

Abstract
Approximately 5–10% of all breast and/or ovarian cancer cases are considered as inherited. BRCA1 and BRCA2 tumor suppressor genes account for a high penetrance of hereditary cases, but familial cases without mutations in these genes can also occur. Despite their low penetrance, other hereditary cancer-related genes are known to be associated with breast and ovarian cancer risk. However, the extent to which these genes prevail in breast and ovarian cancer remains to be elucidated. To estimate the frequency of mutations in these predisposition genes, we analyzed the germline mutations of 25 hereditary cancer-related genes in 155 patients using targeted next-generation sequencing. These subjects included 11 BRCA1/2 mutation-positive cases and 144 negative cases. Of these, three patients (1.9%) had pathogenic mutations in ATM, MRE11A, or MSH6, all of which have a central role in DNA repair and the mismatch repair pathway. The MSH6 splice-site mutation (IVS6+1G>T) was predicted to be pathogenic, as demonstrated by in vitro and immunohistochemical analyses. These results suggested deficiencies in cellular DNA repair functions result in the development of breast and ovarian cancer.
Keywords: Breast and ovarian cancer, DNA repair, gene panel, next-generation sequencing
Introduction
Breast and ovarian cancer are common cancers in women, affecting ∼60,000 and 10,000 new cases, respectively, per year in Japan. Approximately 5–10% of breast and ovarian cancers are hereditary disorders. Germline mutations in BRCA1 and BRCA2 (BRCA1/2) tumor suppressor genes predispose to the development of breast and ovarian cancer (Easton et al. 1993; Narod et al. 1995; King et al. 2003), but BRCA1/2 mutations have been observed in only about 20% of these cases (Couch et al. 2014b). A subset of the remaining cases appears to be caused by germline mutations in other cancer susceptibility genes (Couch et al. 2014b). For instance, TP53, PTEN, CDH1, STK11, ATM, PALB2, and CHEK2 are predisposing genes for breast cancer (Economopoulou et al. 2015). In addition, mutations in DNA mismatch repair genes such as MLH1, MSH2, MSH6, and PMS2, which are responsible for Lynch syndrome (also known as hereditary nonpolyposis colorectal cancer), are associated with not only colon and endometrial cancer but also with ovarian, biliary tract, and stomach cancer (Bonai et al. 2011; Vierkoetter et al. 2014). These observations suggested that analysis of predisposition genes associated with hereditary cancer is useful in screening mutation carriers for cancer surveillance and prevention.
With the advent of high-throughput sequencing technologies, next-generation sequencing (NGS), it is possible to simultaneously analyze multiple genes of interest at low cost (Tung et al. 2015). NGS is gradually making its way into clinical research for diagnostic testing of hereditary disorders (Domchek et al. 2013; Grant et al. 2014; LaDuca et al. 2014; Yorczyk et al. 2014). Previous reports demonstrating NGS analysis of hereditary cancer-related genes identified germline mutations in breast and ovarian cancer patients (Walsh et al. 2011; Couch et al. 2014a; Cybulski et al. 2014; Kurian et al. 2014). However, the extent to which hereditary cancer genes other than BRCA1/2 are responsible for breast and ovarian cancer remains to be fully elucidated. In this study, we selected 25 cancer-predisposing genes, and performed targeted sequencing in 155 breast and/or ovarian cancer subjects to identify mutation carriers and assess the frequency of mutations in these predisposition genes.
Materials and Methods
Patients and sample preparation
Peripheral blood samples were obtained from 155 breast and/or ovarian cancer patients who attended the Yamanashi Prefectural Central Hospital (Yamanashi, Japan) between 2013 and 2014. This cohort included 76 patients with breast cancer, 69 with ovarian cancer, and 10 with both breast and ovarian cancers and the median age of cancer onset was 55 years (range, 16–87 years). According to the National Comprehensive Cancer Network (NCCN) criteria, 146 patients (94%) had genetic risks based on the family history. Lymphocytes were isolated following centrifugation of peripheral blood samples at 820g at 25°C for 10 min. Peripheral blood lymphocytes were stored at −80°C until required for DNA extraction. Total DNA was extracted from lymphocytes using the QIAamp DNA Blood Mini kit (Qiagen, Tokyo, Japan) or QIAamp DNA Blood Mini QIAcube Kit (Qiagen) with the QIAcube (Qiagen). The concentration of DNA was determined using the Nano Drop 2000 spectrophotometer (Thermo Fisher Scientific, Yokohama, Japan). Informed consent was obtained from all subjects, and this study was approved by the Institutional Review Board at Yamanashi Prefectural Central Hospital.
Targeted NGS
For targeted NGS analysis, Ion AmpliSeq designer software (Life Technologies, Tokyo, Japan) was used to design primers, which consisted of 610 primer pairs in two pools covering the exons and exon–intron boundaries of 25 cancer-predisposing genes (APC, ATM, BARD1, BMPR1A, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, MLH1, MRE11A, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD50, RAD51C, RAD51D, SMAD4, STK11, and TP53) (Walsh et al. 2011; Couch et al. 2014b; Economopoulou et al. 2015; Tung et al. 2015). Multiplex polymerase chain reaction (PCR) was performed using 50–100 ng genomic DNA with 17 cycles with a premixed primer pool using Ion AmpliSeq Library Kit 2.0, as previously described (Hirotsu et al. 2014). The PCR amplicons were treated with 2 μL FuPa reagent to partially digest primer sequences and phosphorylate the amplicons. The amplicons were ligated to adapters with the diluted barcodes of the Ion Xpress Barcode Adapters kit (Life Technologies). Adaptor-ligated amplicon libraries were purified using Agencourt AMPure XP reagents (Beckman Coulter, Tokyo, Japan). The library concentration was determined using an Ion Library Quantitation Kit (Life Technologies), then each library was diluted to 8 pM and the same amount of libraries was pooled for one sequence reaction. Next, emulsion PCR was carried out using the Ion OneTouch System and Ion PI Template OT2 200 Kit v2 (Life Technologies) according to the manufacturer’s instructions. Template-positive Ion Sphere Particles were then enriched with Dynabeads MyOne Streptavidin C1 Beads (Life Technologies) using an Ion OneTouch ES system (Life Technologies). Purified Ion Sphere particles were loaded on an Ion PI Chip v2. Massively parallel sequencing was carried out on an Ion Proton System (Life Technologies) using the Ion PI Sequencing 200 Kit v2. Sequencing was performed using 500 flow runs that generated ∼200 bp reads.
Data analysis
The sequence data were processed using standard Ion Torrent Suite Software running on the Torrent Server. Raw signal data were analyzed using Torrent Suite version 4.2. The pipeline included signaling processing, base calling, quality score assignment, adapter trimming, PCR duplicate removal, read alignment to human genome 19 reference (hg19), quality control of mapping quality, coverage analysis, and variant calling. Following data analysis, annotation of single nucleotide variants, insertions, deletions, and splice-site alternations were performed by the Ion Reporter Server System (Life Technologies). Splice-site alternations were analyzed 2 bp upstream or downstream of exon–intron boundaries. Sequence data were visually confirmed with the Integrative Genomics Viewer (IGV) and any sequence, alignment, or variant call error artifacts were discarded. Heterozygous MUTYH mutations were not considered as pathogenic in this study. Public databases used included ClinVar (Landrum et al. 2014), the 1000 Genomes Project database (Abecasis et al. 2012), the 5000 Exome project (http://evs.gs.washington.edu/EVS/), and The Human Genetic Variation Browser (HGVB) (http://www.genome.med.kyoto-u.ac.jp/SnpDB).
Sanger sequencing
PCR was performed using genomic DNA and primer pairs flanking the deleterious variant sites. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen). Sequencing was performed with BigDye Terminator v3.1 (Life Technologies) using forward or reverse primers. PCR products were purified and subsequently analyzed by the 3500 Genetic Analyzer (Applied Biosystems, Tokyo, Japan). Primer sequences are provided in Table S1.
Reverse transcriptase-PCR
Total RNA from peripheral blood was extracted using the NucleoSpin RNA Blood kit (Takara, Shiga, Japan) and reverse transcribed to cDNA using High-Capacity cDNA Reverse Transcription Kits (Applied Biosystems). PCR was performed using Platinum PCR SuperMix High Fidelity (Life Technologies) with primer pairs listed in Table S1. The PCR products were separated on a 2% agarose gel.
Immunohistochemical analysis
The sections were deparaffinized, and antigen retrieval was performed by heat treatment in EDTA (ethylenediaminetetraacetic acid) solution pH 8.0. MSH6 protein expression in tumors and surrounding normal tissue was evaluated on 4-μm-thick, formalin-fixed, paraffin-embedded (FFPE) sections with anti-MSH6 monoclonal antibodies (clone 44; 1:400; BD Biosciences, Tokyo, Japan) using the Ventana BenchMark XT staining system (Roche, Tokyo, Japan). Normal tissue adjacent to tumor tissue served as a positive control. A pathologist determined the tumors to be positive when nuclear staining in tumor tissue was present or negative when the nuclear stain was absent.
MSI analysis
The tissue was stained with hematoxylin–eosin and then microdissected using an ArcturusXT laser-capture microdissection system (Life Technologies). DNA was extracted from FFPE tumor tissue using the QIAamp DNA FFPE Tissue Kit (Qiagen). Peripheral blood DNA was used as a control. For microsatellite instability (MSI) analysis, five microsatellite markers (BAT25, BAT26, D5S346, D2S123, and D17S250) were used to classify the tumor as MSI-high (MSI-H, the presence of at least two markers showing novel alleles compared with normal tissue), MSI-low (defined as one marker with a novel allele), or microsatellite stable (MSS, no marker with novel alleles) (Berg et al. 2000). Fluorescently labeled PCR products were separated by capillary electrophoresis using a 3500 Genetic Analyzer (Applied Biosystems) and the product size was analyzed by GeneMapper Software 5 (Applied Biosystems). Primer sequences are provided in Table S1.
Results
Sequencing multiple genes related to hereditary cancer
We selected 25 hereditary cancer-related genes not including BRCA1/2 and designed a customized gene panel, which covered 97.6% of target regions in these genes (Table S2). Massively parallel sequencing determined the sequence of coding regions and intron–exon boundaries of these genes in lymphocyte DNA from 155 breast and/or ovarian cancer patients. These subjects included 11 BRCA1/2 mutation-positive cases and 144 negative cases (Hirotsu et al. 2014). Target sequencing was conducted using an Ion Proton sequencer. Each library yielded an average of 10.5 Gb of total number of bases aligned to reference sequence (Table S3). The mean sequencing depth was 2805×, and the average uniformity of coverage was 90.2% (Table S4).
As a result, we identified 10 inactivating mutations in ATM, MRE11A, MSH6, and MUTYH genes in nine patients who did not carry BRCA1/2 mutations (Table1). These mutations are expected to cause protein truncations through frameshift insertions or deletions (n = 3), nonsense mutations (n = 1), or splice-site alterations (n = 6) (Table1). Among these variants, MSH6 frameshift mutation (p.K1358fs) is predicted to be nonpathogenic because the mutation causes the deletion of two amino acids at the C-terminal region (MSH6 full length: 1360 amino acids) and may not affect protein function (Fig. S1). Furthermore, a splice site mutation in MSH6 (IVS9+2_+5delGTAAC>G) was also supposed to be nonpathogenic, because the consensus dinucleotide GT was retained despite the deletion of four nucleotides (TAAC) (Fig. S2). The frequencies of the MSH6 p.K1358fs and MSH6 IVS9+2_+5delGTAAC>G variants in a Japanese population were 1.8% and 1.3%, respectively, according to HGVB (Table1), supporting the theory that these variants are relatively common and nonpathogenic. MUTYH is considered as a predisposing gene for MYH-associated polyposis coli (MAP), an autosomal recessive disease (Al-Tassan et al. 2002). In this study, four patients had MUTYH monoallelic mutations in the splice site (Table1), but biallelic mutations were not identified.
Table 1.
Mutations in hereditary cancer-related genes in 155 patients with breast and/or ovarian cancer
| Patient no. | Personal history (age at diagnosis) | Gene | Chr | Position | Designation | Coding | Variant Allele Fraction | 1000 genome MAF | HGVB | ClinVar |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Breast (65) | MRE11A | 11 | 94212003 | p.G148X | c.442G>T | 0.45 | ND | ND | ND |
| 2 | Breast (44) | ATM | 11 | 108203577 | p.I2629fs | c.7878_7882delTTATA | 0.48 | ND | ND | ND |
| 3 | Breast (59) | MSH6 | 2 | 48033981 | p.K1358fs | c.4065_4066insTTGA | 0.49 | 0.8% | 1.8% | ND |
| 4 | Ovary (cc) (57) Breast | MSH6 | 2 | 48033981 | p.K1358fs | c.4065_4066insTTGA | 0.45 | 0.8% | 1.8% | ND |
| 5 | Breast (41) | MSH6 | 2 | 48033791 | IVS9+2_+5delGTAAC>G | splice site | 0.46 | ND | 1.3% | ND |
| Ovary (muc) (56) | ||||||||||
| 6 | Ovary (endo) (47) | MSH6 | 2 | 48032167 | IVS6+1G>T | splice site | 0.43 | ND | ND | ND |
| MUTYH | 1 | 45797760 | IVS10-2A>G | splice site | 0.37 | 0.2% | 2.6% | Likely pathogenic | ||
| Uterine corpus (47) | ||||||||||
| 7 | Breast (46) | MUTYH | 1 | 45797760 | IVS10-2A>G | splice site | 0.44 | 0.2% | 2.6% | Likely pathogenic |
| 8 | Ovary (59) | MUTYH | 1 | 45797760 | IVS10-2A>G | splice site | 0.41 | 0.2% | 2.6% | Likely pathogenic |
| 9 | Breast (43) | MUTYH | 1 | 45797760 | IVS10-2A>G | splice site | 0.39 | 0.2% | 2.6% | Likely pathogenic |
Chr, chromosome; MAF, minor allele frequency; cc, clear cell; muc, mucinous; endo, endometrioid; ND, not documented; HGVB, human genetic variation browser.
Therefore, we concluded that deleterious mutations in BRCA1/2 (7.1%; 11 out of 155) genes and in non-BRCA1/2-predisposing genes (1.9%; three out of 155) were present in our subjects (Fig.1 and Table S5). All deleterious mutations in MRE11A, ATM, and MSH6 genes were confirmed by Sanger sequencing (Fig.1). Taken together, the data suggested a subpopulation of breast and ovarian cancer has a germline mutation related to DNA repair genes.
Figure 1.
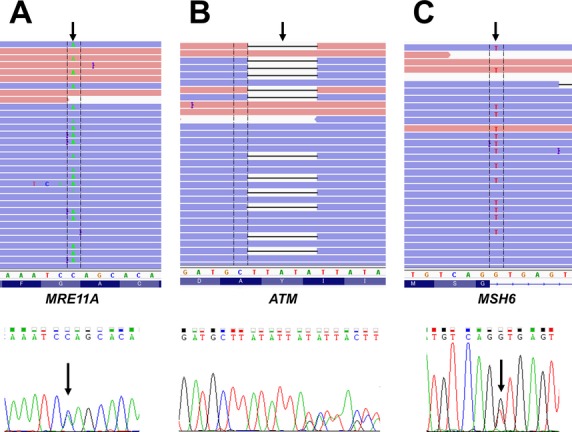
Germline deleterious mutations identified in patients with breast and/or ovarian cancer. Representative image of read alignments visualized with IGV (upper image). Sequencing chromatograms show the mutations and frameshift insertions/deletions in peripheral blood DNA from each patient (lower image). Mutations were identified in MRE11A p.G148X (A), ATM p.I2629fs (B), and MSH6 IVS6+1G>T (C). Arrows indicate the position of the mutations in the patient genome.
Deleterious mutation in predisposing genes
We identified MRE11A nonsense (p.G148X) or ATM frameshift (p.I2629fs) mutations in probands who developed breast cancer (Table1). The mutations in ATM and MRE11A were identified in breast cancer patients (Broeks et al. 2000; Bartkova et al. 2008). Family 1 included a heterozygous MRE11A nonsense mutation (p.G148X) that was present in probands and two relatives with breast cancer (Fig.2A). Family 2 included two aunts and one grandmother who developed breast cancer. A heterozygous ATM frameshift mutation (p.Ile2629fs) was identified in a proband who developed breast cancer at the age of 43 years (Fig.2B).
Figure 2.
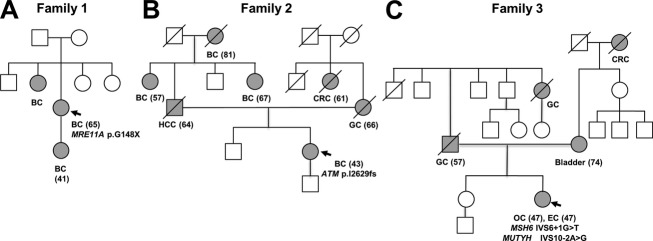
Pedigrees of three families with deleterious mutations. Individuals with any cancer are shown as filled circles. Arrows show a proband with deleterious mutations of (A) MRE11A in family 1, (B) ATM in family 2 and (C) MSH6 in family 3. The age at diagnosis plus the identified mutation is shown under the relevant individuals. BC, breast cancer; OC, ovarian cancer; CRC, colorectal cancer; GC, gastric cancer; HCC, hepatocellular cancer; Bladder, bladder cancer; EC, endometrial cancer.
In addition, a deleterious splice-site mutation in MSH6 was detected in ovarian and endometrial cancer patients, and ovarian cancer was diagnosed with mucinous carcinoma (Table1). Consistent with the evidence that MSH6 is associated with Lynch syndrome, family 3 included one grandfather who developed colorectal cancer (Fig.2C). Germline mutations in other mismatch repair genes such as MLH1, MSH2, or PMS2 were not identified in this study.
Mutation in MSH6 genes (IVS6+1G>T) were pathogenic
The splice-site mutation in MSH6 (IVS6+1G>T) disrupts the canonical GT dinucleotide at the 5′ splice site and has not been reported previously to our knowledge. In addition, loss of MSH6 promotes tumorigenesis in colorectal cancer is well known, but not in ovarian cancer. To examine whether this G to T substitution influences transcriptional processing, we synthesized MSH6 complementary DNA (cDNA) from peripheral blood RNA and performed PCR with primers flanking exon 6 (Fig.3A). The PCR product sizes were ∼356 bp in length, which represented the normal transcript of exon 5–7 expressed in both controls and patients (Fig.3B). However, MSH6 mRNA expression levels were apparently reduced in patients compared with controls (Fig.3B). These results suggested the splice-site mutation introduced premature termination codons and subsequently mRNA was degraded by nonsense-mediated decay. We next performed MSI analysis and immunohistochemistry staining for MSH6. This revealed that the tumors had MSI-high and lacked MSH6 protein expression (Figs.3C, 4). These results showed the MSH6 IVS6+1G>T variant disrupted mismatch-repair function and promoted tumorigenesis.
Figure 3.
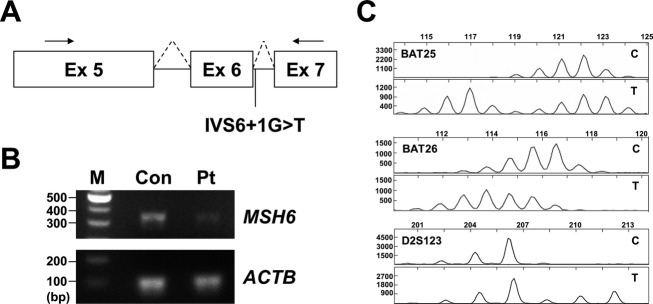
MSH6 splice-site mutation is pathogenic. (A) Schematic of MSH6 transcript. The arrows indicate the primers used in this analysis, located in exon (Ex) 5 and exon 7. (B) Reverse transcriptase polymerase chain reaction analysis of RNA from peripheral blood of a control (Con) and patient (Pt) with ovarian and endometrial cancer harboring the MSH6 IVS6+1G>T splice-site mutation. Gel electrophoresis showing PCR fragments (∼356 bp in length). DNA marker (M) was loaded in the left lane. ACTB was used as an internal control. (C) Representative images of MSI profiles. Ovarian tumor showing microsatellite instability for BAT25, BAT26, and D2S123. C, control; T, tumor tissue; IVS, intervening sequence; MSI, microsatellite instability.
Figure 4.
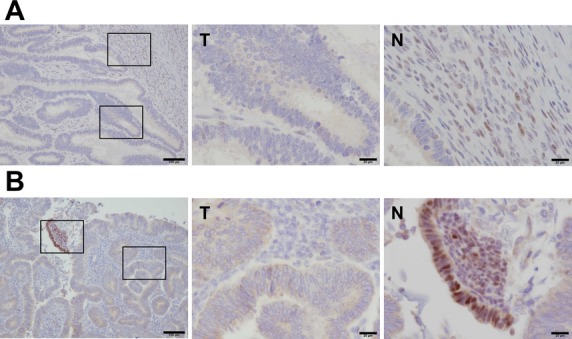
MSH6 proteins are downregulated in ovarian and endometrial cancer. (A and B) Immunohistochemical staining of tumor (T) and surrounding normal (N) tissue with an MSH6 antibody in ovarian (A) and endometrial cancer (B). MSH6 protein accumulated in the nuclei of normal tissue (left image), but not in tumor tissue (middle image). Right-hand images show a high-power magnification of left-hand images. Scale bars, 100 μm for left-hand image, and 20 μm for middle and right-hand image. N, normal tissue; T, tumor tissue.
Discussion
In this study, we performed NGS using a multigene panel to estimate the frequency of germline mutation carriers with breast and/or ovarian cancer. Of 155 patients, three had pathogenic mutations in ATM, MRE11A, or MSH6. These results suggested individuals carrying germline mutations in DNA repair genes were at risk of breast or ovarian cancer. Thus, instead of single-gene testing, multigene panel-based genetic testing is an alternative tool for screening hereditary cancer.
DNA repair genes maintain genomic stability in response to DNA damage to retain cellular homeostasis. The MER11 complex (MRE11–RAD50–NBS1: MRN) binds double-strand break ends and activates ataxia-telangiectasia-mutated (ATM) protein (Lee and Paull 2005). Homozygous or compound heterozygous mutations in ATM or MRE11A cause ataxia-telangiectasia (MIM: 208900) and ataxia-telangiectasia-like disorder-1 (ATLD1) (MIM: 604391) (Savitsky et al. 1995; Stewart et al. 1999). ATM is a predisposing gene for breast and pancreatic cancer (Broeks et al. 2000; Roberts et al. 2012).
Of note, we identified heterozygous germline mutations in these two genes in breast cancer but not ovarian cancer patients, which implies that defects in the MRN–ATM signaling pathway result in the development of breast cancer. A previous study demonstrated ATM heterozygous mutation carriers have a two-fold increased risk of breast cancer compared with the general population, and in particular, women under the age of 50 years have a five-fold increased risk (Thompson et al. 2005). Consistent with this, our analysis showed a proband carrying an ATM germline mutation developed breast cancer at the age of 43 years (Fig.2B). Moreover, this proband had three relatives with second-degree relatives who developed breast cancer. Despite limited evidence, mutations in MRE11A are associated with breast cancer (Bartkova et al. 2008). Our data showed a proband carrying a MRE11A mutation had two relatives who developed breast cancer, reinforcing the evidence that MRE11A is a predisposing gene for breast cancer.
Germline mutations in mismatch repair genes, including MLH1, MSH2, MSH6, and PMS2, predispose to Lynch syndrome. Defects in MLH1 (50%) and MSH2 (40%) account for the majority of Lynch syndrome cases, but mutations in MSH6 (∼7–10%) and PMS2 (<5%) are responsible for the minority of cases (Hegde et al. 2013). This study identified one type of MSH6 germline mutation in ovarian cancer cases. Unlike Lynch syndrome, mutations in MSH2 and MLH1 genes were not observed in our study. Walsh et al. (2011) also reported mutations in MSH6 but not in MLH1 or MSH2 in ovarian cancer patients. Collectively, it is possible that MSH6 among the mismatch repair genes was strongly associated with ovarian cancer.
In conclusion, despite the low frequency of the deleterious mutations in non-BRCA1/2 predisposing genes, multigene panel genetic testing is an alternative tool for screening familial cancer patients in the clinic. Discovery of deleterious mutations by NGS has potential clinical utility both for individuals with cancer and their relatives. For instance, ATM and MRE11A deficiency is sensitive to poly (ADP-ribose) polymerase-1 (PARP-1) inhibition (Williamson et al. 2010; Vilar et al. 2011; Ledermann et al. 2014). In addition, clinical intervention such as salpingo-oophorectomy, which is undertaken in many women with BRCA mutations, may decrease cancer risk in probands and in their relatives with mutations (Munsell et al. 2006; Rebbeck et al. 2009). Therefore, breast and/or ovarian cancer patients without BRCA1/2 mutations should be screened using panel-based analyses for cancer prevention during routine medical checkups.
Acknowledgments
We thank Takuro Uchida and Yumi Kubota for their help. This study was supported by a Grant-in-Aid for Genome Research Project from Yamanashi Prefecture (Y. H. and M. O.).
Conflict of Interest
None declared.
Supporting Information
Table S1. Primer sequencings.
Table S2. Gene list in multi-gene panel.
Table S3. Run summary.
Table S4. Coverage data.
Table S5. Deleterious mutation identified in this analysis.
Figure S1. MSH6 K1358fs (c.4065_4066insTTGA) mutation is suspected to be a nondeleterious mutation. Representative image of read alignments visualized with IGV; the arrow (purple bar in image) indicates the insertion site (upper image). Sequencing chromatograms show the frameshift insertions in peripheral blood DNA (lower image). Nucleotide sequence and amino acid alignment of wild-type and mutant MSH6 (amino acid position: 1353–1360). Underlining indicates the insertion site.
Figure S2. MSH6 splice-site mutation (IVS9+2_+5delGTAAC>G) is suspected to be nondeleterious. Schematic representation of MSH6 wild-type and splice-site variant. The consensus dinucleotide GT is underlined. Representative image of read alignments visualized with IGV; the arrow indicates the deletion site (upper image). Sequencing chromatograms showing deletions in peripheral blood DNA from the patient (lower image).
References
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, et al. Inherited variants of MYH associated with somatic G:C→T: a mutations in colorectal tumors. Nat. Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, et al. Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene. Mol. Oncol. 2008;2:296–316. doi: 10.1016/j.molonc.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KD, Glaser CL, Thompson RE, Hamilton SR, Griffin CA. Eshleman JR. Detection of microsatellite instability by fluorescence multiplex polymerase chain reaction. J. Mol. Diagn. 2000;2:20–28. doi: 10.1016/S1525-1578(10)60611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonai B, Grandjouan S, Huiart L, Caron O, Colas C. Bonai C. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305:2304–2310. doi: 10.1001/jama.2011.743. [DOI] [PubMed] [Google Scholar]
- Broeks A, Urbanus JHM, Floore AN, Dahler EC, Klijn JGM, Rutgers EJT, et al. ATM-heterozygous germline mutations contribute to breast cancer – susceptibility. Am. J. Hum. Genet. 2000;66:494–500. doi: 10.1086/302746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2014a;33:304–311. doi: 10.1200/JCO.2014.57.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch FJ, Nathanson KL. Offit K. Two decades after BRCA: setting care and prevention. Science. 2014b;343:1466–1471. doi: 10.1126/science.1251827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cybulski C, Lubiński J, Wokołorczyk D, Kuźniak W, Kashyap A, Sopik V, et al. Mutations predisposing to breast cancer in 12 candidate genes in breast cancer patients from Poland. Clin. Genet. 2014;1:1–5. doi: 10.1111/cge.12524. [DOI] [PubMed] [Google Scholar]
- Domchek SM, Bradbury A, Garber JE, Offit K, Robson ME, Cornell W, et al. Multiplex genetic testing for cancer susceptibility: out on the high wire without a net ? J. Clin. Oncol. 2013;31:1267–1270. doi: 10.1200/JCO.2012.46.9403. [DOI] [PubMed] [Google Scholar]
- Easton DF, Bishop DT, Ford D, Crockford GP Breast Cancer Linkage Consortium. Genetic linkage analysis in familial breast and ovarian cancer: results from 214 families consortium. Am. J. Hum. Genet. 1993;52:678–701. [PMC free article] [PubMed] [Google Scholar]
- Economopoulou P, Dimitriadis G. Psyrri A. Beyond BRCA: new hereditary breast cancer susceptibility genes. Cancer Treat. Rev. 2015;41:1–8. doi: 10.1016/j.ctrv.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, et al. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology. 2014;148:556–564. doi: 10.1053/j.gastro.2014.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde M, Ferber M, Mao R, Samowitz W. Ganguly A. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis) Genet. Med. 2013;16:101–116. doi: 10.1038/gim.2013.166. [DOI] [PubMed] [Google Scholar]
- Hirotsu Y, Nakagomi H, Sakamoto I, Amemiya K, Mochizuki H. Omata M. Detection of BRCA1 and BRCA2 germline mutations in Japanese population using next-generation sequencing. Mol. Genet. Genomic Med. 2014;3:121–129. doi: 10.1002/mgg3.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King M, Marks JH, Mandell JB, Ben-yishay M, Dutcher JP, Gross SJ, et al. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–647. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J. Clin. Oncol. 2014;32:2001–2009. doi: 10.1200/JCO.2013.53.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaDuca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet. Med. 2014;16:830–837. doi: 10.1038/gim.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum JE, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852–861. doi: 10.1016/S1470-2045(14)70228-1. [DOI] [PubMed] [Google Scholar]
- Lee J-H. Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Munsell MF, Soliman PT, Clark MB, Daniels MS, White KG, Boyd-rogers SG, et al. Prophylactic surgery to reduce the risk of gynecologic cancers in the Lynch syndrome. N. Engl. J. Med. 2006;354:261–269. doi: 10.1056/NEJMoa052627. [DOI] [PubMed] [Google Scholar]
- Narod SA, Ford D, Devilee P, Barkardottir RB, Lynch HT, Smith SA, et al. An evaluation of genetic heterogeneity in 145 breast-ovarian cancer families. Am. J. Hum. Genet. 1995;59:254–264. [PMC free article] [PubMed] [Google Scholar]
- Rebbeck TR, Kauff ND. Domchek SM. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J. Natl. Cancer Inst. 2009;101:80–87. doi: 10.1093/jnci/djn442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts NJ, Jiao Y, Yu J, Kopelovich L, Petersen GM, Bondy ML, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2:41–46. doi: 10.1158/2159-8290.CD-11-0194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitsky K, Bar-shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Stewart GS, Maser RS, Stankovic T, Bressan DA, Kaplan MI, Jaspers NG, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99:577–587. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- Thompson D, Duedal S, Kirner J, McGuffog L, Last J, Reiman A, et al. Cancer risks and mortality in heterozygous ATM mutation carriers. J. Natl. Cancer Inst. 2005;97:813–822. doi: 10.1093/jnci/dji141. [DOI] [PubMed] [Google Scholar]
- Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121:25–33. doi: 10.1002/cncr.29010. [DOI] [PubMed] [Google Scholar]
- Vierkoetter KR, Ayabe AR, VanDrunen M, Ahn HJ, Shimizu DM. Terada KY. Lynch syndrome in patients with clear cell and endometrioid cancers of the ovary. Gynecol. Oncol. 2014;135:81–84. doi: 10.1016/j.ygyno.2014.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011;71:2632–2642. doi: 10.1158/0008-5472.CAN-10-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA. 2011;108:18032–18037. doi: 10.1073/pnas.1115052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson CT, Muzik H, Turhan AG, Zamò A, O’Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol. Cancer Ther. 2010;9:347–357. doi: 10.1158/1535-7163.MCT-09-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorczyk A, Robinson LS. Ross TS. Use of panel tests in place of single gene tests in the cancer genetics clinic. Clin. Genet. 2014 doi: 10.1111/cge.12488. doi: 10.1111/cge.12488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequencings.
Table S2. Gene list in multi-gene panel.
Table S3. Run summary.
Table S4. Coverage data.
Table S5. Deleterious mutation identified in this analysis.
Figure S1. MSH6 K1358fs (c.4065_4066insTTGA) mutation is suspected to be a nondeleterious mutation. Representative image of read alignments visualized with IGV; the arrow (purple bar in image) indicates the insertion site (upper image). Sequencing chromatograms show the frameshift insertions in peripheral blood DNA (lower image). Nucleotide sequence and amino acid alignment of wild-type and mutant MSH6 (amino acid position: 1353–1360). Underlining indicates the insertion site.
Figure S2. MSH6 splice-site mutation (IVS9+2_+5delGTAAC>G) is suspected to be nondeleterious. Schematic representation of MSH6 wild-type and splice-site variant. The consensus dinucleotide GT is underlined. Representative image of read alignments visualized with IGV; the arrow indicates the deletion site (upper image). Sequencing chromatograms showing deletions in peripheral blood DNA from the patient (lower image).