

Abstract
The extended synaptotagmins (E-Syts) are ER proteins that act as Ca2+-regulated tethers between the ER and the plasma membrane (PM) and have a putative role in lipid transport between the two membranes. Ca2+ regulation of their tethering function, as well as the interplay of their different domains in such function, remains poorly understood. By exposing semi-intact cells to buffers of variable Ca2+ concentrations, we found that binding of E-Syt1 to the PI(4,5)P2-rich PM critically requires its C2C and C2E domains and that the EC50 of such binding is in the low micromolar Ca2+ range. Accordingly, E-Syt1 accumulation at ER-PM contact sites occurred only upon experimental manipulations known to achieve these levels of Ca2+ via its influx from the extracellular medium, such as store-operated Ca2+ entry in fibroblasts and membrane depolarization in β-cells. We also show that in spite of their very different physiological functions, membrane tethering by E-Syt1 (ER to PM) and by synaptotagmin (secretory vesicles to PM) undergo a similar regulation by plasma membrane lipids and cytosolic Ca2+.
Keywords: membrane contact sites, Orai1, PLC, STIM1, tricalbin
Introduction
The identification of diverse cellular processes occurring at the interface between the ER and other cellular membranes has prompted intense investigations into the molecular nature, function, and regulation of the tethers that mediate such contacts (Levine & Loewen, 2006; English & Voeltz, 2013; Schuldiner & Weissman, 2013; Stefan et al, 2013; Lahiri et al, 2015). Recently, the three tricalbins (in yeast) and the three extended synaptotagmins (E-Syts) (in mammalian cells) were identified as molecular tethers between the ER and the plasma membrane (PM) (Manford et al, 2012; Toulmay & Prinz, 2012; Giordano et al, 2013; Fernandez-Busnadiego et al, 2015). The E-Syts are embedded in the ER membrane via an N-terminal hairpin that is followed by a cytoplasmic region comprising in sequence a synaptotagmin-like mitochondrial lipid-binding protein (SMP) domain, which is responsible for homo- and heterodimerization, and multiple C2 domains (5 in E-Syt1 and 3 in E-Syt2 and E-Syt3), some of which bind in trans the PM (Giordano et al, 2013; Schauder et al, 2014). Their name reflects a domain organization somewhat similar to the classical synaptotagmins: an N-terminal membrane anchor and cytoplasmic C2 domains (Min et al, 2007). However, the classical synaptotagmins are anchored to synaptic vesicles or other secretory vesicles and participate in Ca2+-dependent fusion (exocytosis) with the PM (Brose et al, 1992; Sudhof, 2013). In contrast, tethering between the ER and the PM mediated by the E-Syts does not lead to fusion. The E-Syts are thought to mediate lipid exchange between the two membranes via their SMP domain dimers, which harbor lipids in a deep hydrophobic groove (Kopec et al, 2011; Toulmay & Prinz, 2012; Schauder et al, 2014).
In spite of these distinct functions, the binding to the PM of the E-Syts, as in the case of the synaptotagmins, requires acidic phospholipids in this membrane and is regulated by cytosolic Ca2+ elevation (Brose et al, 1992; Bai & Chapman, 2004; Giordano et al, 2013; Sudhof, 2013). The cytosolic domains of E-Syt2 and E-Syt3 bind constitutively to the PM via interactions of their C-terminal C2 (C2C) domains with phosphatidylinositol 4,5-bisphosphate, PI(4,5)P2, in the plasma membrane. In contrast, E-Syt1, when expressed alone, is primarily localized throughout the ER and is recruited to the PM in response to cytosolic Ca2+ elevations via the Ca2+-binding properties of its central C2C domain (Giordano et al, 2013). An additional Ca2+ binding site present in the C2A domain of all three E-Syts was shown to mediate Ca2+-dependent, but not acidic phospholipid dependent, membrane binding in vitro, but the significance of this interaction in living cells remains unclear (Min et al, 2007; Xu et al, 2014). As E-Syt1 heterodimerizes with E-Syt2 and E-Syt3 via the SMP domain (Schauder et al, 2014), E-Syt1 containing heterodimers have intermediate properties, with a pool concentrated at ER-PM contact sites under basal conditions and an ER-dispersed pool that can be recruited to the PM upon cytosolic Ca2+ elevation (Giordano et al, 2013).
While the Ca2+-binding properties of the synaptotagmins, and of synaptotagmin 1 (Syt1) in particular, have been extensively characterized (Brose et al, 1992; Sugita et al, 2002; Shin et al, 2009; Wang et al, 2011), the Ca2+-binding properties that mediate the ER-PM tethering functions of the E-Syt remain unknown. Here, we have investigated such properties. We show that E-Syt1 recruitment to the PM requires cytosolic Ca2+ elevation in the micromolar range, that is, concentrations that can be reached by the influx of extracellular Ca2+ via store-operated Ca2+ entry (SOCE), or membrane depolarization in excitable cells. Release of Ca2+ from intracellular stores is insufficient to induce E-Syt1 recruitment if not coupled to store-operated Ca2+ entry. We also show that, in spite of fundamental functional dissimilarities, binding of the cytosolic C2 domains of Syt1 and E-Syt1 to the PM exhibits similar Ca2+- and lipid-binding properties.
Results
A microscopy-based assay reveals that binding of E-Syt1 to the plasma membrane occurs at low micromolar Ca2+
In order to quantify Ca2+-dependent binding of E-Syt1 to the PM, a microscopy-based assay involving semi-intact cells was developed. HeLa cells expressing mCherry-E-Syt1 were loaded with the fluorescent Ca2+-indicator Fluo-4 and mounted on the stage of a microscope (levels of expression of mCherry-E-Syt1 and of other proteins expressed in HeLa cells in this study are shown in Appendix Fig S1). The cells were subsequently permeabilized with Staphylococcus aureus α-toxin and superfused with cytosolic-like buffers, while fluorescence from the basal PM region was recorded. α-toxin generates pores in the PM with a diameter of 1.4 nm, thus allowing free passage of ions and small molecules while proteins are retained within the cell (Fig1A; Bhakdi & Tranum-Jensen, 1991). The buffers contained ATP and calibrated Ca2+-concentrations (between 0.1 and 500 μM). Upon permeabilization, approximately 90% of the Fluo-4 molecules were lost from the cells. The remaining Fluo-4 molecules, probably bound to intracellular membranes, responded in a dose-dependent manner to step-wise increases in the intracellular Ca2+ concentration with half-maximal effect seen at 1.1 ± 0.05 μM Ca2+ (Fig1B–D), a value close to its reported KD of 0.35 μM. Using this assay and total internal reflection fluorescence (TIRF) microscopy, Ca2+ concentration-dependent recruitment of mCherry-E-Syt1 to preexisting ER-PM contacts (as reflected by its appearance within the evanescent field) was observed, with half-maximal (EC50) effect seen at 1.8 ± 0.7 μM (Hill coefficient 1.4; Fig1E–H). Ca2+-induced binding of E-Syt1 to the PM was also observed on lateral surfaces of cells using confocal microscopy (Fig1I), which rules out possible artifacts associated with TIRF microscopy, such as Ca2+-induced changes in cell adherence leading to changes of the fluorescence within the evanescent field.
Figure 1.

Ca2+-induced plasma membrane binding of E-Syt1 leading to expansion of ER-PM contacts
- Schematic representation of the in situ binding assay based on fluorescence microscopy of α-toxin-permeabilized HeLa-M cells expressing fluorescent fusion proteins. Insets shows ER-PM contact sites as seen by confocal microscopy (top) or TIRF microscopy (bottom). In this and all other black and white micrographs, fluorescence is show in black. Scale bar, 10 μm.
- TIRF microscopy images of a HeLa cell loaded with the Ca2+ indicator Fluo-4 following α-toxin permeabilization and exposure to buffers with the indicated Ca2+ concentrations.
- Fluo-4 fluorescence intensity changes following permeabilization and the addition of Ca2+ buffers as in (B). Scale bar, 10 μm.
- Dose–response curve for Ca2+-induced Fluo-4 fluorescence change (means ± SEM for 59 cells from 9 separate experiments).
- Drawings of the E-Syts and synaptotagmin 1 (Syt1). C2 domains with greater structural similarity to each other are indicated by the same shade of gray.
- TIRF microscopy images showing expansion of ER-PM contacts labeled by mCherry-E-Syt1 (top) or the luminal ER marker ER-oxGFP (bottom) in response to Ca2+. Scale bar, 2 μm.
- TIRF microscopy recording of mCherry-E-Syt1 fluorescence from a cell following permeabilization and exposure to buffers with the indicated Ca2+ concentrations.
- Dose–response curve for Ca2+-induced mCherry-E-Syt1 plasma membrane binding (means ± SEM for 38 cells from 6 separate experiments).
- TIRF (top) and confocal (bottom) microscopy images of HeLa cells expressing mCherry-E-Syt1 following permeabilization and exposure to Ca2+. Scale bar, 2 μm. Red arrows points to the plasma membrane region.
- TIRF microscopy recording of mCherry-E-Syt1 fluorescence from a cell following permeabilization and exposure to buffers with the indicated Ca2+ concentrations.
- Confocal microscopy images of HeLa cells showing the distribution of the PI(4,5)P2 biosensor GFP-PH-PLCδ1 before and after permeabilization and exposure to buffers with the indicated Ca2+ concentrations. Scale bar, 10 μm.
- PM GFP-PH-PLCδ1 fluorescence changes following permeabilization and the addition of buffers with the indicated Ca2+ concentrations.
- Dose–response curve for Ca2+-induced GFP-PH-PLCδ1 PM fluorescence change (means ± SEM for 12 cells from 3 separate experiments).
- TIRF micrographs from a permeabilized cell expressing mCherry-E-Syt1 and GFP-PH-PLCδ1. Scale bar, 2 μm.
- PM fluorescence change for the cell in (N).
Ca2+ concentrations above 50 μM resulted in dissociation of E-Syt1 from the PM (Fig1I and J). Since Ca2+-dependent PM binding of E-Syt1 requires PI(4,5)P2 (Giordano et al, 2013), the impact on the levels of this phospholipid after permeabilization and step-wise increases in the Ca2+ concentration was assessed using the PH domain of PLCδ1 (GFP-PH-PLCδ1) as a reporter and confocal (Fig1K) or TIRF (Fig1L–O) microscopy. At basal Ca2+ levels, GFP-PH-PLCδ1 was primarily localized at the PM, reflecting robust presence of PI(4,5)P2 even under these lysis conditions due to the availability of ATP for PI(4,5)P2 synthesis in the buffer (Thore et al, 2007). Ca2+ induced the dose-dependent dissociation of the PH domain from the PM (EC50 24 ± 2 μM Ca2+; Fig1K–O), most likely due at least in part to phospholipase C activation (Rhee, 2001), with maximal effect (complete loss) seen at 50 μM, that is, the concentration range of Ca2+ that induced E-Syt1 dissociation from the PM (Fig1I and J). The persistence of some E-Syt1 at the cell cortex at Ca2+ concentrations that result in complete loss of the PI(4,5)P2 reporter (Fig1N) may reflect binding of E-Syt1 to other acidic lipids, primarily phosphatidylserine (PS).
Cooperation between the C2C and C2E domains in the Ca2+-dependent binding of E-Syt1 to the plasma membrane
Structural predictions indicate the presence of Ca2+-binding loops in the C2A and C2C domains of E-Syt1 (Giordano et al, 2013; Xu et al, 2014). The one in the C2C domain has been confirmed by the importance of this domain, and its predicted Ca2+-binding residues, in the Ca2+-dependent recruitment of E-Syt1 to the PM (Chang et al, 2013; Giordano et al, 2013). The one in the C2A domain is predicted by sequence alignments with the highly similar C2A domain of E-Syt2, which binds liposomes in a Ca2+-dependent way irrespective of the presence of acidic phospholipids in the bilayer and whose binding to Ca2+ has been confirmed by crystallography (Min et al, 2007; Xu et al, 2014). The role of this domain in E-Syt1 recruitment to the PM remains unclear. To further address the contribution of different portions of the cytosolic region of E-Syt1 to PM binding, deletion constructs were generated and tested using the semi-intact cell assay described above (Fig2A).
Figure 2.

Ca2+-dependent interactions of E-Syt1 with plasma membrane PI(4,5)P2 require the C2C and C2E domains
- Schematic representation of E-Syt1 and Syt1 domain structures and of fusion proteins used in the study.
- Confocal microscopy images (left) and changes in plasma membrane fluorescence (right) following α-toxin permeabilization and exposure to increasing Ca2+ concentrations for mCherry-E-Syt1-SMP-C2ABCDE.
- Quantification of the relative fluorescence change at the plasma membrane for the indicated E-Syt1 fusion proteins after exposure to 1, 10, 50, 200, and 500 μM Ca2+ (means ± SEM for 24 (C2AB), 31 (C2ABC), 27 (C2ABCD), 38 (C2C), 33 (C2CD), 21 (C2E), 44 (C2CDE), 45 (C2ABCDE), and 48 (SMP-C2ABCDE) cells from > 3 separate experiments). The Ca2+-induced dissociation of mCherry-E-Syt1-C2E from the PM is shown to the right on a separate y-axis, with example confocal micrographs to illustrate the distribution at low and high Ca2+ concentration shown above.
- Normalized dose–response curves for Ca2+-induced mCh-E-Syt1 and mCh-E-Syt1-SMP-C2ABCDE PM binding.
- Normalized dose–response curves for mCh-E-Syt1-C2C and mCh-E-Syt1-C2E PM binding at different Ca2+ concentrations.
- Confocal microscopy images (left) and changes in plasma membrane fluorescence (right) following α-toxin permeabilization and exposure to increasing Ca2+ concentrations for mCherry-Syt1-C2AB.
- Quantification of the fluorescence change at the plasma membrane for the indicated Syt1 fusion proteins after exposure to 1, 10, 50, and 200 μM Ca2+ (means ± SEM for 31 and 11 cells from 6 and 2 separate experiments, *P < 0.05).
- Dose–response curves for Ca2+-induced PM binding of mCh-Syt1-C2AB and mCh-HPE-Syt1-C2ABSyt1.
- Estimated EC50 values for Ca2+-induced plasma membrane binding (means ± SEM). As discussed in the text, these values are approximate. They represent an underestimation for those fusion proteins whose PM binding is biphasic at high Ca2+ concentrations (see FigEV1 for representative curves from which these values were derived).
Data information: All images have been inverted to show fluorescence in black. Scale bars, 10 μm.
mCherry-E-Syt1-SMP-C2ABCDE, a cytosolic protein containing all five C2 domains and the SMP domain but not the ER anchor, exhibited Ca2+ concentration-dependent binding to the PM (Figs2B and EV1A). Following the step increase to 50 μM Ca2+, there was a biphasic response of this construct (Fig2B, right), with an initial increase in the fluorescence at the PM followed by a decrease, most likely reflecting PI(4,5)P2 hydrolysis. This biphasic response, observed also in some of the following experiments, complicates accurate EC50 estimations of the Ca2+-dependent binding to the PM, as the peak PM binding does not reflect a steady-state binding, but a balance between binding affinity and PI(4,5)P2 hydrolysis. In spite of this limitation, approximate EC50 values derived from measuring maximal PM binding at each step increase in Ca2+ up to 50 μM provide an indication of the relative affinity of different constructs for PI(4,5)P2 in the PM. The approximate EC50 for the (soluble) mCherry-E-Syt1-SMP-C2ABCDE construct was 11 ± 2 μM Ca2+. Further deletion of this construct revealed no contribution of either the C2AB domains or the SMP domain to this binding, but key roles for the C2C domain, as expected (Giordano et al, 2013), and, surprisingly, also of the C2E domain (Figs2C and EV1B–I). In particular, deletion of the C2E domain from this cytosolic C2ABCDE construct resulted in both reduced affinity and magnitude of Ca2+-induced binding. The residual binding appeared to be largely PI(4,5)P2 independent, as it occurred also at very high Ca2+ concentrations (> 50 μM) (Figs2C and EV1D). Accordingly, the C2E domain, when expressed alone, was found to bind the PM and to exhibit Ca2+-induced dissociation similar to that observed for the PH domain of PLCδ1 (EC50 for dissociation: 42 ± 6 μM Ca2+; Fig2C and E). The C2C domain alone or together with the C2D domain bound the PM only very weakly and required the cooperative action of the C2E domain for efficient PM binding (Figs2C and E, and EV1F and H).

Contribution of the different C2 domains to E-Syt1 plasma membrane binding (relates to Figure 2)
- A–C TIRF micrographs (left) and single-cell TIRF recordings of mCherry fluorescence for the indicated E-Syt1 domain deletion protein binding the plasma membrane PI(4,5)P2 in a Ca2+-dependent manner.
- D–H TIRF micrographs (left) and single-cell TIRF recordings of mCherry fluorescence for the indicated E-Syt1 domain deletion protein binding the plasma membrane in a PI(4,5)P2-independent and Ca2+-dependent manner.
- I TIRF micrographs (left) and single-cell TIRF recording of mCherry fluorescence for the indicated E-Syt1 domain deletion protein binding the plasma membrane in a PI(4,5)P2-dependent and Ca2+-independent manner.
Data information: The clustering or aggregation of fluorescent protein seen in (D, E and H) is only observed at 200 μM Ca2+ and reflects cytosolic aggregates. All scale bars, 10 μm.
Comparison of the C2 domains of E-Syt1 with the C2 domains of E-Syt2 and E-Syt3, based on amino acid sequence similarity and structural predictions, helped to explain these findings. The C2E domain of E-Syt1 is most similar to the C2C domains of E-Syt2 and E-Syt3, that is, the domains that mediated constitutive binding to the PM of these two E-Syts. Structural modeling revealed that all these C-terminal C2 domains have a basic acidic phospholipid binding surface and no putative Ca2+-binding loops (Appendix Fig S2) (Kelley & Sternberg, 2009). The C2C-C2D domains of E-Syt1, based on their amino acid sequences, appear to represent a duplication of the C2A-C2B domain pair (Jimenez & Davletov, 2007). The absence of the C2C-C2D module in E-Syt2 and E-Syt3, and thus of regulatory interactions of this module, likely accounts for the constitutive binding of these two proteins to the PM.
For comparison, we expressed the well-characterized cytosolic tandem C2A and C2B (C2AB) fragment of classical synaptotagmin 1 (Syt1) and found, using the same assay, an EC50 (7 ± 2 μM Ca2+) roughly similar to that (11 ± 2 μM, see above) of the cytosolic portion of E-Syt1. We also observed a similar biphasic response upon the 50 μM Ca2+ step and further decrease in PM binding at higher Ca2+ levels, indicating interactions with PI(4,5)P2 (Fig2F–H; Radhakrishnan et al, 2009). Some residual bindings at 200 μM likely reflect phosphatidylserine binding (Shin et al, 2009; Honigmann et al, 2013), as in the case of E-Syt1.
The observation that the estimated EC50 for PM binding was about one order of magnitude higher for the cytosolic portion of E-Syt1 compared to full-length E-Syt1 indicates an impact of the proximity of its C2 domains to the ER membrane on its property to bind the PM. Perhaps this is due to cooperative actions, so that formation of E-Syt1 tethers facilitates the accumulation of other E-Syt1 molecules at the same ER-PM contact sites as they diffuse within the ER membrane. Interestingly, appending the hairpin region of E-Syt1 to the N-terminus of the C2AB domain of classical synaptotagmin 1 also enhanced its EC50 for Ca2+-dependent PM binding (2.1 ± 0.2 μM Ca2+; Fig2G and H).
Deletion studies were also carried out on E-Syt1 constructs that retained the N-terminal ER anchor (Fig3A). Removal of the SMP domain right shifted the Ca2+-dependence of PM binding (accumulation at ER-PM contacts as revealed by TIRF microscopy) to an approximate EC50 of 9.6 ± 0.8 μM. This possibly reflects the reduced avidity caused by the inability of this mutant to form dimers (Figs3B and C and EV2B). Removal of the three C-terminal C2 domains resulted in significantly reduced accumulation of E-Syt1 at the PM at all tested Ca2+ concentrations, consistent with cooperation between the Ca2+-binding C2C domain and the PI(4,5)P2-binding C2E domain in such accumulation (Figs3B and EV2C).
Figure 3.

Presence of the SMP domain is required for efficient E-Syt1 plasma membrane binding
- A Schematic representation of fusion proteins used in the study.
- B Quantification of the fluorescence change at the plasma membrane for the indicated fusion proteins after exposure to 1, 10, 50, and 200 μM Ca2+ presented as means ± SEM for 51 (E-Syt1), 34 (E-Syt1(ΔSMP)), and 36 (E-Syt1(ΔC2CDE)) cells from > 3 separate experiments. *P < 0.01 compared to WT. #P < 0.01 (ΔSMP) or P < 0.01 (ΔC2CDE) compared to WT. §P < 0.01 compared to ΔSMP. ¶P < 0.01 compared to ΔSMP. %P < 0.01 compared to ΔSMP.
- C Dose–response curves for Ca2+-induced PM binding of mCh-E-Syt1 and mCh-E-Syt1(ΔSMP).
- D Dose–response curve for Ca2+-induced PM dissociation of GFP-E-Syt2 (means ± SEM for 12 cells from 3 separate experiments).
- E Dose–response curves for Ca2+-induced plasma membrane binding of GFP-E-Syt2 (yellow) and mCherry-E-Syt1 (black) in cells co-expressing the two constructs (means ± SEM for 13 cells from 3 separate experiments).
- F Immunoblot showing E-Syt2 expression levels in control (C) and E-Syt2 (E2) siRNA-treated HeLa cells.
- G, H Dose–response curves for Ca2+-induced PM binding of mCherry-E-Syt1 (G) or mCherry-E-Syt1(ΔSMP) (H) in control and E-Syt2 KD cells (means ± SEM for 15 cells from 3 separate experiments).
Data information: Estimated EC50 values for Ca2+-induced PM binding (means ± SEM) are given next to each trace. As discussed in the text, these values are approximate. They represent an underestimation for those fusion proteins whose PM binding is biphasic at high Ca2+ concentrations (see FigEV2 for representative curves from which these values were derived).
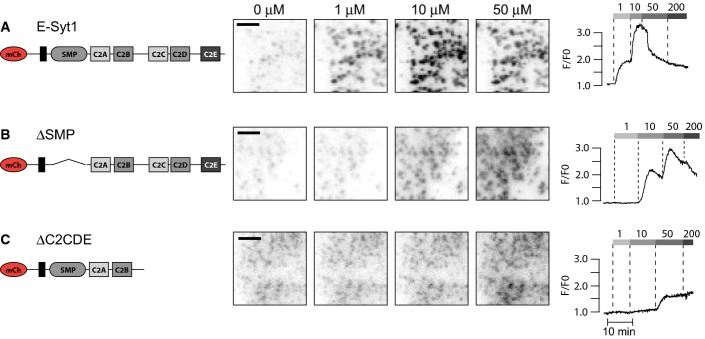
Contribution of the SMP domain to E-Syt1 plasma membrane binding (relates to Figure3)
- A–C TIRF micrographs (left) and single-cell TIRF recordings of mCherry fluorescence (right) for the indicated, ER-localized E-Syt1 domain deletion proteins. All scale bars, 2 μm.
Since it was shown that members of the E-Syt protein family form heterodimers (Giordano et al, 2013), we tested the possibility that interactions with E-Syt2 can impact the dynamics of E-Syt1. When expressed alone (thus resulting in E-Syt2 overexpression), mCherry-E-Syt2 was constitutively concentrated at the PM as reported (Giordano et al, 2013) and exhibited a Ca2+ concentration-dependent dissociation from the PM (Fig3D). The EC50 of this reaction (20 ± 4 μM) was similar to that of the PI(4,5)P2 biosensor PH-PLCδ1 (Fig1M), confirming the requirement of PI(4,5)P2 for its membrane binding. Co-expression of GFP-E-Syt2 and mCherry-E-Syt1 had little effect on the Ca2+-dependent PM binding of E-Syt1 (1.7 ± 0.4 μM), but conferred a Ca2+-dependent component to E-Syt2 PM binding with an EC50 identical to that of E-Syt1 (1.4 ± 0.2 μM; Figs3E and EV3A–C). This strongly indicates the formation of E-Syt1-E-Syt2 heterodimers. In cells where E-Syt2 expression was reduced by siRNA (Fig3F), full-length E-Syt1 still bound the PM in a Ca2+-dependent manner, but the EC50 of this reaction was right shifted compared to control cells (1.7 ± 0.4 and 5.1 ± 0.7 μM Ca2+, Figs3G and EV3D–G). Interestingly, E-Syt2 knockdown was without effect on the Ca2+-induced PM binding of E-Syt1 lacking the SMP domain, that is, the heterodimerization domain (Schauder et al, 2014; 11.8 ± 0.6 and 10.8 ± 1.0 μM Ca2+, Fig3H), confirming that an interaction between the SMP domain of E-Syt1 and E-Syt2 stabilizes E-Syt1 at ER-PM contacts due to the strong constitutive binding of E-Syt2 to the PM.

E-Syt1-E-Syt2 heterodimerization has an impact on plasma membrane binding (relates to Figure3)
- A, B TIRF micrographs of GFP-E-Syt2 (A) or GFP-E-Syt2 and mCherry-E-Syt1 (B) fluorescence from a permeabilized cell exposed to buffers with the indicated Ca2+ concentrations.
- C Single-cell TIRF recording of mCherry-E-Syt1 (black) and GFP-E-Syt2 (yellow) fluorescence following exposure to buffers with the indicated Ca2+ concentrations.
- D, E TIRF micrographs (D) and single-cell TIRF recording (E) of mCherry-E-Syt1 fluorescence in control cells after permeabilization and exposure to buffers with the indicated Ca2+ concentrations.
- F, G TIRF micrographs (F) and single-cell TIRF recording (G) of mCherry-E-Syt1 fluorescence in E-Syt2 knockdown cells after permeabilization and exposure to buffers with the indicated Ca2+ concentrations.
Data information: All scale bars, 2 μm.
E-Syt1 binding to the plasma membrane requires Ca2+ influx from the extracellular medium
As the experiments described above involved semi-intact cells, it was important to determine whether the EC50 values of the Ca2+-dependent PM binding of full-length E-Syt1 (low micromolar), as determined by the experiments described above, were consistent with the dynamics of E-Syt1 in intact cells. It has already been shown that stimulation of cells with a muscarinic receptor agonist to induce phospholipase C (PLC) activation, and thus IP3-dependent Ca2+ release from the ER, can induce PM binding of E-Syt1 (Giordano et al, 2013). Likewise, drugs that directly mobilize Ca2+ from the ER by inhibiting the sarco-endoplasmatic reticulum Ca2+ ATPase (SERCA pump), such as thapsigargin (irreversible SERCA pump inhibitor; Lytton et al, 1991), were shown to induce E-Syt1 PM binding. However, these experiments were all performed in the presence of extracellular Ca2+, that is, under conditions where release of Ca2+ from the ER is closely coupled to massive influx of Ca2+ from the extracellular space via STIM1-Orai1-mediated store-operated Ca2+ entry (SOCE; Wu et al, 2007). To assess whether the smaller increase in cytosolic Ca2+ induced by SERCA pump inhibition or IP3 generation in the absence of SOCE is sufficient to trigger E-Syt1 redistribution, these experiments were repeated in the absence of extracellular Ca2+.
Addition of reversible (2,5-di-t-butyl-1,4-benzohydroquinone; BHQ) or irreversible (thapsigargin) SERCA inhibitors (Llopis et al, 1991) or of the muscarinic receptor agonist oxotremorine-M in the absence of extracellular Ca2+ induced release of Ca2+ from the ER, as expected (Figs4A and EV4A). However, such release was not accompanied by the translocation of E-Syt1 to the PM, as detected by TIRF microscopy (Figs4B and EV4B and C). This is consistent with the micromolar Ca2+ requirement for E-Syt binding to the PM, as cytosolic Ca2+ levels achieved by the addition of SERCA inhibitors are in the several 100s’ nanomolar range (Broad et al, 1999). As also detected by TIRF microscopy, the depletion of Ca2+ from the lumen of the ER occurring under these conditions led to a progressive accumulation of ER-anchored STIM1 at the PM, where it activates Orai Ca2+ channels (Fig4C). Subsequent addition of 1–10 mM Ca2+ to the extracellular medium, either in the absence or in the presence of SERCA inhibitors, caused a massive increase in cytosolic Ca2+ (SOCE) through the activated Ca2+ channels (Figs4A and EV4D–G). Such increase induced a very robust translocation of E-Syt1 to the PM (Figs4B and EV4D–G), which, in the absence of SERCA inhibition (i.e., when a reversible inhibitor of the SERCA pump had been washed out), preceded the dissociation of STIM1 and the inactivation of SOCE (Fig4D). Inspection of TIRF microscopy images during the manipulation showed that E-Syt1 does not form new contacts, but populates and expands contacts previously occupied by STIM1 and eventually replaces STIM1 at these sites (Fig4E). Ruling out the possibility of potential interference or competition between E-Syt1 and STIM1 for PM binding, we also found that sequential PM recruitment of E-Syt1 induced first by the photolysis of caged Ca2+ (a manipulation that does not generate STIM1 accumulation at contact sites) and then by the addition of thapsigargin (a manipulation that induces STIM1 accumulation at contact sites) resulted in expansion of the same ER-PM contacts, demonstrating that this property of E-Syt1 is independent of STIM1 (Fig EV4H and I).
Figure 4.

E-Syt1 plasma membrane binding is triggered by SOCE
- A–C TIRF microscopy recordings of GCaMP5G (A), mCherry-E-Syt1 (B), and mRFP-STIM1 (C) fluorescence change in HeLa cells in response to release of Ca2+ from the ER induced by 50 μM BHQ and subsequent activation of SOCE by the addition of 3 mM Ca2+. Data are presented as means ± SEM for 55 (GCaMP5G), 39 (E-Syt1), and 39 (STIM1) cells from 4 separate experiments.
- D Simultaneous TIRF microscopy recordings of GFP-E-Syt1 and mRFP-STIM1 fluorescence from a HeLa cell during release of Ca2+ from the ER (BHQ) and activation of SOCE (3 Ca2+). The indicated region is shown below on an expanded time scale.
- E TIRF microscopy images of the HeLa cell in (D). Scale bar is 2 μm.
- F Immunoblot for STIM1 in control (Ctrl) and STIM1 siRNA-treated HeLa cells.
- G TIRF microscopy recordings of mCherry-E-Syt1 fluorescence in response to 1 μM thapsigargin (TG) in control (black) and STIM1 siRNA-treated (yellow) HeLa cells (means ± SEM for 31 and 28 cells in 3 separate experiments).
- H TIRF microscopy recording from a single HeLa cell expressing mCherry-E-Syt1 and the Ca2+ reporter GCaMP5G and exposed in sequence to the SERCA inhibitor CPA to activate SOCE and then to 1 μM Gd3+ to stop SOCE without disrupting STIM1-Orai1 contacts.
- I Quantification of mCh-E-Syt1 PM binding in response to SOCE and the addition of Gd3+. Means ± SEM for 24 cells in 3 separate experiments. *P < 0.001 (vs Basal); #P < 0.01 (vs 3 mM Ca2+).
- J, K TIRF microscopy recordings of GCaMP5G (J) and mCherry-E-Syt1 (K) fluorescence from HeLa cells treated with 0.1% DMSO (v/v) (control, black), 1 μM EGTA-AM (light blue), or 1 μM BAPTA-AM (dark blue) followed by activation of SOCE (means ± SEM for 43 (control), 52 (EGTA), and 51 (BAPTA) cells in 3 separate experiments).
- L Immunoblot for E-Syt1 in control (Ctrl) and E-Syt1 siRNA-treated HeLa cells.
- M Ca2+ recordings from control (black) or E-Syt1 siRNA-treated (red) HeLa cells loaded with Fluo-4 in response to release of Ca2+ from the ER (BHQ) and activation of SOCE (3 Ca2+) (means ± SEM for 425 (control) and 325 (E-Syt1 KD) cells in 7 separate experiments).
Data information: All black and white images have been inverted to show fluorescence in black.

E-Syt1 PM binding requires triggered Ca2+ influx (relates to Figure4)
- A, B TIRF microscopy recordings of GCaMP5G (A) and mCherry-E-Syt1 (B) fluorescence following stimulation with 10 μM oxo-M, in the absence (red) or presence (black) of extracellular Ca2+. The HeLa cells also overexpressed an M1 muscarinic receptor.
- C Quantification of the peak Ca2+ increase (left) and E-Syt1 PM binding (right) from the experiments in (A) and (B). Data show means ± SEM for 19 cells from 3 separate experiments (**P < 0.01 for difference from 1.3 mM Ca2+).
- D, E TIRF microscopy recordings of GCaMP5G (green) and mCherry-E-Syt1 (purple) fluorescence following exposure to 1 μM thapsigargin in the absence of extracellular Ca2+, followed by the addition of Ca2+ at different concentrations (0.2–10 mM).
- F, G Quantification of the peak fluorescence increase in mCherry-E-Syt1 (F) and GCaMP5G (G) measured by TIRF microscopy in response to the application of the indicated Ca2+ concentration in the presence of 1 μM thapsigargin. Data show means ± SEM for 19 cells from 4 experiments.
- H Confocal micrographs of mCherry-E-Syt1 fluorescence from a HeLa cell maintained in a Ca2+-containing buffer and exposed to flash photolysis of caged Ca2+ and subsequent addition of 1 μM thapsigargin (TG). Scale bar, 1 μm.
- I Means ± SEM (n = 4 cells from 2 experiments) of mCherry-E-Syt1 fluorescence change at ER-PM contacts following flash photolysis of Ca2+ and subsequent addition of 1 μM thapsigargin (TG).
To confirm the requirement of STIM1 and SOCE in the recruitment of E-Syt1, the cellular levels of STIM1 were reduced by siRNA (Fig4F). This resulted in complete inhibition of SOCE (achieved by thapsigargin) and prevented E-Syt1 PM binding (Figs4G and EV5A and B). Conversely, induction of SOCE by the SERCA inhibitor cyclopiazonic acid (CPA) followed by pharmacological blockage of SOCE by 1 μM Gd3+, a treatment known to prevent Ca2+ influx without dispersing STIM1-Orai clusters (Putney, 2010), reversed E-Syt1 PM accumulation (Fig4H and I). These observations show that PM binding of E-Syt1 only occurs after the massive increases in the cellular Ca2+ concentration that result from influx of extracellular Ca2+, which is consistent with the EC50 values (micromolar) revealed by experiments in semi-intact cells (see Fig1). To further assess the response of E-Syt1 to Ca2+ entry, SOCE was triggered in mCherry-E-Syt1-expressing HeLa cells loaded with the Ca2+ chelators EGTA or BAPTA. EGTA is a slow Ca2+ chelator that is unable to efficiently buffer rapid changes in Ca2+ concentration, whereas BAPTA is a fast chelator that can rapidly buffer even large rapid Ca2+ changes. As expected, BAPTA was more potent than EGTA in antagonizing SOCE (Fig4J). A similar difference was seen between the effects of the two chelators on E-Syt1 PM recruitment (Fig4K).
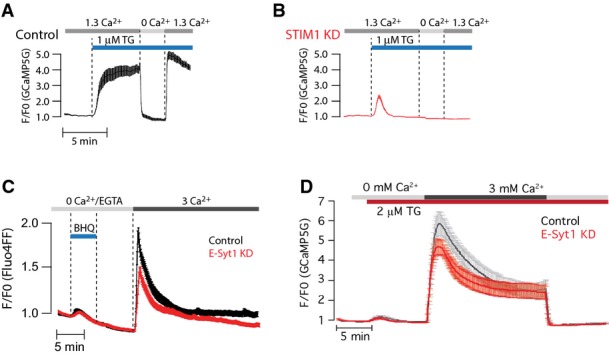
E-Syt1 knockdown has little impact on SOCE (relates to Figure4)
- A, B TIRF microscopy recordings of GCaMP5G fluorescence in response to 1 μM thapsigargin (TG) in control (A, black) and STIM1 siRNA-treated (B, red) HeLa cells (means ± SEM for 31 (control) and 28 (STIM1 KD) cells in 3 separate experiments).
- C Ca2+ imaging of control or E-Syt1 KD HeLa cells loaded with the low-affinity Ca2+ indicator Fluo-4FF during release of Ca2+ from the ER and activation of SOCE. Curves are presented as means ± SEM for 38 (control, red) and 33 (E-Syt1 KD, black) cells from 4 separate experiments.
- D Fluorescence microscopy recordings of GCaMP5G fluorescence in control or E-Syt1 knockdown HeLa cells exposed to 1 μM thapsigargin (TG) in Ca2+-free buffer, followed by the re-addition of 3 mM Ca2+. Data are presented as means ± SEM for 21 (control) and 23 (E-Syt1-KD) cells from 3 experiments.
In agreement with earlier observations (Giordano et al, 2013), we did not observe a major effect of the knockdown of E-Syt1 on either the activation or inactivation of SOCE, as measured by both high (Fluo4)- and low (Fluo4FF)-affinity Ca2+ indicators (Figs4L and M and EV5C and D).
Ca2+-induced plasma membrane binding of E-Syt1 and expansion of ER-PM contacts in excitable cells
We next explored whether Ca2+ concentrations achieved by depolarization-dependent Ca2+ entry in excitable cells could also trigger the PM association of E-Syt1. To this aim, we used the electrically excitable pancreatic β-cell that secretes insulin in response to depolarization-induced influx of Ca2+ through L-type Ca2+ channels (Tengholm & Gylfe, 2009). Clonal MIN6 β-cells expressing GFP-E-Syt1 and the red-shifted Ca2+-indicator R-GECO (Zhao et al, 2011) were imaged with TIRF microscopy upon depolarization by 30 mM K+. The depolarization resulted in a rapid increase in cytosolic Ca2+ that was accompanied by E-Syt1 PM binding (Fig5A and B). Chelation of intracellular Ca2+ by BAPTA prevented such recruitment (Fig5C). As expected, based on the experiments described in Fig2, depolarization also induced the accumulation at the PM of the mCherry-tagged C2CDE domains of E-Syt1 and of the C2AB domains of classical synaptotagmin 1, both of which are cytosolic fragments containing C2 domains that bind PM PI(4,5)P2 in a Ca2+-dependent way (Fig5C–E). Also consistent with data obtained in permeabilized cells, depolarization-induced PM binding of both E-Syt1 and the C2AB fragment of classical synaptotagmin 1 was counteracted by optogenetic depletion of PI(4,5)P2 in the PM (Fig5D and E; Idevall-Hagren et al, 2012). We also observe that the binding of the cytosolic fragments to the PM was more rapid than that observed for ER-localized fragments, consistent with restricted diffusion within the ER membrane (Fig EV6; Nehls et al, 2000).
Figure 5.

Depolarization-induced Ca2+ influx triggers E-Syt1 plasma membrane binding and expansion of ER-PM contacts in insulin-secreting β-cells
- A TIRF microscopy images of a MIN6 β-cell expressing GFP-E-Syt1 (shown) and R-GECO before and during depolarization by 30 mM K+. Scale bar, 10 μm.
- B TIRF microscopy recordings of the Ca2+-indicator R-GECO (red) and GFP-E-Syt1 (black) fluorescence change during depolarization of a MIN6 β-cell with 30 mM K+. Numbers correspond to images in (A).
- C Means ± SEM for the peak response to 30 mM K+ for the indicated mCherry fusion proteins (n = 16 (E-Syt1), 12 (BAPTA), 16 (C2CDE), and 17 (Syt1-C2AB) cells in 2 separate experiments).
- D, E TIRF microscopy recording of mCherry-E-Syt1 (D) or mCherry-Syt1-C2AB (E) fluorescence change during K+ depolarization before and after blue light-induced PI(4,5)P2 dephosphorylation. Bar graphs show means ± SEM for the peak response to 30 mM K+ in the presence or absence of PI(4,5)P2 (n = 16 and 18 cells in 3 separate experiments). *P < 0.05, ***P < 0.001.
- F Confocal micrographs of a MIN6 β-cell expressing mCherry-E-Syt1 and ER-oxGFP (a luminal ER marker) 30 s before (left panel) and 30 s after (right panel) depolarization by 30 mM K+. Note the partial redistribution of E-Syt1 and ER-oxGFP to the cell periphery after depolarization. Scale bar, 5 μm.
- G TIRF microscopy recordings of ER-oxGFP fluorescence from a MIN6 β-cell during K+ depolarization. Images from the time points indicated by numbers are shown below. Scale bar, 10 μm.
- H Quantification of the change in PM area occupied by ER-oxGFP fluorescence for the indicated treatments (means ± SEM for 15 cells from 3 separate experiments).
- I TIRF micrographs of GFP-E-Syt1 fluorescence from a MIN6 β-cell exposed to 30 mM K+ followed by 1 μM latrunculin A. Graph below shows GFP-E-Syt1 fluorescence change at the PM (means ± SEM for 15 cells from 3 separate experiments). Scale bar, 10 μm.
- J Simultaneous TIRF microscopy recordings of R-GECO (red) and GFP-E-Syt1 (black) fluorescence during stimulation of a MIN6 β-cell with 20 mM glucose.
- K Means ± SEM for the peak GFP-E-Syt1 response to 20 mM glucose alone, in the absence of extracellular Ca2+ or in the presence of 100 μM of the L-type Ca2+-channel inhibitor methoxyverapamil (n = 29 (Control), 30 (0 mM Ca2+), and 36 (Verapamil) cells from 3 separate experiments).
Data information: All black and white images have been inverted to show fluorescence in black.
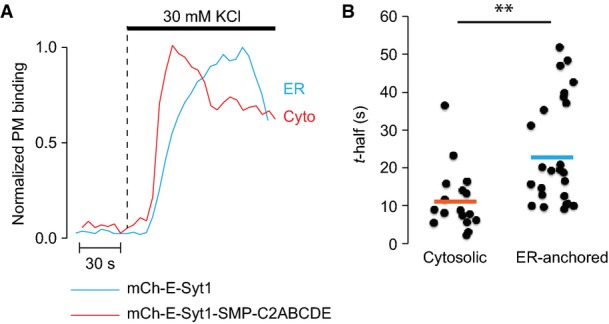
Different PM-binding kinetics of cytosolic and ER-localized E-Syt1
- Representative TIRF microscopy recordings from two MIN6 cells expressing mCherry-E-Syt1 (blue) or mCherry-E-Syt1-SMP-C2ABCDE (red) following depolarization with 30 mM K+.
- Scatter plot showing time to half-maximal PM binding for cells stimulated as in (A). N = 17 cells (mCherry-E-Syt1) and N = 24 cells (mCherry-E-Syt1-SMP-C2ABCDE) from 2 separate experiments (**P < 0.01).
The depolarization-induced binding of E-Syt1 was accompanied by the expansion of ER-PM contacts (Fig5F), as shown by monitoring the accumulation at the cell cortex of ER-oxGFP, a fluorescent luminal ER protein. This expansion of ER-PM contacts in MIN6 β-cells occurred even in the absence of exogenous, and thus overexpressed, E-Syt1, proving that the ER recruitment to the PM in response to Ca2+ is not only due to the overexpression of E-Syt1 (Fig5G and H). Importantly, the expansion of E-Syt1-dependent ER-PM contacts was not due to a loss of cortical F-actin caused by the elevation of cytosolic Ca2+, as loss of F-actin induced by latrunculin A did not result in such expansion (Fig5I).
Ca2+ increases leading to insulin secretion can also be triggered under physiological conditions by an increase in the extracellular glucose concentration. This signaling pathway involves uptake of the sugar and its conversion to ATP, which in turn closes ATP-sensitive K+ channels in the plasma membrane, leading to membrane depolarization and Ca2+ influx through L-type channels (Rorsman et al, 2011). Exposure of MIN6 β-cells expressing GFP-E-Syt1 and R-GECO to a step increase in the glucose concentration from 3 to 20 mM resulted in pronounced increases in cytosolic Ca2+, often in the form of oscillations. Most oscillations were followed by E-Syt1 PM binding and were prevented by the omission of extracellular Ca2+ or by blocking L-type voltage-dependent Ca2+ channels with methoxyverapamil (Fig5J and K).
Discussion
We have assessed the cytosolic Ca2+ concentration required to recruit ER-anchored E-Syt1 to the PI(4,5)P2-rich plasma membrane where it participates in the formation of ER to PM tethers. To do so, we have monitored the redistribution of mCherry-E-Syt1 in α-toxin-permeabilized cells exposed to ATP containing cytosolic buffers of different Ca2+ concentration. This method, in contrast to more traditional assays involving purified proteins and artificial membranes, detects protein membrane interactions under conditions that mimic more closely the physiological intracellular environment and provides sub-cellular spatial information. We have found that E-Syt1 binding to the PI(4,5)P2-rich PM requires Ca2+ levels in the low micromolar range. Accordingly, we have also found that elevations of the cytosolic Ca2+ concentration induced by the release of Ca2+ from intracellular stores under conditions that prevent SOCE—PLC-triggered IP3 receptor activation or the addition of SERCA pump inhibitors in absence of extracellular Ca2+—were not sufficient to induce E-Syt1 binding to the PM in non-permeabilized cells. Typically, upon such manipulations, Ca2+ reaches 100s’ nanomolar concentrations (Broad et al, 1999), but not micromolar concentrations. E-Syt1, however, was rapidly and reversibly recruited to the PM in intact cells under conditions that result in massive influx of extracellular Ca2+, such as during SOCE or during depolarization-dependent Ca2+ entry in excitable cell, when Ca2+ concentrations, at least in the cortical cell region, rise to the micromolar range (Marsault et al, 1997).
We note that the Ca2+ concentration that matters for E-Syt accumulation at ER-PM contact sites is the one that occurs adjacent to the cytosolic leaflet of the PM, as E-Syt1 is not actively translocated to the PM, but simply gets trapped at such sites as it diffuses in the ER (Giordano et al, 2013). The faster recruitment of cytosolic fragments compared to full-length E-Syt or other ER-anchored proteins is explained by the possibility of such fragments to diffuse in three dimensions, while the ER-anchored proteins only travel in two dimensions along the ER membrane (Nehls et al, 2000).
The interaction of C2 domains with membranes can be regulated by two separate sites, the Ca2+-binding loops and an adjacent cationic surface patch. Different C2 domains exhibit different Ca2+ sensitivity and lipid selectivity due to varying properties of the two sites (Rizo & Sudhof, 1998). Analysis of the contribution of the different domains of E-Syt1 in its Ca2+-dependent binding to the PM confirmed a critical role of its C2C domain in such redistribution (Giordano et al, 2013). However, while presence of this domain was required, it did not to any large extent engage in direct binding to PM PI(4,5)P2, contrary to our earlier suggestion (Giordano et al, 2013). It appeared instead to require the cooperation of the C2E domain, which shares the properties of the C2C domain of E-Syt2 and E-Syt3, that is, the domains that mediate the constitutive binding of these other two E-Syts to the PI(4,5)P2-rich PM. We therefore propose a model where the C2C and C2E domains function as intra-molecular coincidence detectors. Both domains require the other in a mutually exclusive manner for efficient Ca2+-induced PM binding to occur within the physiological range of the ion. Additionally, our deletion analysis of E-Syt1 did not indicate a role of its C2A domain in PM binding in response to Ca2+ elevations. This was in spite of the predicted property of this domain (based on its homology to the C2A domain of E-Syt2) to bind artificial lipid membranes in a Ca2+-dependent way, irrespective of the presence of acidic phospholipids in the bilayer (Jimenez & Davletov, 2007; Min et al, 2007; Xu et al, 2014). Perhaps the Ca2+-dependent interaction of this domain with membranes occurs in cis with the ER membrane to facilitate SMP domain positioning for lipid extraction/exchange.
Other factors that regulate the binding of E-Syt1 to the PM include intermolecular interactions and its ER anchorage. Physiologically, E-Syt1 can occur as a heterodimer with E-Syt2 (Giordano et al, 2013). As a consequence, E-Syt2, when co(over)expressed with E-Syt1, is recruited to the PM in a Ca2+-dependent way, with an EC50 of the heterodimer similar to that of E-Syt1 homodimers. On the other hand, the knockdown of endogenous E-Syt2 resulted in a right shift (lower affinity) of the EC50 of E-Syt1, possibly reflecting a contribution of endogenous E-Syt2 to E-Syt1 binding to the PM. ER anchorage has an important impact on the EC50 of E-Syt1 recruitment to the PM, as the EC50 of the entire cytosolic fragment of E-Syt1 without the membrane anchor is about one order of magnitude higher than that of full-length E-Syt1. One possibility is that Ca2+-dependent in cis interactions of the C2A domain may be involved in this regulation. The potential contribution of other proteins in the PM, the ER, or the cytosol (Jean et al, 2012) to the tethering function of the E-Syts should also be considered.
The approximately 2 μM EC50 value for the Ca2+-dependent recruitment of E-Syt1 to the PM is in the same range of EC50 values reported for the Ca2+-dependent binding of classical synaptotagmin 1 to acidic phospholipids (Davletov & Sudhof, 1993; Sugita et al, 2002; Shin et al, 2009). Accordingly, when the tandem C2A-C2B domains of synaptotagmin were appended to the ER anchor of E-Syt1, this construct was recruited to the PM with dynamics similar to what was observed for E-Syt1. We also found that the cytosolic fragments of both E-Syt1 and classical Syt1 exhibit some PM binding at Ca2+ concentrations associated with PI(4,5)P2 depletion from the PM in our binding assay, most likely reflecting phosphatidylserine binding (Shin et al, 2009).
A key finding of this study is that cytosolic Ca2+ levels required to recruit E-Syt1 to the PM can be reached in response to physiological stimulation. Thus, recruitment of E-Syt1 to the PM with resulting expansion of ER-PM contacts occurs in model excitable cells, pancreatic β-cells, upon the elevation of cytosolic Ca2+ produced not only by K+-induced membrane depolarization, but also by glucose, a physiological stimulus that also acts through membrane depolarization. Both stimuli lead to Ca2+ influx through the opening of L-type voltage-dependent Ca2+ channels and thus to insulin secretion via Ca2+ binding to synaptotagmin, a component of the granule membrane (Lang et al, 1997). Accordingly, we found that depolarization-induced Ca2+ influx in β-cells expressing cytosolic fragments of these proteins—C2CDE domains of E-Syt1 and the C2AB domains of synaptotagmin—results in PM recruitment of both fragments. In both cases, recruitment is abolished by the depletion of PI(4,5)P2 in the PM indicating the cooperative action of Ca2+ and PI(4,5)P2 (Appendix Fig S3).
It remains to be understood why an interaction between two membranes (secretory vesicles and the PM) leading to Ca2+-dependent exocytosis (synaptotagmin) shares strong similarities to an interaction between two membranes (ER and PM) that does not participate in fusion (E-Syt) but appears to be required to facilitate lipid exchanges between the two bilayers. Elucidating the relation between the functions of these two classes of proteins represent an interesting avenue for future research.
Materials and Methods
Reagents
Fluo4-AM, Fluo-4FF-AM, NP-EGTA-AM, EGTA-AM, BAPTA-AM, and thapsigargin were from Life Technologies. BHQ, CPA, EGTA, NTA, latrunculin A, α-toxin, methoxyverapamil, Mg-ATP and Na-GTP, and all other salts were from Sigma-Aldrich. Cell culture reagents were from Life Technologies. Primers and siRNA were obtained from Integrated DNA Technologies and MWG-Eurofins. Primary antibodies used in this study were as follows: anti-ESYT1 (HPA016858, Sigma-Aldrich), anti-ESYT2 (HPA002132, Sigma-Aldrich), anti-STIM1 (G555, Cell Signaling), anti-mCherry (ab167453, Abcam), and anti-β-actin (sc-81178, Santa Cruz Biotechnology).
cDNA constructs and siRNAs
mCherry-E-Syt1 and GFP-E-Syt1 as well as a Ca2+-binding-deficient version have been previously described (Giordano et al, 2013). The following cDNA constructs were used in this study: ER-oxGFP (EL Snapp, Albert Einstein College of Medicine), SP-mRFP-STIM1 (B Baird, Cornell University) and SP-YFP-STIM1 (Addgene plasmid 18857), GFP-Orai1 (RS Lewis, Stanford University), CIBN-GFP-CAAX, CRY2-5ptaseOCRL (Idevall-Hagren et al, 2012), GFP-PH-PLCδ1 (T Meyer, Stanford University), R-GECO (Addgene plasmid 45494), GCaMP5G (Addgene plasmid 31788), and Muscarinic receptor (M1R; B Hille, University of Washington). All other cDNA constructs used in this study were made using standard molecular cloning approaches followed by sequence verification.
ER-localized E-Syt1 deletion constructs
mCherry-E-Syt1(Δhairpin): Human E-Syt1 lacking the 82 N-terminal amino acids, including the ER-anchoring sequence, was amplified using the following primers: SMP-C2_BsrG1_F, CCGTGTACAGTTTCGGCCTCGCCCTCTA, and SMP-C2_SacII_R, GTCACCGCGGCTAGGAGCTGCCCTTGTCCTT. The fragment was ligated to the mCherry-C1 vector at the BsrG1/SacII sites.
mCherry-E-Syt1(ΔC2E): Human E-Syt1 lacking the C-terminal C2E domain was amplified with the following primers: OI445_SacII_F, TATATACCGCGGGAATGGAGCGATCTCCAGGAGA, and
ESyt1-dC2E_SacII_R, TATATACCGCGGCTACAGAGGCCCGGCTGGAGC. The PCR fragment was ligated to the mCherry-C1 vector at the SacII site.
mCherry-E-Syt1-ΔSMP: The hairpin region of E-Syt1 was PCR amplified using the following primers: E1-TM_Pvu1-F: TACGATCGCAATGGAGCGATCTCCAGGA, and E1-TM_Pvu1-R: TACGATCGTCAGGTAGAGGGCGAGGC. The PCR fragment was subsequently ligated to mCherry-E-Syt1-C2ABCDE at a Pvu1 site generated between the fluorophore and the C2A domain.
mCherry-E-Syt1(ΔC2CDE): The N-terminal portion of human E-Syt1 including the hairpin region, SMP domain, and the C2AB domains was amplified with the following primers: ESyt1-ER-Sal1-F, TATATAGTCGACatggagcgatctccaggaga, and ESyt1-ER-SacII-R, TATATACCGCGGtcgaggtggggcatccaca. The PCR product was ligated between the Sal1/SacII sites in the mCherry-C1 vector.
Cytosolic E-Syt1 deletion constructs
mCherry-E-Syt1-C2ABCDE: Human E-Syt1 C2ABCDE-domains were amplified using the following primers: E1-ABCDE-Pvu1-BsrG1-F, CCGTGTACACGATCGCACGATTACTGGTGCCCCTTGT, and E1-ABCDE-Stop-SacII-R, GTCACCGCGGCTAGGAGCTGCCCTTGTCCTT. The PCR fragment was ligated to the mCherry-C1 vector at the BsrG1/SacII sites.
mCherry-E-Syt1-C2CDE: The C2CDE domains of human E-Syt1 was amplified using the following primers: E-Syt1-C2C-Xho1-FWD, CCGCTCGAGGACTGCCTCTGGCCCGCCT, and E-Syt1-C2CDE-Stop-Kpn1-REV, GGCTCGAGCTAGGAGCTGCCCTTGTCCTT. The fragment was ligated between Xho1/Kpn1 sites in the mCherry-C1 vector.
mCherry-E-Syt1-C2AB: The C2AB domains of E-Syt1 was amplified with the following primers: ESyt1_C2AB_SacII_F, TATATACCGCGGGAATGGAGCGATCTCCAGGAGA, and ESyt1_C2AB_SacII_R, TATATACCGCGGTTATCGAGGTGGGGCATCCACA. The fragment was subsequently ligated to the mCherry-C1 vector at the SacII site.
mCherry-E-Syt1-C2ABC: The C2ABC domains of E-Syt1 was amplified with the following primers: E1-ABCDE-BsrG1_F, CCGTGTACACGATCGCACGATTACTGGTGCCCCTTGT, and ESyt1_ΔC2DE_SacII_R, TATATACCGCGGTTAATTCACCTGCAGCACCT. The fragment was subsequently ligated to the mCherry-C1 vector between the BsrG1 and SacII sites.
mCherry-E-Syt1-C2ABCD: The C2ABCD domains of E-Syt1 was amplified with the following primers: E1-ABCDE-BsrG1_F, CCGTGTACACGATCGCACGATTACTGGTGCCCCTTGT, and ESyt1_ΔC2E_SacII_R, TATATACCGCGGCTACAGAGGCCCGGCTGGAGC. The fragment was subsequently ligated to the mCherry-C1 vector between the BsrG1 and SacII sites.
mCherry-E-Syt1-C2CD: The C2CD domains of E-Syt1 was amplified with the following primers: ESyt1-C2C_EcoR1_F, CGGAATTCACTGCCTCTGGCCCGCCT, and ESyt1_C2CD_SacII_R, TATATACCGCGGCTACAGAGGCCCGGCTGGA. The fragment was subsequently ligated to the mCherry-C1 vector between the EcoR1 and SacII sites.
mCherry-E-Syt1-C2C: The C2C domain of E-Syt1 was amplified with the following primers: ESyt1-C2C_EcoR1_F, CGGAATTCACTGCCTCTGGCCCGCCT, and ESyt1_C2C_SacII_R, TATATACCGCGGCTAATTCACCTGCAGCACCT. The fragment was subsequently ligated to the mCherry-C1 vector between the EcoR1 and SacII sites.
mCherry-E-Syt1-C2E: The C2E domain of E-Syt1 was amplified with the following primers: ESyt1-C2E_EcoR1_F, CGGAATTCACTGCCCCTCTCAGAGCT, and ESyt1_C2E_SacII_R, TATATACCGCGGCTAGGAGCTGCCCTTGTCCT. The fragment was subsequently ligated to the mCherry-C1 vector between the EcoR1 and SacII sites.
Synaptotagmin-1 constructs
mCherry-Syt1-C2AB: The C2AB domains from human synaptotagmin-1 was amplified using the following primers: OI435_Syt1_HindIII-F, CCCAAGCTTCGAAGAAATGTTTGTTCAAAAAG, and OI436_Syt1_BamH1-R, CGGGATCCTTACTTCTTGACGGCCA. The PCR product was ligated to the mCherry-C1 vector between HindIII/BamH1 sites.
mCherry-E-Syt1-HP-Syt1-C2AB: The 275 bp N-terminal region of E-Syt1 (containing the ER-hairpin) was amplified using the following primers: OI457_E1-HP-BsrG1-F, CCGtgtacaAGATGGAGCGATCTCCAGGA, and OI459_E1-HP-HindIII-R, CCCaagcttGCAGGTAGAGGGCGAGGC. The PCR product was ligated between mCherry and the C2AB fragment of Syt1 in mCherry-Syt1-C2AB using the BsrG1 and HindIII sites.
siRNA
siRNAs used in this study were as follows: E-Syt1 (J-010652-06-0010, Dharmacon), E-Syt2 (HSC.RNAI.N020728.12.5, IDT), STIM1 (HSC.RNAI.N003156.12.3, IDT), and universal negative control siRNA (DS NC1, IDT).
Cell culture and transfection
HeLa-M and COS-7 cells were kept in Dulbecco’s Modified Eagles Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1× L-glutamine. MIN6 cells were kept in DMEM supplemented with 15% FBS, 1× L-glutamine, and 50 μM β-mercaptoethanol. Plasmid DNA transfections were performed in 1 ml OptiMEM-I together with 0.5–1.5 μg and 1–2 μl Lipofectamine 2000 according to the manufacturer’s instructions. Knockdown using siRNA was performed in 1 ml OptiMEM-I together with 20 nM siRNA duplexes and 2 μl Lipofectamine RNAiMAX. Knockdown efficiency was assessed 60–72 h post-transfection.
Immunoblotting
HeLa cells treated with siRNA for 60–72 h were washed in ice-cold PBS and lysed on ice in lysis buffer containing 50 mM NaCl, 1% Triton X-100, 10 mM EDTA pH 7.2, and protease inhibitor cocktail. Cell lysates were then centrifuged at 21,000 g for 20 min at 4°C, and supernatants were boiled for 2 min in SDS sample buffer and separated on a 8% SDS–PAGE gel. Immunoblotting was carried out as described in Giordano et al (2013).
α-toxin permeabilization
Transfected HeLa-M cells, grown on 25-mm poly-L-lysine-coated glass coverslips, were incubated with 5 μM of the AM ester form of Fluo-4 for 30 min at 37°C. The coverslips were subsequently used as exchangeable bottoms in a modified Sykes-Moore open superfusion chamber that was mounted on the stage of a confocal or TIRF microscope setup (described below) and connected to a peristaltic pump that allow rapid exchange of the buffer surrounding the cells. Following exchange from an extracellular-like (described above) to an intracellular-like buffer (see below), the superfusion was interrupted and α-toxin was added directly to the chamber (final concentration ≈ 0.2 mg/ml). Permeabilization was considered complete when the Fluo-4 fluorescence had decreased by 90%, which typically took 20–30 min. Superfusion was then started again, and the cells were exposed to intracellular-like buffers containing calibrated Ca2+ concentrations while fluorescence from both remaining Fluo-4 and mCherry-tagged fusion proteins was recorded. These experiments were performed at ambient temperature (21–23°C).
Intracellular-like buffers
Intracellular-like buffers used in α-toxin permeabilization experiments contained the following: 6 mM Na+, 140 mM K+, 1 mM (free) Mg2+, 0–0.5 mM (free) Ca2+, 1 mM Mg-ATP (required to maintain PI(4,5)P2-levels in the PM), 0.25 mM Na-GTP, 10 mM HEPES, 2 mM (total) EGTA, and 2 mM (total) nitrilotriacetic acid (NTA) with pH adjusted to 7 at 22°C with 2 M KOH. The total concentration of Ca2+ and Mg2+ was calculated using the online version of MaxChelator (http://www.stanford.edu/~cpatton/webmaxcS.htm). Buffers were made fresh on the day of experiment and kept on ice until used. To validate the buffer composition, cells loaded with the Ca2+ indicator Fluo-4 were mounted on a TIRF microscope, permeabilized, and exposed to buffers with increasing Ca2+ concentrations. From these data, a dose–response curve was generated and the EC50 for Ca2+ binding to Fluo-4 was estimated to be 1.1 μM, which compares favorably with the reported KD of 0.35 μM (see Fig1).
Fluorescence microscopy
Before each experiment, 25-mm glass coverslips with attached cells were transferred to experimental buffer and incubated for 30 min at 37°C. Where indicated, this buffer was supplemented with Ca2+ indicators (see below) or 1.5 μM of EGTA-AM or BAPTA-AM. The buffer contained 125 mM NaCl, 4.8 mM KCl, 1.3 mM CaCl2, 1.2 mM MgCl2, 25 mM HEPES, 3 mM D-glucose, and 0.1% (w/v) bovine serum albumin with pH adjusted to 7.4 with NaOH.
Confocal microscopy: Spinning-disk confocal microscopy was performed using the Improvision UltraVIEW VoX system (PerkinElmer) built around a Nikon Ti-E inverted microscope, equipped with PlanApo objectives (60× 1.45-NA) and controlled by Volocity (Improvision) software as previously described. All imaging was performed at 37°C except for experiments on permeabilized cells, which were performed at room temperature.
TIRF microscopy: Protein biosensor and Ca2+ indicator fluorescence were measured with a setup consisting of an Eclipse Ti microscope (Nikon, Tokyo, Japan) with a total internal reflection fluorescence (TIRF) illuminator and a 60× 1.45 NA objective. 491-nm and 561-nm diode-pumped solid state lasers (Cobolt AB, Solna, Sweden) provided excitation light for GFP/Fluo-4 and mCherry/RFP, respectively. Fluorescence was detected with a back-illuminated DU-897 EMCCD camera (Andor Technology, Belfast, UK) controlled by MetaFluor software (Molecular Devices Corp., Downington, PA, USA). Emission wavelengths were selected with a 527/27-nm half-bandwidth filter for GFP/Fluo-4 and a 584-nm long-pass filter for mCherry/RFP (Semrock) mounted in a wheel (Sutter Instruments). Images or image pairs were acquired every 2–5 s. For the Ca2+ imaging shown in Figs4K and EV6, a prism-based TIRF microscope (Dyachok et al, 2008) equipped with a 16× (0.8-NA) water-immersion objective was used, which enabled simultaneous recordings of responses from a large number of cells at the expense of spatial resolution.
Ca2+ imaging
For Ca2+ imaging, HeLa cells grown on glass coverslips were washed once with experimental buffer followed by 45 min incubation at 37°C in experimental buffer supplemented with 5 μM of the acetoxymethylester of Fluo-4 or Fluo-4FF. Cells were subsequently washed and imaged with TIRF or confocal microscopy as described above.
Photolysis of caged Ca2+
HeLa cells were washed once with experimental buffer containing 1.3 mM Ca2+, followed by 30 min incubation with experimental buffer supplemented with 5 μM NP-EGTA-AM at 37°C. The cells were subsequently washed twice with experimental buffer, followed by further incubation at 37°C for 20 min to allow hydrolysis of the AM ester bond. Flash photolysis of the caged Ca2+ was achieved by 1 s illumination by the 405-nm laser line on the confocal microscope described above, with the laser power set to maximum (25 mW out of the laser head).
Optogenetic depletion of PI(4,5)P2 in the plasma membrane
Blue light-induced 5′-dephosphorylation of plasma membrane PI(4,5)P2 was performed as previously described (Kennedy et al, 2010; Idevall-Hagren et al, 2012). Briefly, the 5-phosphatase domain of human OCRL (residues 234–539) was ligated to the C-terminus of the PHR domain of Arabidopsis thaliana cryptochrome 2 (CRY2) to generate CRY2-5-ptaseOCRL and Arabidopsis thaliana CIBN was fused to a C-terminal CAAX-box motif for plasma membrane anchoring (CIBN-CAAX). When expressed in cells, blue light (491 nm) illumination induces immediate binding of CRY2 to CIBN, thus bringing the 5-ptase to the plasma membrane. For TIRF imaging, dimerization was induced by 50 × 100 ms 491 nm light pulses delivered through the evanescent field.
Image and statistical analysis
All image analyses were done offline using Fiji (Schindelin et al, 2012). Typically, fluorescence changes in the basal plasma membrane region was quantified by manually generating a region of interest around each cell, followed by measurement of the fluorescence intensity within the region for each frame, making sure there were no saturated pixels within the region. The obtained values were normalized to the initial fluorescence after background correction (F/F0). In some instances (Figs1F, 4D and E), individual ER-PM contacts were manually identified and the fluorescence change within these structures were tracked over time. At least 20 contacts (of varying fluorescence intensities and sizes) from each cell were chosen for analysis. Dose–response curves were generated from normalized data, where the minimum was set to the relative fluorescence at 0 μM Ca2+ and the maximum to the highest recorded fluorescence value for each cell or the fluorescence value obtained at 50 μM Ca2+. The reason for choosing 50 μM as the cut-off was that values obtained above this Ca2+ concentration were underestimated due to Ca2+-induced loss of plasma membrane PI(4,5)P2 (see Fig1J–L). The normalized data were plotted, and a sigmoidal curve was fitted to the data points using the Hill equation: base + (max – base)/(1 + (x-half/x)rate), with base = 0 and max = 1. Curve fitting was made using IgorPro (Version 6.34A, WaveMetrics). The x-half values were extracted and used to calculate EC50 values. Throughout the paper, data are presented as means ± SEM and statistical comparisons between groups were assessed with two-tailed Student’s t-test.
Acknowledgments
We thank Yiying Cai for help and discussion, and Yasunori Saheki, Anders Tengholm, and Erik Gylfe for discussion. This work was supported in part by grants from the Swedish Research Council (D0029801) and the Göran Gustafsson foundation to OIH and from the NIH (NS36251, DK082700 and DK45735) to PDC.
Author contributions
OI-H developed and AL performed the in situ binding assay; OI-H performed imaging experiments; OI-H and BX performed the molecular biology work; OI-H and PDC designed the experiments, analyzed and interpreted the data; and OI-H and PDC wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Appendix
Expanded View Figures PDF
Review Process File
References
- Bai J, Chapman ER. The C2 domains of synaptotagmin–partners in exocytosis. Trends Biochem Sci. 2004;29:143–151. doi: 10.1016/j.tibs.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Bhakdi S, Tranum-Jensen J. Alpha-toxin of Staphylococcus aureus. Microbiol Rev. 1991;55:733–751. doi: 10.1128/mr.55.4.733-751.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad LM, Armstrong DL, Putney JW., Jr Role of the inositol 1,4,5-trisphosphate receptor in Ca(2+) feedback inhibition of calcium release-activated calcium current (I(crac)) J Biol Chem. 1999;274:32881–32888. doi: 10.1074/jbc.274.46.32881. [DOI] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Chang CL, Hsieh TS, Yang TT, Rothberg KG, Azizoglu DB, Volk E, Liao JC, Liou J. Feedback regulation of receptor-induced Ca2+ signaling mediated by E-Syt1 and Nir2 at endoplasmic reticulum-plasma membrane junctions. Cell Rep. 2013;5:813–825. doi: 10.1016/j.celrep.2013.09.038. [DOI] [PubMed] [Google Scholar]
- Davletov BA, Sudhof TC. A single C2 domain from synaptotagmin I is sufficient for high affinity Ca2+/phospholipid binding. J Biol Chem. 1993;268:26386–26390. [PubMed] [Google Scholar]
- Dyachok O, Idevall-Hagren O, Sagetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjarvi G, Gylfe E, Tengholm A. Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab. 2008;8:26–37. doi: 10.1016/j.cmet.2008.06.003. [DOI] [PubMed] [Google Scholar]
- English AR, Voeltz GK. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb Perspect Biol. 2013;5:a013227. doi: 10.1101/cshperspect.a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Busnadiego R, Saheki Y, De Camilli P. Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites. Proc Natl Acad Sci USA. 2015;112:E2004–E2013. doi: 10.1073/pnas.1503191112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano F, Saheki Y, Idevall-Hagren O, Colombo SF, Pirruccello M, Milosevic I, Gracheva EO, Bagriantsev SN, Borgese N, De Camilli P. PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013;153:1494–1509. doi: 10.1016/j.cell.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, Mullar S, Diederichsen U, Fasshauer D, Grubmuller H, Hell SW, Eggeling C, Kuhnel K, Jahn R. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nat Struct Mol Biol. 2013;20:679–686. doi: 10.1038/nsmb.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idevall-Hagren O, Dickson EJ, Hille B, Toomre DK, De Camilli P. Optogenetic control of phosphoinositide metabolism. Proc Natl Acad Sci USA. 2012;109:E2316–E2323. doi: 10.1073/pnas.1211305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean S, Tremblay MG, Herdman C, Guillou F, Moss T. The endocytic adapter E-Syt2 recruits the p21 GTPase activated kinase PAK1 to mediate actin dynamics and FGF signalling. Biol Open. 2012;1:731–738. doi: 10.1242/bio.2012968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez JL, Davletov B. Beta-strand recombination in tricalbin evolution and the origin of synaptotagmin-like C2 domains. Proteins. 2007;68:770–778. doi: 10.1002/prot.21449. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nat Methods. 2010;7:973–975. doi: 10.1038/nmeth.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec KO, Alva V, Lupas AN. Bioinformatics of the TULIP domain superfamily. Biochem Soc Trans. 2011;39:1033–1038. doi: 10.1042/BST0391033. [DOI] [PubMed] [Google Scholar]
- Lahiri S, Toulmay A, Prinz WA. Membrane contact sites, gateways for lipid homeostasis. Curr Opin Cell Biol. 2015;33C:82–87. doi: 10.1016/j.ceb.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang J, Fukuda M, Zhang H, Mikoshiba K, Wollheim CB. The first C2 domain of synaptotagmin is required for exocytosis of insulin from pancreatic beta-cells: action of synaptotagmin at low micromolar calcium. EMBO J. 1997;16:5837–5846. doi: 10.1093/emboj/16.19.5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine T, Loewen C. Inter-organelle membrane contact sites: through a glass, darkly. Curr Opin Cell Biol. 2006;18:371–378. doi: 10.1016/j.ceb.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Llopis J, Chow SB, Kass GE, Gahm A, Orrenius S. Comparison between the effects of the microsomal Ca(2+)-translocase inhibitors thapsigargin and 2,5-di-(t-butyl)-1,4-benzohydroquinone on cellular calcium fluxes. Biochem J. 1991;277(Pt 2):553–556. doi: 10.1042/bj2770553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytton J, Westlin M, Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991;266:17067–17071. [PubMed] [Google Scholar]
- Manford AG, Stefan CJ, Yuan HL, Macgurn JA, Emr SD. ER-to-plasma membrane tethering proteins regulate cell signaling and ER morphology. Dev Cell. 2012;23:1129–1140. doi: 10.1016/j.devcel.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Marsault R, Murgia M, Pozzan T, Rizzuto R. Domains of high Ca2+ beneath the plasma membrane of living A7r5 cells. EMBO J. 1997;16:1575–1581. doi: 10.1093/emboj/16.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Chang WP, Sudhof TC. E-Syts, a family of membranous Ca2+-sensor proteins with multiple C2 domains. Proc Natl Acad Sci USA. 2007;104:3823–3828. doi: 10.1073/pnas.0611725104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehls S, Snapp EL, Cole NB, Zaal KJ, Kenworthy AK, Roberts TH, Ellenberg J, Presley JF, Siggia E, Lippincott-Schwartz J. Dynamics and retention of misfolded proteins in native ER membranes. Nat Cell Biol. 2000;2:288–295. doi: 10.1038/35010558. [DOI] [PubMed] [Google Scholar]
- Putney JW. Pharmacology of store-operated calcium channels. Mol Interv. 2010;10:209–218. doi: 10.1124/mi.10.4.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Stein A, Jahn R, Fasshauer D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2009;284:25749–25760. doi: 10.1074/jbc.M109.042499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem. 2001;70:281–312. doi: 10.1146/annurev.biochem.70.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC. C2-domains, structure and function of a universal Ca2+-binding domain. J Biol Chem. 1998;273:15879–15882. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Eliasson L, Kanno T, Zhang Q, Gopel S. Electrophysiology of pancreatic beta-cells in intact mouse islets of Langerhans. Prog Biophys Mol Biol. 2011;107:224–235. doi: 10.1016/j.pbiomolbio.2011.06.009. [DOI] [PubMed] [Google Scholar]
- Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, De Camilli P, Reinisch KM. Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature. 2014;510:552–555. doi: 10.1038/nature13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Weissman JS. The contribution of systematic approaches to characterizing the proteins and functions of the endoplasmic reticulum. Cold Spring Harb Perspect Biol. 2013;5:a013284. doi: 10.1101/cshperspect.a013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin OH, Xu J, Rizo J, Sudhof TC. Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc Natl Acad Sci USA. 2009;106:16469–16474. doi: 10.1073/pnas.0908798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan CJ, Manford AG, Emr SD. ER-PM connections: sites of information transfer and inter-organelle communication. Curr Opin Cell Biol. 2013;25:434–442. doi: 10.1016/j.ceb.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y, Sudhof TC. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tengholm A, Gylfe E. Oscillatory control of insulin secretion. Mol Cell Endocrinol. 2009;297:58–72. doi: 10.1016/j.mce.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Thore S, Wuttke A, Tengholm A. Rapid turnover of phosphatidylinositol-4,5-bisphosphate in insulin-secreting cells mediated by Ca2+ and the ATP-to-ADP ratio. Diabetes. 2007;56:818–826. doi: 10.2337/db06-0843. [DOI] [PubMed] [Google Scholar]
- Toul may A, Prinz WA. A conserved membrane-binding domain targets proteins to organelle contact sites. J Cell Sci. 2012;125:49–58. doi: 10.1242/jcs.085118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liu H, Gu Y, Chapman ER. Reconstituted synaptotagmin I mediates vesicle docking, priming, and fusion. J Cell Biol. 2011;195:1159–1170. doi: 10.1083/jcb.201104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MM, Luik RM, Lewis RS. Some assembly required: constructing the elementary units of store-operated Ca2+ entry. Cell Calcium. 2007;42:163–172. doi: 10.1016/j.ceca.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Bacaj T, Zhou A, Tomchick DR, Sudhof TC, Rizo J. Structure and Ca(2)(+)-binding properties of the tandem C(2) domains of E-Syt2. Structure. 2014;22:269–280. doi: 10.1016/j.str.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca(2+) indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File