within chromosome 12q23 that encompasses part of CHST11 (encoding chondroitin-4-sulfotransferase 1) and an embedded microRNA (MIR3922). The deletion was homozygous in the proband but not in each of three unaffected siblings. Genotyping data from the 1000 Genomes Project suggest that deletions inclusive of both CHST11 and MIR3922 are rare events. Given that CHST11 deficiency causes severe chondrodysplasia in mice that is similar to human limb malformation, these results underscore the importance of chondroitin modification in normal skeletal development. Our findings also potentially reveal an unexpected role for CHST11 and/or MIR3922 as tumor suppressors whose disruption may contribute to malignant lymphoproliferative disease.
Keywords: CHST11, inherited lymphoproliferative disorder, malignant lymphoproliferative disorder, MIR3922, skeletal malformation
Introduction
Glycosaminoglycans (GAGs) such as chondroitin and dermatan are a heterogeneous group of unbranched polysaccharides of variable length that consist of repeating disaccharides. With the exception of hyaluronic acid, GAGs are synthesized in the Golgi and then modified by enzymes including sulfotransferases. Chondroitin (the most prevalent GAG), heparin, dermatan, and keratin are all sulfated and subsequently appended to newly synthesized proteins via O- or N-linked glycosylation to form mature proteoglycans. Proteoglycans function at the cell membrane or as secreted components of the extracellular matrix (Theocharis et al. 2010).
Although GAG-modifying genes such as sulfotransferases are required for normal GAG and proteoglycan synthesis, the role of these genes in human development and disease is less well understood. Here, we report a previously undescribed syndrome whose cardinal features include bilateral bony deformities of upper and lower extremities and susceptibility to malignant lymphoproliferative disease. The phenotype was present in two of seven siblings but in neither parent, suggestive of a genetic disorder with an autosomal recessive mode of inheritance. Whole-Genome Sequencing (WGS) of the proband revealed a biallelic 55 kb deletion at chromosome 12q23 spanning part of the locus for CHST11 and including an embedded MIR3922. Nullizygosity for the deletion was observed in the proband and not in three unaffected living siblings who subsequently underwent testing. When considered together with the known role for CHST11 in murine cartilage growth plate formation during development, our findings strongly suggest that chondroitin sulfation is required for normal development of the bony skeleton in humans. Our findings also support the hypothesis that CHST11 and/or MIR3922, a microRNA whose function has not previously been described, may act as tumor suppressors in the lymphocyte lineage.
Clinical history
The proband is a 46-year-old woman who initially presented to the adult genetics clinic at Brigham and Women’s Hospital in 2005 for evaluation of a personal and family history of congenital hand/foot malformations and peripheral T-cell lymphoma. At 61 in. in height (10th percentile, CDC growth curves), she is shorter than all of her first-degree relatives except one similarly affected sister. She was born with bony abnormalities of the fingers and toes bilaterally, for which she underwent multiple surgical repairs in early childhood. On clinical exam, her fingers demonstrated brachydactyly with disproportionately short index fingers and either valgus or varus deformity at many of the interphalangeal (IP) joints (Fig.1A). Similar findings were observed in the toes. X-ray images were taken of the hands and feet, with multiple notable findings. On the left hand, the index finger displayed a long proximal phalanx, a fused distal phalanx and subluxation of the index metacarpophalangeal (MCP) joint with remodeling of the bony surfaces; the third digit had four phalanges and a horizontal articulation at the base of the proximal phalanx; the fourth and fifth fingers each had long proximal phalanges; and the fifth finger middle phalanx had a curved articulation distally (Fig.1B). The metacarpals appeared more normal with the exception of a hypoplastic index metacarpal. The thumbs and wrist bones appeared normal. Similar findings were observed in radiographs of the right hand (Fig.1C).
Figure 1.
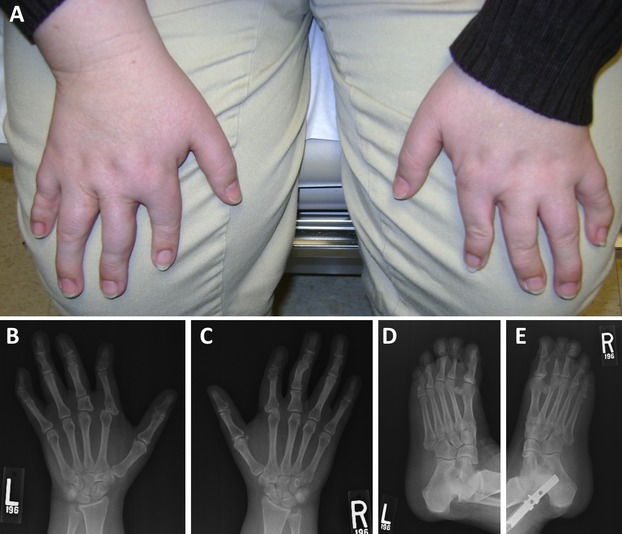
(A) At present, the hands of the proband are notable for malformed digits. (B and C) X-rays of the hands at present. See text for detailed description. (D and E) X-rays of the feet at present. See text for detailed description.
In the left foot, plain films revealed fusion across the great toe IP joint; small middle phalanges of the second and third toes; and only two phalanges of the fourth toe with a well-defined lucency at the base of the proximal phalanx of the fourth toe, thought to be a separate ossicle (Fig.1D). The fifth toe showed a faint lucency with sclerotic margins across the proximal phalanx. The metatarsals demonstrated no abnormalities. The right foot displayed similar abnormalities (Fig.1E).
The proband’s medical history was further complicated at 44 years of age, when she developed several weeks of fever, flu-like symptoms, and pulmonary infiltrates unresponsive to antibiotics that culminated in respiratory failure and mechanical ventilation. Extensive testing for an infectious etiology was negative and she was diagnosed with acute respiratory distress syndrome (ARDS) of uncertain etiology. Her symptoms ultimately improved with high-dose steroids but recurred when steroids were discontinued. Surgical lung biopsy revealed an atypical lymphoid infiltrate consisting primarily of CD3+ T cells but with a minor component of CD20+ B cells. Immunoglobulin heavy chain polymerase chain reaction (PCR) did not identify any evidence of B-cell clonality. The histologic pattern of an angiocentric lymphoid infiltrate was suggestive of lymphomatoid granulomatosis, an Epstein–Barr virus (EBV)-associated lymphoproliferative disorder. However, additional evidence supporting this diagnosis including atypical lymphoid cells reactive for B-cell antigens or EBV-associated mRNAs was not identified. Bone marrow biopsy was unremarkable. The patient was restarted on antibiotics and steroids and improved clinically. Her lung biopsy was reviewed at the National Cancer Institute and the overall impression was that of a reactive process, possibly an organizing pneumonia, rather than a primary lymphoproliferative disorder such as lymphomatoid granulomatosis, although the latter could not be definitively ruled out. The patient’s condition ultimately went without diagnosis.
She subsequently underwent a second lung biopsy, again revealing nodular lymphoid infiltrates of primarily CD3+ T cells with scattered CD20+ B cells and CD68+ histiocytes. PCR for rearranged immunoglobulin heavy chain and T-cell receptor (TCR) genes did not demonstrate any evidence of B- or T-cell clonality, respectively. Flow cytometry of peripheral blood lymphocytes revealed no abnormalities. Overall, definitive diagnostic features of a T- or B-cell lymphoma were not observed. Based on clinical suspicion of malignancy, the patient was treated with rituximab followed by four cycles of chemotherapy with cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP). The patient’s clinical symptoms improved in concert with partial regression of her pulmonary infiltrates. New wedge biopsies of the right middle and upper lobes revealed a nodular, angiocentric and bronchocentric atypical T-cell lymphoid infiltrate extending into intra-alveolar spaces. The infiltrate was composed of medium size and large atypical lymphoid cells admixed with small lymphocytes and histiocytes. Immunohistochemical studies revealed that the majority of large atypical lymphoid cells stained positively for the T-cell markers CD3, CD4, and CD8 (minority). CD5 expression was absent but for a small number of cells. The infiltrating T cells were also positive for CD43 and TIA-1 in a granular cytoplasmic pattern. EBV LMP-1 was negative, as were B-cell markers CD20 and CD79a and the natural killer cell marker CD56. Additional negative markers included CD30, CD15, CD34, MPO, and CD1a. Ki-67 stained approximately 20–30% of cells. EBV mRNA by in situ hybridization was also negative. On this occasion, TCR gamma gene rearrangement studies revealed evidence of clonality. The patient’s progressive clinical course, the cytologically atypical T-cell infiltrate, the immunophenotypic aberrancy (absence of CD5), the lack of EBV mRNA, and molecular evidence of clonality with an identical pattern observed at two different anatomical sites (right middle lobe, right upper lobe) were most consistent with a diagnosis of peripheral T-cell lymphoma. She received additional chemotherapy prior to undergoing a matched unrelated allogeneic bone marrow transplant from a male donor. Her clinical course has been complicated only by grade 1 graft versus host disease and several non-life-threatening infections. She continues to have no evidence of disease on periodic restaging positron emission tomography–computed tomography (PET-CT) scans.
The proband’s family history is most notable for an older sister with similar congenital malformation of the hands/feet who succumbed to complications of an unnamed lymphoproliferative disorder in her early 20s. At a height of sixty inches, the deceased sister was also shorter than all other first-degree relatives. A review of her medical records from the 1980s revealed that she presented at 17 years of age with abdominal pain, nausea, and vomiting, subsequently found to be caused by a small bowel obstruction complicated by perforation and pneumoperitoneum. She also described a several week history of skin lesions of uncertain origin, fever, shortness of breath, and tachypnea. Radiographic imaging of the lungs revealed diffuse bilateral nodular infiltrates. Management of the small bowel perforation ultimately included partial ileal resection, right-sided colectomy, and jejunostomy. Per report, pathologic evaluation of the resected small bowel revealed dense lymphocytic infiltrates. Skin biopsy similarly revealed a lymphocytic infiltrate. Biopsy of the lung was notable for an angiocentric lymphocytic infiltrate resembling lymphomatoid granulomatosis, the same EBV-related condition initially suspected to underlie the proband’s pulmonary lymphoid infiltrates. A bone marrow biopsy was reportedly normal on two occasions. No additional molecular diagnostic testing was available and/or performed at the time. She was treated with cyclophosphamide, azathioprine, and prednisone with resolution of both pulmonary infiltrates and cutaneous manifestations. However, her course was subsequently complicated by new-onset seizures. Computed tomography (CT) imaging suggested involvement of the central nervous system (CNS) with the lymphoproliferative disorder. A brain biopsy was performed and the pathology was consistent with malignant lymphoma of the CNS, not otherwise specified. She ultimately died from complications of her disease at age 22.
The proband and her deceased sister have five unaffected living siblings (Fig.2). No other individuals in the extended family are reported to have congenital abnormalities of the hands or feet. With the exception of the proband’s father, who was diagnosed with chronic lymphocytic leukemia (CLL) at age 59 and died at age 64, no other individuals have developed any lymphoproliferative disorders. The proband’s parents were both immigrants to the US from the same region of Ireland, so the possibility of distant consanguinity cannot be ruled out. The family history is most consistent with a rare monogenic disorder of autosomal recessive inheritance.
Figure 2.

The pedigree demonstrates a large sibship with two affected individuals, one deceased. The table below the pedigree indicates the genotyping results for the proband and the three siblings who underwent testing. The data include number of CHST11 deletion alleles (homozygous null, heterozygous, or homozygous wild type) as well as results of droplet digital PCR (exon 2, intron 1), including mean and standard deviation. STR genotyping results (for chimerism) are also indicated.
A search of online databases including OMIM and PubMed did not identify any named syndrome associated with both skeletal dysplasia and any type of lymphoproliferative disorder. Therefore, no specific candidate gene rose to a level of suspicion to warrant clinical testing. The results of chromosome breakage analysis with diepoxybutane (DEB) were normal and therefore did not support a diagnosis of Fanconi anemia (FA). Array-based comparative genomic hybridization using a custom-designed platform with 180,880 probes throughout the human genome did not reveal any clinically significant genomic gains or losses. Therefore, we performed WGS on genetic material obtained from the proband to identify rare homozygous variants that could potentially explain the phenotype in this family.
Methods
Consent
Consent to perform WGS under Clinical Laboratory Improvement Amendments (CLIA) conditions was obtained under protocols approved by the Institutional Review Board (IRB) of Partners HealthCare, Inc. Explicit permission to publish the findings contained herein was obtained from the proband.
Skin biopsy and fibroblast culture
Due to the patient’s prior allogeneic bone marrow transplant, which resulted in reconstitution of the entire hematopoietic system by donor cells, we isolated and sequenced genomic DNA from cultured skin fibroblasts rather than from circulating (donor) hematopoietic cells (Valencia et al. 2014). The skin biopsy was rinsed, minced, and then cultured in P-60 dishes containing 5 mL complete minimal essential media medium with 10% fetal bovine serum (FBS), penicillin and streptomycin (100 units/mL), and gentamicin (50 μg/mL). The dishes were incubated at 37°C with 5% CO2 and examined daily for cell growth. When the cells became 90–100% confluent in culture, genomic DNA was isolated from fibroblasts using the Gentra Puregene Cell Kit (QIAGEN, Venlo, Netherlands) and subsequently used for the WGS sequencing.
Whole-genome sequencing
WGS was performed by the Illumina Clinical Services Laboratory (Illumina, Inc; San Diego, CA). Briefly, genomic DNA was randomly fragmented and then sequenced using 50 nt paired-end reads on an Illumina HiSeq 2000 Sequencer at an average depth of 30× coverage (Bentley et al. 2008). Resulting BAM files were converted to FASTQ format and realigned to the human reference sequence [University of California Santa Cruz (UCSC) hg19/GRCh37 assembly] using the Burroughs-Wheeler Alignment tool (BWA) (Li and Durbin 2009) in paired-end mode. Duplicated reads resulting from PCR overamplification or optical duplication were flagged and discarded by the MarkDuplicates module of Picard tools (version 1.67). Targeted local realignment was then performed near known short insertions and deletions (indels) and by base quality recalibration using the Genome Analysis Toolkit (GATK, version v2.1-8-g5efb575) (McKenna et al. 2010; DePristo et al. 2011). Single-nucleotide variants (SNVs) and indels were identified using batch calling with a set of unrelated individuals by using the Unified Genotyper module in the GATK in multisample calling mode. Suspected sequencing artifacts that were present across the samples of several unrelated individuals were removed. Variant quality score recalibration was performed using the GATK to identify a set of high-confidence candidate variants. The functional consequence of each variant was predicted using Variant Effect Predictor (Release 71) (Yourshaw et al. 2014). Variants not found in dbSNP (Single Nucleotide Polymorphism Database) build 135 (Sherry et al. 2001), in the 1000 Genomes Project (1KG) and the Exome Sequencing Project (ESP, N = 6503) data sets were considered for analysis (NIH 2012) (1000 Genomes Project FTP Server 2012). A recessive inheritance scenario was most probable in this case, so a stringent threshold of 0.1% population frequency in ESP and 1KG was used, resulting in rare or private candidate variants. Homozygous and compound heterozygous sequence variants were evaluated. Structural variants (SV) were identified using a panel of WGS from 946 unrelated individuals by comparing the average coverage for controls and the proband’s sample over the same exons. The resulting SV variants were genotyped across individuals from the 1000 Genomes Project to assess the SV’s population frequency, and SVs with minor allele frequency over 1% were filtered using GenomeSTRiP (Handsaker et al. 2011).
Droplet digital PCR (ddPCR) assay
The homozygous deletion of CHST11 and MIR3922 identified by WGS of DNA from the proband’s cultured fibroblast cells was confirmed by testing the proband’s DNA from both her cultured fibroblasts and buccal epithelia simultaneously by ddPCR. TaqMan assays recognizing target genes were designed for a region on intron 1 (also overlapping MIR3922), exon 2 of CHST11, along with reference genes RPP30 and AP3B1 (sequences and other information can be found in Table S1). Each assay employed forward and reverse primers and an MGB-TaqMan probe labeled with either FAM- or VIC- dyes custom-designed using PrimerExpress software (Life Technologies, Waltham, MA) and synthesized by Life Technologies. Assays were designed specifically to exclude a MseI cutting site within the amplicon since MseI was used to fragment DNA into smaller pieces and to avoid copy number polymorphisms in the general population in the Database of Genomic Variants (MacDonald et al. 2013). The detailed workflow for ddPCR has been described previously (Hindson et al. 2011). Raw data analysis was automatically performed by QuantaSoft software (Bio-Rad, Hercules, CA), followed by manual normalization of the raw copy number values using a correction factor to yield a copy number of 2.0 to the RPP30 reference gene.
Identity testing
To confirm that deletion of CHST11 and MIR3922 was present in the constitutional genome of the proband and not an artifact induced by culturing skin fibroblasts, we performed genotyping of highly polymorphic short tandem repeats (STRs) using genomic DNA from the proband’s buccal epithelia and cultured fibroblast cells. A multiplex PCR reaction was performed for 16 STR loci using the commercially available AmpFlSTR® IdentifilerTM PCR Amplification kit (Applied Biosystems; Carlsbad, CA), following the manufacturer’s instructions. Resulting STR amplicons were separated and visualized using an ABI Prism® (Waltham, MA) Genetic Analyzer. STR alleles were scored using GeneMarker software v1.5 (Softgenetics, State College, PA).
Results
Given the recessive mode of inheritance observed for the phenotype in this family, we searched the proband’s WGS data for genes harboring rare biallelic single-nucleotide and compound heterozygous variants (Table1). We identified compound heterozygous variants in MBD3L5 and PRND. A homozygous missense variant in EPHA10 and a homozygous variant in OLA1 3′UTR were also identified. While the variants in these four genes appeared to be technically valid, there was insufficient evidence based on known expression and/or biological function to link these variants to the observed phenotype.
Table 1.
Other variants identified during whole-genome sequencing
| Gene | Chromosome | Coordinate | Ref | Alt | Protein Chg | Notes |
|---|---|---|---|---|---|---|
| Compound heterozygous variants | ||||||
| MBD3L5 | chr19 | 7032665 | A | G | R129R | Synonymous |
| MBD3L5 | chr19 | 7032688 | T | G | I137S | Missense |
| PRND | chr20 | 4706780 | C | T | UTR 3′ | Possible artifact |
| PRND | chr20 | 4706782 | A | T | UTR 3′ | Possible artifact |
| SIRT6 | chr19 | 4174745 | C | A | A241S | Missense |
| SIRT6 | chr19 | 4174758 | G | C | N236K | Missense |
| Homozygous alternate variants | ||||||
| EPHA10 | chr1 | 38218738 | G | C | C389W | Missense |
| OLA1 | chr2 | 175113308 | G | A | UTR 3′ | UTR 3′ |
Other compound heterozygous and homozygous alternative variants were identified during whole-genome sequence interpretation. These variants have been ruled out as causal in this case using our interpretation pipeline.
We next examined the proband’s WGS data for homozygous deletions too small to be detected by array-based comparative genomic hybridization. Three such deletions were identified: chr12:104,948,000-105,003,000 (55 kb), chr19:55,253,424-55,295,014 (41.5 kb), and chr19:54,801,000-54,807,500 (6.5 kb). The first deletion included exon 2 of the gene CHST11 as well as the embedded microRNA MIR3922. By computational prediction, MIR3922 may target ZNF585A (Zinc Finger Protein 585A [19q13.12]), RNF214 (Ring Finger Protein 214 [11q23.3]), RAB10 (RAS-Related Protein RAB-10 [2p23.3]) and/or ERBB2 (erythroblastic leukemia viral oncogene homolog 2) (Maragkakis et al. 2009). The two deletions of chromosome 19 included the genes KIR2DL3, KIR2DL1, and LILRA3.
The population frequency of these homozygous deletions was determined using genotyping data generated by Genome STRiP in the 1KG cohort (Fig.3). The two deletions on chromosome 19 have many individuals with deletions in the 1KG samples (>1%), while the deletion on chromosome 12 that includes CHST11 and MIR3922 appears to be a rare variant, with no deletions observed in the 1KG project. The parents of the proband were born in the same region in Ireland, suggesting that the rare homozygous deletion could be a part of a region where there is identity by descent (IBD). Indeed, a potential IBD region of approximately 180 kb overlaps the entire deletion, spanning chr12:104,920,000-105,100,000 (Fig.4).
Figure 3.
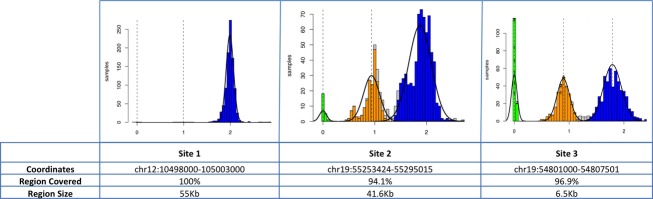
Three homozygous deletions were identified in genomic DNA from the proband. The population frequency of these deletions was determined using genotyping data generated by Genome STRiP in the 1000 genomes cohort. As shown in the histograms, the two deletions on chromosome 19 have many individuals with deletions in the 1KG samples (>1%), while the deletion on chromosome 12 that includes CHST11 and MIR3922 appears to be a rare variant, with no deletions observed in the 1KG project.
Figure 4.

There are very few observed sequence reads in the region between chr12:104,948,000-105,005,000, providing much more precise resolution on the boundaries of the deletion.
To rule out the possibility of culture artifact causing homozygous deletion of CHST11 and MIR3922, saliva was obtained from the proband and genomic DNA was isolated from buccal epithelial cells. Droplet digital PCR (ddPCR) was performed to analyze the copy number of CHST11 and MIR3922 alleles. The average CHST11 copy number was confirmed to be 0 in the fibroblast cells and 1.23 in buccal epithelial cells (σ = 0.01). This unexpected result suggested the possibility of genomic heterogeneity secondary to a mixed population of host and donor-derived cells/genomes or due to chimerism from variable fusion of CHST11-null host DNA with CHST11-diploid donor genomic DNA (Ferrand et al. 2010). To evaluate for genomic heterogeneity, we performed identity testing by microsatellite genotyping and compared the results from genomic material isolated from fibroblasts to those from buccal epithelium (Table S2). Analysis of microsatellite markers in genomic DNA isolated from fibroblast culture was consistent with origin from a single individual. The same analysis of genomic DNA isolated from buccal epithelium, however, demonstrated evidence of chimerism at 13/16 loci and the presence of a Y chromosome marker within the female proband, confirming the presence of genetic material originating from the male bone marrow donor. When chimerism was calculated on 10 of 13 informative loci the mean value was found to be 62%, consistent with 1.23 alleles contributed by the CHST11-diploid donor as determined by ddPCR. When taken together, these findings strongly suggest that homozygous deletion of CHST11 and MIR3922 in skin-derived fibroblasts is representative of the proband’s germline and not the result of a culture artifact.
To determine whether deletion of CHST11 and/or MIR3922 is likely to underlie the combined phenotype of skeletal malformation and T-cell lymphoma in this family, we attempted to obtain biological material from the proband’s affected (deceased) sister but were ultimately unsuccessful. Instead, we analyzed the copy number of CHST11 and MIR3922 in the proband’s unaffected siblings. A ddPCR analysis performed for the CHST11 and MIR3922 loci did not reveal homozygous deletion of CHST11 and MIR3922 in any of the three unaffected siblings who were tested (Fig.2).
To determine whether somatic copy number changes or mutations in CHST11 and/or MIR3922 are commonly found in sporadic hematologic malignancies, we searched the Broad TumorPortal and Catalog of Somatic Mutations in Cancer (COSMIC) databases (Koboldt et al. 2012). No copy number variants in CHST11 (and by extension MIR3922) were reported for the available sequences from hematopoietic or lymphoid tumor samples. However, somatic copy number variants in CHST11 were identified in several solid tumor samples. In the COSMIC database (accessed December 2013), 511 samples representing 8 of 27 solid tumor types were found to harbor copy number loss in the CHST11 locus. CHST11 copy number loss was most common in ovarian cancer (31.2% of samples). In addition, 28 and 33 nonsynonymous SNVs in CHST11 are reported in the Broad TumorPortal and COSMIC, respectively. Notable variants include missense substitutions in diffuse large B-cell lymphoma (p.P99Q) and CLL (p.R42L), a frameshift (p.F31 fs) in clear cell renal carcinoma, and nonsense variants in esophageal cancer (p.Q329*), lung adenocarcinoma (p.E325*), and liver cancer (p.E239*). The functional significance of these variants is presently unknown. No variant data are reported for MIR3922. Of note, a somatic translocation involving CHST11 (t(12;14)(q23;q32)) was previously identified in a patient with B-cell chronic lymphocytic leukemia (B-CLL) (Schmidt et al. 2004).
Discussion
In this study, we identified biallelic deletion of CHST11/MIR3922 in the proband as the putative cause of an unnamed autosomal recessive syndrome presenting with congenital malformations of the digits of the upper and lower limbs and malignant lymphoproliferative disease. Homozygosity for the deletion allele was not observed in three unaffected living siblings who were available for testing. Unfortunately, biological material from the deceased affected sister was not available. Although the proband’s clonal lymphoproliferative disorder was immunophenotypically classified as T-cell lymphoma, the lack of molecular classification of her affected sister’s lymphoma in the 1980s prevents us from definitively associating CHST11/MIR3922 deletion specifically with abnormalities of the T-cell lineage.
The rare homozygous deletion identified in the proband is unlikely to be an artifact. While ddPCR of buccal epithelium from saliva demonstrated hemizygosity for the CHST11/MIR3922 locus, extensive chimerism at 13 of 16 informative STR markers (including a donor Y chromosome) was detected in saliva from our female proband. This suggests the contribution of donor cell-derived genomic DNA to the proband’s gastrointestinal tract epithelium, as has been previously described (Ferrand et al. 2010; Rennert et al. 2013).
Our analysis of existing genomic databases suggests that the CHST11 locus is relatively intolerant of deletions and loss-of-function variants. Although a study using genome-wide BAC aCGH relative expression levels across 95 samples reported a hemizygous deletion near the beginning of the CHST11 gene with 4% frequency (Wong et al. 2006), no deletion events at this locus were observed using higher quality whole-genome sequence data across 946 samples in the 1KG data set. The same study also reported a complex structural rearrangement encompassing CHST11 and MIR3922, but similar rearrangements were neither observed in the 1KG data set nor would necessarily preclude wild-type expression and function. It seems implausible to observe 5 true SVs in 95 samples but no such events in 946 samples analyzed using higher quality data and more sensitive methods of detection. Therefore, we believe that CHST11/MIR3922 SVs are rare. In more than 63,000 exomes cataloged at the Broad Institute, no homozygous loss-of-function variants were observed in either CHST11 or MIR3922, further supporting the hypothesis that biallelic deletion of the locus best accounts for the proband’s phenotype (exome data available at http://exac.broadinstitute.org/gene/ENSG00000171310).
Our review of the literature suggests that the combined phenotype of skeletal dysplasia and malignant lymphoproliferative disease is rare. A single case report describes an infant with short-limbed skeletal dysplasia and severe combined immunodeficiency who developed EBV-associated B-cell lymphoma at 10 months of age (van den Berg et al. 1997). Another report describes two sisters with skeletal dysplasia and Hodgkin’s lymphoma arising in late adolescence (Gokhale et al. 1995). However, no specific genetic etiology was identified to account for the clinical presentation.
Notably, the phenotype observed in our proband resembles other rare Mendelian disorders that combine skeletal anomalies with specific hematopoietic derangements. The recessive disorder thrombocytopenia-absent radius (TAR) syndrome is characterized by low platelets and anomalies of the radius and may also present with eosinophilia, anemia, various other skeletal abnormalities, and disturbances of other systems (skin, cardiovascular, neurologic). TAR has been associated with genetic variants in the gene RBM8A, a protein that serves as one component of the multiprotein exon junction complex bound to mRNA at splice junctions. Fanconi anemia is a genetically heterogeneous recessive disorder that presents with pancytopenia (or isolated cytopenias) and skeletal anomalies such as radial aplasia and thumb aplasia or hypoplasia, in addition to disturbances of several additional systems (genitourinary, skin, cardiac, neurologic). The common molecular defect shared by all FA complementation groups is in DNA repair, resulting in hypersensitivity to DNA cross-linking agents.
Chondroitin sulfate is the most common proteoglycan in cartilage and is present on the surface of cells in many tissue types as well as in the extracellular matrix. Chondroitin is an unbranched GAG that consists of alternating sugars of glucuronic acid and N-acetylgalactosamine (GalNAc). Sulfate moieties added at position 4 or 6 of GalNAc residues alter the physiochemical properties of chondroitin and chondroitin glycoconjugates. The CHST11 gene encodes a chondroitin 4-sulfotransferase localized in the Golgi apparatus that catalyzes the transfer of sulfate to position 4 of GalNAc in chondroitin. CHST11 is one of several known carbohydrate sulfotransferases, sharing four regions of homology including 5′ and 3′ phosphosulfate binding sites and two C-terminal alpha helical domains. The gene CHST3, for example, encodes a chondroitin 6-sulfotransferase that catalyzes the transfer of sulfate to position 6 of GalNAc in chondroitin.
The association of germline variation in CHST11 with a clinical phenotype underscores the importance of carbohydrate sulfotransferase function in human biology. Biallelic loss-of-function mutations in genes encoding other carbohydrate sulfotransferases have been previously described in rare syndromes with autosomal recessive inheritance. Homozygous or compound heterozygous mutations in CHST3, encoding a chondroitin 6-sulfotransferase, are associated with recessive spondyloepiphyseal dysplasia and congenital joint dislocations (OMIM 143095) (Thiele et al. 2004; Unger et al. 2010). Inactivating mutations and deletions of CHST6, encoding a keratan 6-sulfotransferase expressed in the human cornea, have been associated with macular corneal dystrophy types I and II (Akama et al. 2000; Aldave et al. 2004). Loss-of-function mutations in CHST14, a dermatan 4-sulfotransferase, have been associated with Ehlers-Danlos syndrome musculocontractural type 1 (OMIM 601776) (Dündar et al. 2009).
The digital malformations exhibited by our proband and reportedly present in her affected sister suggest an important role for CHST11 in normal bone development. Differentiation of chondrocytes is responsible for development of a cartilage model of future bone, and chondrodysplasias in mice are known to be associated with bone and joint abnormalities (McLean and Olsen 2001). In mice, CHST11 is expressed in the apical ectodermal ridge of developing limbs and subsequently in the cartilage growth plate during embryogenesis (Klüppel et al. 2002). Mice homozygous for a loss-of-function mutation in the murine CHST11 homolog die within hours of birth and exhibit severe chondrodysplasia characterized by disorganized cartilage growth plates (Klüppel et al. 2005). This phenotype is associated with imbalance of chondroitin sulfation, mislocalization of chondroitin sulfate in growth plates, upregulation of TGF-beta and downregulation of BMP signaling. These perturbations disturb growth plate morphogenesis and ultimately lead to the abnormal development of bones formed by endochondral ossification, resulting in abnormal development of the feet after E15 and small stature. This appears consistent with the observed human skeletal phenotype, however, as the sisters were adults, there are no data available to demonstrate whether the human developmental mechanism is the same.
A possible functional role for CHST11 in lymphocyte development and/or differentiation is less well understood. In adult human tissues, CHST11 expression is enriched in hematopoietic cells including peripheral leukocytes, the thymus, lymph nodes, and the spleen (BioGPS; Hiraoka et al. 2000). During the late stages of murine embryogenesis (E15), CHST11 is expressed in a punctate fashion in the liver, suggestive of expression in embryonic hematopoietic cells (Klüppel et al. 2002). Notably, the CHST11−/− mouse model was not reported to develop abnormalities of lymphoid tissue or lymphocyte proliferation or differentiation. However, the early death of CHST11−/− mice may have precluded the later development of a lymphoproliferative disorder. Alternatively, a phenotype was present in some form but was not evaluated, given the lack of any prior evidence suggesting a functional role for CHST11 in lymphocytes. Finally, there may be true differences between the function of CHST11 in humans and mice.
Nonetheless, prior experimental evidence implicates a role for proteoglycan sulfation in lymphocyte differentiation, supporting a plausible contribution of CHST11 deficiency to malignant lymphoproliferative disease. The enzymes required for biosynthesis of chondroitin sulfate and heparin sulfate appear to be developmentally regulated in B-cell progenitors, resting B cells, and activated B cells (Duchez et al. 2011). Overexpression of the biosynthetic enzyme for heparin sulfate in the B-cell lineage of transgenic mice, which diminished the chondroitin sulfate/heparin sulfate ratio, resulted in a partial differentiation block at the pro-B- to pre-B-cell transition (Duchez et al. 2011). In a two-step culture system, chondroitin sulfate B (CS B) was found to promote IL-4/5 driven murine B-cell differentiation in a dose-dependent fashion, as assayed by IgM production, the number of IgM-secreting cells, and the number of CD138-positive cells (Yoshihara et al. 2007). While similar studies investigating T lymphocyte differentiation do not appear to have been performed, the findings for B lymphocytes support a probable role for chondroitin sulfate in normal lymphocyte differentiation. Notably, chondroitin sulfate was also found to be the major secreted GAG in three leukemic cell lines (Jurkat, a T-cell leukemia model; Daudi, a Burkitt’s or B-cell leukemia model; and THP-1, an acute monocytic leukemia cell line) and the major GAG retained at the membrane in two of the cell lines (Jurkat, Daudi) as assessed by chromatography (Makatsori et al. 2001). Whether chondroitin is abnormally sulfated in these cells is presently unclear.
In addition to impaired differentiation, another hallmark of the transformed phenotype is dysregulated proliferation. Emerging evidence suggests that CHST11 may regulate cellular proliferation downstream of RAS/MAPK signaling, a pathway dysregulated in both congenital syndromes with risk of malignancy and many sporadic cancers (Rauen 2013). In fibroblasts isolated from a patient with Costello syndrome, a disorder caused by germline activating mutations in HRAS, CHST11 mRNA, and protein expression and chondroitin-4-sulfate were all found to be markedly reduced (Klüppel et al. 2012). Moreover, misexpression of oncogenic HRAS in normal fibroblasts repressed CHST11 expression, indicating that CHST11 is a negatively regulated target gene downstream of HRAS signaling. Furthermore, overexpression of wild-type CHST11 in the HRAS-mutant fibroblasts rescued the excessive proliferation associated with HRAS gain of function, demonstrating that restoration of CHST11 is sufficient to reverse an important cellular phenotype associated with malignancy. It is unknown at the present time whether somatic activating MAPK pathway mutations commonly identified in a range of sporadic malignancies also downregulate CHST11, and whether a change in expression of the latter contributes to the malignant phenotype.
Based on these data, we propose the hypothesis that loss of normal chondroitin sulfation may predispose affected individuals to a clonal lymphoproliferative disorder. It is unclear whether manifestation of the phenotype requires the contribution of other germline variants, environmental factors, or somatic genetic mutations in the lymphocyte lineage. It is tempting to speculate that loss of MIR3922, the microRNA that lies within the CHST11 locus at 12q23 and is nullizygous in our proband, contributes to the lymphoproliferative phenotype. Unfortunately, no functional data are available at the present time for this microRNA.
In summary, we have identified CHST11/MIR3922 deletion as the presumptive cause of the complex phenotype observed in our proband. While CHST11 deletion alone could account for the patient’s bony malformations, the contribution of the deletion to her T-cell lymphoproliferative disorder is less clear. Concomitant loss of MIR3922 and perhaps additional events (genetic or environmental) may be required for full expression of the combined phenotype, or it is possible that the genetic etiology of the lymphoproliferative disorder in each sister is distinct. Future studies could aim to directly test whether CHST11 expression is functionally required for the normal differentiation and/or proliferation of T lymphocytes, using cell lines or other in vitro models of lymphocyte differentiation. Moreover, it would be useful to assess the expression and function of MIR3922 in lymphocytes, both alone and in combination with reduced expression of CHST11. By analyzing mRNA and protein levels, it would also be of interest to assess whether CHST11 expression is reduced in sporadic hematologic malignancies, particularly because somatic dysregulation at the genetic level was not commonly observed in the Broad TumorPortal or the COSMIC database. Finally, additional evidence linking CHST11/MIR3922 deletion to our proband’s phenotype may be derived from ascertainment and genetic testing of additional families sharing a similar phenotype.
Acknowledgments
We acknowledge support from the Brigham Genomic Medicine Program for bioinformatics and clinical support, as well as critical feedback recommendations on this case. This research was supported by NIH grants HG007229, HG007690, CA009172 and the Dana-Farber Leadership Council. [Correction added on 26 May 2015, after first online publication: The first statement in the Acknowledgments section has been corrected to ‘We acknowledge support from the Brigham Genomic Medicine Program for bioinformatics and clinical support, as well as critical feedback recommendations on this case’.]
Conflict of Interest
None declared.
Supporting Information
Table S1. Primer sequences and transcript information for ddPCR assays.
Table S2. (A) Results of microsatellite genotyping of fibroblasts and buccal epithelial cells. (B) Chimerism in the buccal epithelia.
References
- Akama T, Nishida K, Nakayama J, Watanabe H, Ozaki K, Nakamura T, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat. Genet. 2000;26:237–241. doi: 10.1038/79987. [DOI] [PubMed] [Google Scholar]
- Aldave A, Yellore V, Thonar E, Udar N, Warren J, Yoon M, et al. Novel mutations in the carbohydrate sulfotransferase gene (CHST6) in American patients with macular corneal dystrophy. Am. J. Ophthalmol. 2004;137:465–473. doi: 10.1016/j.ajo.2003.09.036. [DOI] [PubMed] [Google Scholar]
- Bentley D, Balasubramanian S, Swerdlow H, Smith G, Milton J, Brown C, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg H. Wage K, Burggraaf JD, Peters M. Malignant B cell lymphoma in an infant with severe combined immunodeficiency with short limbed skeletal dysplasia. Acta Paediatr. 1997;86:778–780. doi: 10.1111/j.1651-2227.1997.tb08588.x. [DOI] [PubMed] [Google Scholar]
- DePristo M, Banks E, Poplin R, Garimella K, Maguire J, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchez S, Pascal V, Cogné N, Jayat-Vignoles C, Julien R. Cogné M. Glycotranscriptome study reveals an enzymatic switch modulating glycosaminoglycan synthesis during B-cell development and activation. Eur. J. Immunol. 2011;41:3632–3644. doi: 10.1002/eji.201140865. [DOI] [PubMed] [Google Scholar]
- Dündar M, Müller T, Zhang Q, Pan J, Steinmann B, Vodopiutz J, et al. Loss of dermatan-4-sulfotransferase 1 function results in adducted thumb-clubfoot syndrome. Am. J. Hum. Genet. 2009;85:873–882. doi: 10.1016/j.ajhg.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrand J, Noël D, Lehours P, Prochazkova-Carlotti M, Chambonnier L, Ménard A, et al. Human bone marrow-derived stem cells acquire epithelial characteristics through fusion with gastrointestinal epithelial cells. PLoS ONE. 2010;6:e19569. doi: 10.1371/journal.pone.0019569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1000 Genomes Project FTP Server. 2012. Consortium 1000 genomes project. 1000 genomes phase 1. Version 3, release date 4/30.
- Gokhale D, Evans D, Crowther D. Woll P. Molecular genetic analysis of a family with a history of Hodgkin’s disease and dyschondrosteosis. Leukemia. 1995;9:826–833. [PubMed] [Google Scholar]
- Handsaker R, Korn J, Nemesh J. McCarroll S. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 2011;43:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindson B, Ness K, Masquelier D, Belgrader P, Heredia N, Makarewicz A, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka N, Nakagawa H, Ong E, Akama T, Fukuda M. Fukuda M. Molecular cloning and expression of two distinct human chondroitin 4-O-sulfotransferases that belong to the HNK-1 sulfotransferase gene family. J. Biol. Chem. 2000;275:20188–20196. doi: 10.1074/jbc.M002443200. [DOI] [PubMed] [Google Scholar]
- Klüppel M, Vallis K. Wrana J. A high-throughput induction gene trap approach defines C4ST as a target of BMP signaling. Mech. Dev. 2002;118:77–89. doi: 10.1016/s0925-4773(02)00198-3. [DOI] [PubMed] [Google Scholar]
- Klüppel M, Wight T, Chan C, Hinek A. Wrana J. Maintenance of chondroitin sulfation balance by chondroitin-4-sulfotransferase 1 is required for chondrocyte development and growth factor signaling during cartilage morphogenesis. Development. 2005;132:3989–4003. doi: 10.1242/dev.01948. [DOI] [PubMed] [Google Scholar]
- Klüppel M, Samavarchi-Tehrani P, Liu K, Wrana J. Hinek A. C4ST-1/CHST11-controlled chondroitin sulfation interferes with oncogenic HRAS signaling in Costello syndrome. Eur. J. Hum. Genet. 2012;20:870–877. doi: 10.1038/ejhg.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt D, Fulton R, McLellan M, Schmidt H, Kalicki-Veizer J, McMichael J, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald J, Ziman R, Yuen R, Feuk L. Scherer S. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2013;42:D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makatsori E, Karamanos N, Papadogiannakis N, Hjerpe A, Anastassiou E. Tsegenidis T. Synthesis and distribution of glycosaminoglycans in human leukemic B- and T-cells and monocytes studied using specific enzymic treatments and high-performance liquid chromatography. Biomed. Chromatograp. 2001;15:413–417. doi: 10.1002/bmc.91. [DOI] [PubMed] [Google Scholar]
- Maragkakis M, Reczko M, Simossis V, Alexiou P, Papadopoulos G, Dalamagas T, et al. DIANA-microT web server: elucidating microRNA functions through target prediction. Nucleic Acids Res. 2009;37:W273–W276. doi: 10.1093/nar/gkp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean W. Olsen BR. Mouse models of abnormal skeletal development and homeostasis. Trends Genet. 2001;17:S38–S43. doi: 10.1016/s0168-9525(01)02458-1. [DOI] [PubMed] [Google Scholar]
- NIH. 2012. N. NHLBI GO Exome Sequencing Project.
- Rauen KA. The RASopathies. Annu. Rev. Genomics Hum. Genet. 2013;14:355–369. doi: 10.1146/annurev-genom-091212-153523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennert H, Leonard D, Cushing M, Azurin C. Shore T. Avoiding pitfalls in bone marrow engraftment analysis: a case study highlighting the weakness of using buccal cells for determining a patient’s constitutional genotype after hematopoietic stem cell transplantation. Cytotherapy. 2013;15:391–395. doi: 10.1016/j.jcyt.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Dyomin V, Palanisamy N, Itoyama T, Nanjangud G, Pirc-Danoewinata H, et al. Deregulation of the carbohydrate (chondroitin 4) sulfotransferase 11 (CHST11) gene in a B-cell chronic lymphocytic leukemia with a t(12;14)(q23;q32) Oncogene. 2004;23:6991–6996. doi: 10.1038/sj.onc.1207934. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharis A, Skandalis S, Tzanakakis G. Karamanos N. Proteoglycans in health and disease: novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 2010;277:3904–3923. doi: 10.1111/j.1742-4658.2010.07800.x. [DOI] [PubMed] [Google Scholar]
- Thiele H, Sakano M, Kitagawa H, Sugahara K, Rajab A, Höhne W, et al. Loss of chondroitin 6-O-sulfotransferase-1 function results in severe human chondrodysplasia with progressive spinal involvement. Proc. Natl. Acad. Sci. USA. 2004;101:10155–10160. doi: 10.1073/pnas.0400334101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unger S, Lausch E, Rossi A, Mégarbané A, Sillence D, Alcausin M, et al. Phenotypic features of carbohydrate sulfotransferase 3 (CHST3) deficiency in 24 patients: congenital dislocations and vertebral changes as principal diagnostic features. Am. J. Med. Genet. A. 2010;152A:2543–2549. doi: 10.1002/ajmg.a.33641. [DOI] [PubMed] [Google Scholar]
- Valencia C, Indugula S, Mathur A, Wei C, Brown J, Cole I, et al. Misleading results from saliva samples of patients post-BMT in exome analyses. Blood. 2014;124:660–661. doi: 10.1182/blood-2014-04-572941. [DOI] [PubMed] [Google Scholar]
- Wong K, deLeeuw R, Dosanjh N, Kimm L, Cheng Z, Horsman D, et al. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2006;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara R, Aoyama E, Kadota Y, Kawai S, Goto T, Zhong M, et al. Differentiation of murine B cells induced by chondroitin sulfate B. Cell. Immunol. 2007;250:14–23. doi: 10.1016/j.cellimm.2007.12.002. [DOI] [PubMed] [Google Scholar]
- Yourshaw M, Taylor S, Rao A, Martín M. Nelson S. Rich annotation of DNA sequencing variants by leveraging the Ensembl Variant Effect Predictor with plugins. Brief. Bioinform. 2014;16:255–264. doi: 10.1093/bib/bbu008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences and transcript information for ddPCR assays.
Table S2. (A) Results of microsatellite genotyping of fibroblasts and buccal epithelial cells. (B) Chimerism in the buccal epithelia.