

Abstract
During cortical development, synaptic competition regulates the formation and adjustment of neuronal connectivity. It is unknown whether synaptic competition remains active in the adult brain and how inhibitory neurons participate in this process. Using morphological and electrophysiological measurements, we show that expressing a dominant-negative form of the TrkB receptor (TrkB.T1) in the majority of pyramidal neurons in the adult visual cortex does not affect excitatory synapse densities. This is in stark contrast to the previously reported loss of excitatory input which occurs if the exact same transgene is expressed in sparse neurons at the same age. This indicates that synaptic competition remains active in adulthood. Additionally, we show that interneurons not expressing the TrkB.T1 transgene may have a competitive advantage and obtain more excitatory synapses when most neighboring pyramidal neurons do express the transgene. Finally, we demonstrate that inhibitory synapses onto pyramidal neurons are reduced when TrkB signaling is interfered with in most pyramidal neurons but not when few pyramidal neurons have this deficit. This adjustment of inhibitory innervation is therefore not a cell-autonomous consequence of decreased TrkB signaling but more likely a homeostatic mechanism compensating for activity changes at the population level.
Keywords: BDNF, cell autonomous, inhibition, parvalbumin, TrkB
Introduction
The brain has a strong capacity to learn and adapt to the environment. This adaptation is mainly mediated by the continuous gain, loss, and functional adjustment of synapses based on neuronal activity patterns. Synaptic competition, whereby inputs that are more capable of activating the postsynaptic neuron are strengthened while those that are less capable are lost, is crucial for such adaptation in the developing nervous system. A classic example is ocular dominance plasticity in the primary visual cortex (V1) where monocular deprivation during development causes retraction of thalamic projections serving the closed eye and expansion of those serving the open eye (Hubel et al. 1977). This competitive process operates through 2 separate mechanisms. Correlation-based plasticity weakens the deprived eye responses, while homeostatic synaptic scaling normalizes total postsynaptic drive by increasing the responses to both eyes (Frenkel and Bear 2004; Mrsic-Flogel et al. 2007; Kaneko, Stellwagen et al. 2008; Kaneko, Hanover et al. 2008; Kaneko et al. 2012).
Besides presynaptic competition for postsynaptic targets, postsynaptic competition for presynaptic inputs also occurs in the cortex. When expression of neuroligin-1 (Kwon et al. 2012) or BDNF (English et al. 2012) is reduced in a small subset of excitatory neurons, they become less attractive postsynaptic targets and end up receiving fewer excitatory synaptic inputs than their neighbors. In contrast, if the genetic deficit is present in all excitatory neurons, synapse numbers are unaffected. This implies that presynaptic axons choose their postsynaptic targets based on relative differences in the expression of specific signaling proteins and that some form of homeostatic process is in place to normalize synapse numbers.
Until now, studies on synaptic competition have only been performed during development. An important question that remains unanswered is therefore whether this type of synaptic competition is still operational in adulthood, after the majority of synaptic connections have already been established. Additionally, it is unknown whether and how interneurons take part in such competitive regulation of synaptic connectivity.
To address these questions, we made use of mice carrying a cre-dependent transgene driving expression of a dominant-negative BDNF receptor (TrkB.T1). We have previously shown that expressing TrkB.T1 in a low percentage (<1%) of pyramidal neurons of young adult V1 strongly reduces the density and head size of dendritic spines (Chakravarthy et al. 2006). By making use of another cre-transgenic line, we now expressed the exact same TrkB.T1 transgene in most (>80%) pyramidal neurons, but not in interneurons, of the cortex starting around the same age (Heimel et al. 2010). We find that broad expression of TrkB.T1 does not reduce the size or numbers of excitatory synapses. This difference between sparse and broad expression shows that competition for postsynaptic targets continues into adulthood. In contrast to excitatory synapses, inhibitory synapses onto pyramidal neurons are reduced only when the TrkB.T1 transgene is expressed broadly but not when expressed sparsely. This suggests that population-based rather than cell-autonomous processes regulate inhibitory innervation in the adult cortex.
Materials and Methods
Transgenic Mice
We made use of mice carrying a cre-dependent transgene driving expression of either the TrkB.T1 receptor fused to the enhanced green fluorescent protein (EGFP) (mouse line TLT-817) or a membrane associated EGFP-F-transgene (mouse line TLG-498) under the control of the Thy-1 promoter. For sparse expression, these lines were crossed to the CaMKIIα-cre-transgenic mouse line Cre 3487. All the above-mentioned lines were described previously (Chakravarthy et al. 2006, 2008). Broad expression of the TrkB.T1-EGFP transgene in most cortical pyramidal neurons was achieved by crossing the TLT-817 line to G35-3 cre mice which express cre under the control of the KA1 promoter (Sawtell et al. 2003; Heimel et al. 2010). Mice were crossed to C57BL/6JOlaHsd background for at least 6 generations. We will refer to the broadly expressing TLT-817+G35-3-cre+ mice as “TrkB.T1-broad,” and to the sparsely expressing TLT-817+Cre-3487+ mice as “TrkB.T1-sparse.” TLG-498+Cre-3487+ double-positive mice are referred to as “EGFP-sparse.” All experiments were approved by the institutional animal care and use committee of the Royal Netherlands Academy of Arts and Sciences.
Histology and Immunohistochemistry
Mice that were 8–10 weeks of age were anaesthetized with 0.1 mL/g bodyweight sodium pentobarbital (Janssen Laboratories, Beerse, Belgium) and perfused with 4% paraformaldehyde in phosphate buffered saline (PBS). Brains were dissected, postfixed for 2 h, and subsequently stored at 4°C in PBS. Coronal sections of 50 µm were made using a vibratome (Leica VT1000S, Leica Microsystems, Wetzlar, Germany). Free-floating sections were double-stained using mouse anti-NeuN or anti-GFP antibodies (1:500, Chemicon) and rabbit anti-parvalbumin (PV) (1:1000, Swant) followed by Alexa 488-conjugated goat anti-mouse antibodies (1:500, Molecular Probes) and Alexa 568 conjugated goat anti-rabbit antibodies (1:500, Molecular Probes).
Diolistics
DiI-coated Tefzel tubing was prepared as described earlier (Chakravarthy et al. 2006). Briefly, a mixture of 0.15 mg DiI (Molecular Probes), 50 μL methylene chloride, and 12 mg of 1.1 μm tungsten particles (Bio-Rad, Veenendaal, the Netherlands) was spread on a glass slide and air-dried. Subsequent to resuspension in 1 mL distilled water and sonication, the mixture was sucked into Tefzel tubing (Bio-Rad). The suspension was withdrawn after 2 min and nitrogen gas was passed to dry the tube. The tube was cut into 13-mm pieces and was used in a Helios Gene Gun (Bio-Rad) to shoot 50-μm coronal sections (prepared as described for immunohistochemistry) at 100 psi through a membrane filter of 3-μm pore size and 8 × 105 pores/cm2 (Corning, Acton, MA, USA). Sections were stored in PBS at room temperature for least 12 h to ensure good filling of the labeled neurons.
Confocal Microscopy
DiI-labeled neurons from layer II/III of V1 were imaged using a Carl Zeiss CLSM 510 Meta confocal microscope (Zeiss, Göttingen, Germany) with HeNe (543 nm) laser. Only those DiI-labeled neurons that were also positive for TrkB.T1 were imaged in TrkB.T1-broad mice. In the littermate controls, all DiI-labeled pyramidal neurons from layer II/III were imaged. The dendritic segments after the first branch point of basal dendrites were imaged with a ×63 oil-immersion objective and at an optical zoom of ×2.5. Each image was a stack of 50 planes with 200-nm steps. Care was taken to ensure that the acquisition parameters were so adjusted that the entire detector range was used for the spines. PV-NeuN or PV-GFP double-stained sections were imaged with HeNe (543 nm) and Argon (488 nm) lasers. The images were acquired as nonoverlapping single planes with a ×63 oil-immersion objective using a pinhole of 80 µm.
Spine Classification and Image Analysis
The image stacks were subjected to 3D reconstruction using Zeiss CLSM 510 Meta. Dendritic protrusions were classified as spines (mushroom, long, stubby, and thin) and filopodia (Chakravarthy et al. 2006). All dendritic protrusions in a 15-µm segment of proximal basal dendrites 5 µm after the first branching point were counted and thus classified. Zeiss CLSM Image browser 5 overlay tools was used for size determinations. The head diameter was counted by using the longest straight line in the spine head. A bent-line tool was used to measure the length of the spine neck. Subsequent to image acquisition, all analysis was done blind to the genotype of the mice. Statistical significance was determined by using a multilevel analysis to accommodate nested data.
PV Bouton Analysis
PV boutons around NeuN- or GFP-positive neurons of layer II/III cells from V1 were counted using ImagePro after confocal images were converted into 8-bit RGB TIFF files. The NeuN- or GFP-positive cells were marked using a free-drawing tool. Using customized macros, a 2-µm-wide ring was drawn around all NeuN- or GFP-positive cells. All cells whose edges were <2 µm from the border of the image were omitted from analysis. Automated counting was carried out to determine the number of PV boutons within the 2-µm ring. In short, the puncta were marked by applying median- and low-pass filters to inverted images of the red channel. A minimized image was created by selecting the minimum pixel intensity value for each pixel in median- and low-pass filtered images, providing a better edge selection. Low-pass filtered images were then subtracted from the resultant image and a fixed value of 150 was added to the intensity. Puncta were defined in images with a constraint of size, and were further defined by watershed transform (e.g., a dumbbell-shaped punctum was demarcated as 2 separate puncta). Finally, the number of puncta was counted between initial and expanded boundaries (within the 2-µm perisomatic ring) for each cell. Analysis was carried out blind to the identity of the genotype. Statistical significance was determined by using a multilevel analysis.
Slice Preparation and Electrophysiology
All slice electrophysiology was performed on >3-month-old transgenic mice and age-matched controls. For miniature excitatory postsynaptic current (mEPSC) recordings from pyramidal neurons, mice were anaesthetized using isoflurane and then decapitated, and their brains removed and chilled in ice-cold carbogenated (95% O2/5% CO2) sucrose-based artificial cerebrospinal fluid (ACSF) containing 3.5 mM KCl, 2.4 mM CaCl2, 1.3 mM MgSO4, 1.2 mM KH2PO4, 10 mM glucose, 26 mM NaHCO3, and 212.5 mM sucrose. Slices were stored in carbogenated normal ACSF containing 126 mM NaCl, 3 mM KCl, 2 mM MgSO4, 2 mM CaCl2, 10 mM glucose, 1.20 mM NaH2PO4, and 26 mM NaHCO3 (300 mOsm and pH 7.3). For recordings of miniature inhibitory postsynaptic currents (mIPSCs) in pyramidal neurons and of mEPSCs in fast-spiking interneurons, mice were anaesthetized by intraperitoneal injection of sodium pentobarbital (40 mg/kg) and perfused with 5 mL ice-cold carbogenated choline chloride-based ACSF containing 110 mM choline chloride, 7 mM MgCl2, 0.5 mM CaCl2, 2.5 mM KCl, 11.6 mM Na-ascorbate, 3.10 mM Na-pyruvate, 1.25 NaH2PO4, 25 mM d-glucose, and 25 mM NaHCO3 (Bureau et al. 2006). Brains were chilled for 5 min and cut in this ACSF. Coronal slices (300–330 µm) of V1 were cut using a Microm HM650V vibrating microtome. After 30–45 min incubation in normal ACSF at 36°, the slices were used for recording. For recording, slices were transferred to a submerged recording chamber with constant perfusion of carbogenated ACSF and visualized using an Axioskop FS upright microscope equipped with Hofmann modulation contrast optics (Zeiss). For whole-cell patch-clamp recordings, borosilicate glass patch-pipettes (3–4 MΩ) were filled with intracellular solution. Whole-cell recordings were made using a patch-clamp amplifier (Multiclamp 700a, Axon Instruments, Molecular Devices), and signals were low-pass filtered at 4 kHz and digitized at 10 kHz with a digidata 1440A (Axon Instruments, Molecular Devices) operated by PClamp 10 software. Care was taken that series resistance remained ≤15 MΩ. Injection of current and recording the firing pattern of neurons, in current clamp mode, was used to verify selection of pyramidal neurons. Recordings in which the input resistance fluctuated more than 20% during the experiment, indicating leakiness of the membrane and instability of the whole-cell patch-clamp recording, were discarded.
mEPSC/mIPSC Recording
For recordings in TrkB.T1-sparse and EGFP-sparse mice, transgene-expressing pyramidal neurons were identified based on EGFP fluorescence. For TrkB.T1-broad mice and their wild-type littermates, pyramidal neurons were identified based on their pyramidal morphology and typical regular firing pattern. Fast-spiking interneurons were identified based on their round morphology and typical fast-spiking firing pattern with strong after-hyperpolarization. To isolate action potential independent events, recordings were performed in the presence of 1 µM tetrodotoxin citrate (Ascent Scientific LTD, Bristol, UK). For spontaneous EPSC (sEPSC) and mEPSC recordings, the pipettes were filled with K-gluconate internal solution containing 110 mM K-gluconate, 10 mM KCl, 10 mM Hepes, 4 mM Mg-ATP, 10 mM K2 phosphocreatine, 0.3 mM GTP. pH was set to 7.3, and osmolarity to 290 mOsm. The reversal potential for chloride was at the holding potential, ensuring no interference from incoming mIPSCs. For experiments on fast-spiking interneuron mEPSCs, the GABAA receptor antagonist GABAzine (1 uM; Ascent Scientific LTD) was added to the bath solution. The intracellular solution for recording mIPSCs contained 70 mM K-gluconate, 70 mM KCl, 0.5 mM EGTA, 10 mM Hepes, 4 mM Mg-ATP, 4 mM K2 phosphocreatine, 0.4 mM GTP (PH 7.3 and 290 mOsm). Here, the α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor blocker 6,7-dinitroquinoxaline-2,3-dione disodium salt (DNQX; Ascent Scientific LTD), was applied in the extracellular solution (10 µM).
mEPSC/mIPSC Analysis
Mini Analysis (Synaptosoft, Inc., Decatur, GA, USA) was used for analyzing mEPSCs and mIPSCs. For mEPSCs and mIPSCs onto pyramidal neurons, the amplitude threshold was set at 9 pA, which was >3× root-mean-square (RMS) noise in all recordings. For mEPSCs and sEPSCs onto fast-spiking interneurons, the amplitude threshold was set at 8 pA. After automatic detection of events by the software, each event was visually inspected. Recordings with a systematic drift in average mEPSC and mIPSC rise time of >10% were excluded. mEPSCs with rise times >3 ms were omitted. Interevent intervals and amplitude averages of mEPSCs and mIPSCs, and number of action potentials in response to depolarizing current injections in TrkB.T1-expressing neurons versus their controls were compared for statistical significance by using a multilevel analysis and repeated measures, two-way ANOVA, respectively.
AMPA/NMDA Ratios
For recording AMPA/N-Methyl-D-aspartate (NMDA) ratios, layer II/III pyramidal neurons were patched with intracellular solution containing (mM): 120 CsMeSO3, 8 NaCl, 15 CsCl2, 10 TEA-Cl, 10 HEPES, 2 QX-314, 4 MgATP, 0.3 Na2GTP (pH 7.3). GABAzine (1 µM) was added to the extracellular solution to block GABAA receptors, and glycine (10 µM) was added to saturate the NMDA receptor co-agonist binding site. EPSCs were evoked by extracellular stimulation using a monopolar stimulating pipette positioned 50–100 µm next to the soma of the recorded neuron. Stimulation position and strength (∼20 µA for 0.2 ms) were adjusted to obtain an evoked EPSC with a smooth rise and decay. Then, EPSCs were recorded every 10 s with the neuron alternating between a holding potential of −80 and +40 mV. For analysis, 10–20 EPSCs at each holding potential were averaged. The size of the AMPA receptor component was defined as the peak amplitude of the EPSC recorded at −80 mV, while the NMDA receptor component was defined as the response 30 ms later at +40 mV.
Statistics
Comparisons between control and transgenic groups were conducted using multilevel analysis to account for dependency of the observations in each mouse. In this case, a two-level multilevel model, where cell measurements are nested in mice was used (Aarts et al. 2014).
Results
Mouse Models to Investigate Differential Effects of Sparse or Broad Expression of the Dominant-Negative TrkB.T1 Receptor
To study the differential effects of TrkB.T1 expression in sparse or most cortical pyramidal neurons in the young adult visual cortex, we made use of mouse line TLT-817, which carries a transgene containing the Thy1-promoter-driving expression of TrkB.T1 fused to EGFP in a cre-dependent fashion. This line was crossed to 2 different cre-transgenic lines, which dictated whether the TrkB.T1-EGFP transgene was activated in sparse or most cortical pyramidal neurons. Both crosses have been described previously (Chakravarthy et al. 2006; Heimel et al. 2010). Crossed with the first line, the CaMKIIα-promoter-driven cre-expressing line cre-3487, TrkB.T1-EGFP expression was limited to <1% of all neurons in a Golgi staining-like fashion and started around 6 weeks after birth, increasing further over the next few weeks (Chakravarthy et al. 2006) (Fig. 1A,B). At this age, neurite growth and retraction and synapse turnover have decreased to mature levels. Crossed with the second line, the KA1-promoter-driven cre-expressing line (G35-3),TrkB.T1-EGFP was expressed in 80–90% of all pyramidal neurons of the neocortex and hippocampus (Heimel et al. 2010) (Fig. 1C,D). Expression could first be observed around 5–6 weeks after birth in all extragranular layers of the neocortex and reached adult levels a few weeks later. Thus, these mouse lines showed expression of the same TrkB.T1-EGFP transgene, starting at practically the same age, but in different numbers of pyramidal neurons allowing us to directly compare morphological and electrophysiological properties of the transgene-expressing cells.
Figure 1.

Sparse and broad expression of TrkB.T1-EGFP in the double transgenic mice. (A and B) A representative image of sparse expression of TrkB.T1 and schematic of the constructs used to generate the double transgenic mice. Cre-dependent, Thy1-promoter-driven TrkB.T1-EGFP transgene, and the CamKIIα-promoter-driven cre-recombinase transgene. (C and D). A representative image of broad expression of TrkB.T1 and schematic of the constructs used to generate the double transgenic mice. Cre-dependent, Thy1-promoter-driven TrkB.T1-EGFP transgene and the KA1-promoter-driven cre-recombinase transgene. The resulting offspring showed TrkB.T1-EGFP expression in all layers of the cortex except the granular layer (Layer IV). Scale bars in A and C are 75 and 250 µm, respectively.
Broad Expression of TrkB.T1 in Adult V1 Does Not Induce Changes in Spine Morphology or Density
We have previously shown that expressing TrkB.T1 in sparse pyramidal neurons from layer II/III of adult V1 (8–10 weeks of age) results in the loss of large dendritic spines. This was characterized by a 60% reduction in large mushroom spines and 85% reduction in stubby spines while there was a 2- to 3-fold increase in long- and thin spines and a 22-fold increase in filopodia. The average head size of spines was reduced by 40%, while spine length was increased by 30% (Chakravarthy et al. 2006). To investigate whether this was caused by a cell-autonomous process or induced by relative differences in TrkB signaling between cortical neurons, we analyzed spine morphology in adult V1 of TrkB.T1-broad mice also 8–10 weeks of age. Scattered neurons were labeled with DiI by diolistics (Benediktsson et al. 2005) in fixed slices of V1 (Fig. 2A). EGFP fluorescence was checked to verify that DiI-labeled cells also expressed TrkB.T1. In stark contrast to the situation in TrkB.T1 sparse mice, proximal basal dendrites of layer II/III pyramidal neurons of TrkB.T1-broad mice and littermate controls revealed no differences in the head size (0.67 ± 0.01 μm, n = 424 spines from 3 mice vs. 0.63 ± 0.01 μm, n = 543 spines from 5 mice P = 0.12) and length (0.79 ± 0.02 μm, n = 428, vs. 0.75 ± 0.02 μm, n = 543, P = 0.40) of dendritic spines (Fig. 2B). Also the densities and shapes of the spines (Fig. 2C) were similar in TrkB.T1-broad mice and littermate controls. TrkB.T1-broad mice and littermate controls had similar densities (all indicated as number of protrusions per 15 μm) of mushroom (10 ± 0.42, n = 37 dendrites, vs. 10.5 ± 0.34, n = 47, P = 0.32), stubby (0.22 ± 0.12 vs. 0.28 ± 0.11, P = 0.72), long (1.46 ± 0.22 vs. 1.19 ± 0.26, P = 0.44), and thin spines (0.27 ± 0.09 vs. 0.26 ± 0.08, P = 0.88) and filopodia (0.14 ± 0.06 vs. 0.04 ± 0.04, P = 0.32). The total protrusion density was also unchanged (12.05 ± 0.39, vs. 12.06 ± 0.36, P = 0.99; Fig. 2C). We conclude that, in the adult cortex, loss of dendritic spines is caused by relative, not absolute differences in TrkB signaling. These findings indicate that synaptic competition continues into adulthood.
Figure 2.

Overexpression of TrkB.T1-EGFP in most neurons does not result in structural changes of excitatory synapses. (A) A basal dendrite from a TrkB.T1-broadmouse was labeled with diolistics to quantify various spine classes. Scale bar 5 µm. (B) There were no changes in the spine neck length (P = 0.40) or head diameter (P = 0.12) between TrkB.T1-broad mice and littermate controls. (C) No changes were observed in densities of mushroom (P = 0.32), stubby (P = 0.72), long (P = 0.44), or thin spines (P = 0.88) and filopodia (P = 0.32). Total protrusion densities were also unchanged (P = 0.99). (D) There were no significant differences between the average interevent intervals for TrkB.T1-broad mice (n = 9 cells) compared with littermate controls (n = 10) as shown in cumulative distribution of the interevent intervals in the 2 groups (P = 0.60). (E) Cumulative distributions of the amplitudes of mEPSCs in both TrkB.T1-broad mice and littermate controls are similar (P = 0.77).
Miniature Excitatory Postsynaptic Currents Are Unchanged upon Broad Expression of TrkB.T1
We previously observed that the loss of large spines on pyramidal neurons in TrkB.T1-sparse mice was reflected at the electrophysiological level as a reduction in mEPSC frequency. To test whether the apparent lack of morphological effects of broad TrkB.T1-expression was also reflected at the electrophysiological level, we compared mEPSCs in TrkB.T1-broad mice and their littermate controls by whole-cell recordings in layer II/III pyramidal neurons in V1. In line with the morphological analysis, we saw no significant difference between TrkB.T1-broad mice and littermate controls in the frequency and amplitude of mEPSCs (Fig. 2D,E). The average interevent interval for TrkB.T1-broad mice was 139 ± 16.7 ms, n = 9 cells from 7 mice, compared with 161 ± 19.3 ms, n = 10 from 8 mice, in controls, P = 0.60. The cumulative histograms of interevent intervals in TrkB.T1-broad mice and their littermates are shown in Figure 2D. This lack of change in mEPSC frequency indicates that the number of postsynaptic excitatory structures and the probability of presynaptic vesicle release in TrkB.T1-expressing neurons are unaltered. The amplitude of mEPSCs in layer II/III pyramidal neurons of TrkB.T1-broad mice was 15.9 ± 0.47 pA, n = 9 from 7 mice, compared with 15.9 ± 0.52 pA, n = 10 from 8 mice, for control littermates, P = 0.77. Also the cumulative histograms of the mEPSC amplitudes in both TrkB.T1-broad mice and littermate controls showed very similar distributions (Fig. 2E). These data indicate that, in TrkB.T1-broad mice, there is no difference in postsynaptic AMPA-receptor levels or presynaptic vesicle content. These findings are thus compatible with the lack of morphological changes of dendritic spines in the mice with broad TrkB.T1 expression.
Increased Excitatory Synapses but Unchanged Spontaneous Activity in Fast-Spiking Interneurons
The fact that spine densities and mEPSC frequencies and amplitudes in TrkB.T1-broad mice were normal, while they were strongly impaired in TrkB.T1-sparse mice suggests that competition for presynaptic partners still occurs in the adult visual cortex. We then asked the question whether inhibitory neurons participate in this competitive process. In TrkB.T1-broad mice, inhibitory neurons do not express the transgene and may thus have a competitive advantage over excitatory neurons, which could be reflected in an increase in the excitatory inputs they receive. We recorded mEPSCs in electrophysiologically defined fast-spiking interneurons (predominantly PV-expressing interneurons), identified by their high-frequency action potential firing upon current injection and characteristic action potential shape, of TrkB.T1-broad mice and control littermates (Fig. 3A). Indeed, we found that the average distribution of interval times between events was lower in TrkB.T1-broad mice (62.2 ± 4.9 ms, n = 8 cells from 5 mice) compared with controls (79.8 ± 5.3 ms, n = 10 cells from 5 mice, P = 0.02; Fig. 3B,C). mEPSC amplitudes were unchanged (21.3 ± 0.6 vs. 21.7 ± 1.1 pA, P = 0.76; Fig. 3D). These findings suggest that fast-spiking interneurons in TrkB.T1-broad mice have a larger number of excitatory synapses or that the probability of presynaptic vesicle release is increased.
Figure 3.

Miniature EPSCs in fast-spiking interneurons show an increase in frequency, while spontaneous EPSCs remain unchanged. (A) Sample recording of high-frequency action potentials from a fast-spiking (FS) interneuron elicited by current injection. (B) Representative mEPSCs, recorded from FS interneurons in TrkB.T1-broad mice and littermate controls. (C) Averages and cumulative distributions of interevent intervals in TrkB.T1-broad mice (n = 8 cells) and littermate controls (n = 10) show a significant difference (P = 0.02). (D) Averages and cumulative distributions of amplitudes of mEPSCs into FS interneurons in TrkB.T1-broad mice and littermate controls are not different (P = 0.76). (E) Representative spontaneous EPSCs in TrkB.T1-broad mice and their littermate controls. (F and G) Averages of interevent intervals (P = 0.61) and amplitudes (P = 0.37) of spontaneous EPSCs in TrkB.T1-broad mice (n = 8 cells) and their littermate controls (n = 10) are not significantly different.
We have previously shown that cortical activity in TrkB.T1-broad mice is reduced (Heimel et al. 2010). To understand what the combination of increased excitatory input and reduced network activity meant for the activity of fast-spiking interneurons, we assessed the spontaneous EPSCs onto fast-spiking interneurons, recorded in the absence of TTX. We found that there was no change in the interevent interval (TrkB.T1-broad: 57.1 ± 4.9 ms, n = 8 cells from 5 mice, vs. control littermates: 54.1 ± 3.8 ms, n = 10 from 5 mice, P = 0.61; Fig. 3E,F) or amplitude (TrkB.T1-broad: 26.6 ± 1.0 pA, control littermates: 29.0 ± 2.1 pA, P = 0.37, Fig. 3G) of sEPSCs. It should be noted that network activity in vitro is not necessarily a reflection of in vivo network activity. However, these findings do suggest that spontaneous excitatory activity in the visual cortex of TrkB.T1-broad mice is indeed decreased, causing the spontaneous activity of fast-spiking interneurons to be unaltered despite increased numbers of excitatory synapses.
Intrinsic Firing Properties and AMPA/NMDA Ratios Are Unchanged upon Broad Expression of TrkB.T1
To obtain a better understanding of the reduced network activity in TrkB.T1-broad mice that we observed previously (Heimel et al. 2010), we investigated whether intrinsic firing properties of pyramidal neurons in these animals were changed. We did however not find any evidence for changes in the intrinsic properties of L2/3 pyramidal neurons or their likelihood to fire action potentials (Fig. 4). In TrkB.T1-broad mice, the mean number of action potentials in response to depolarizing current steps were not significantly different compared with their controls (TrkB.T1: n = 20 cells from 10 mice, vs. control: n = 10 cells from 7 mice, F1,28 = 0.15, P = 0.67; Fig. 4A). Also the resting membrane potential was unaltered (TrkB.T1: −70.8 ± 0.6 mV, n = 17 cells from 10 mice, vs. control: −71.0 ± 0.7 mV, n = 6 cells from 4 mice, P = 0.83; Fig. 4B), as were the threshold for initiation of action potential (TrkB.T1: −35.1 ± 0.91 mV, n = 13 cells from 8 mice, control: −34.2 ± 0.49 mV, n = 6 cells from 4 mice, P = 0.45; Fig. 4C), after-hyperpolarization (TrkB.T1: 6.24 ± 0.35 mV, n = 19 cells from 10 mice, vs. control: 6.62 ± 0.52 mV, n = 10 cells from 7 mice, P = 0.48; Fig. 4D), and time from spike onset to the peak of after-hyperpolarization (TrkB.T1: 3.0 ± 0.09 ms, n = 19 cells from 10 mice, vs. control: 3.12 ± 0.08 ms, n = 10 cells from 7 mice, P = 0.65; Fig. 4E). Input resistance did not differ between TrkB.T1-broad mice and control littermates (TrkB.T1: 90.2 ± 5.5 MΩ, n = 19 cells from 10 mice vs. controls: 85.5 ± 12.9 MΩ, n = 10 cells from 7 mice, P = 0.60).
Figure 4.

Overexpression of TrkB.T1-EGFP in most pyramidal neurons does not affect their intrinsic properties. In layer II/III pyramidal neurons of TrkB.T1-broad mice, (A) the mean number of action potentials in response to depolarizing pulses is similar to those of their littermate controls (P = 0.67); Down: Sample recordings of action potentials elicited by injection of a 400-pA current step for 165 ms. Also (B) resting membrane potential, (C) firing threshold potential, (D) after-hyperpolarization, and (E) time from the onset of action potential to peak of after-hyperpolarization are not different compared with littermate controls. (F) Right, example traces from representative experiments showing averaged evoked EPSCs at -80 mV [α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) component] and +40 mV [N-Methyl-D-aspartate (NMDA) and AMPA component) from TrkB.T1-broad mice (gray, bottom) and wild-type littermates (black, top). Gray areas indicate the time points used for quantifying AMPA and NMDA responses. Left, bar graph showing that NMDA/AMPA ratios in TrkB.T1-broad mice are unchanged.
Another possible explanation for the reduced network activity could be a lower relative NMDA receptor contribution in excitatory synapses, which could reduce the supralinear integration of strong evoked stimulation (Larkum et al. 2009) while leaving weak mEPSCs unaffected. To investigate whether this was the case, we recorded AMPA/NMDA ratios in these mice. We found that the AMPA/NMDA ratio was unchanged in broadly expressing TrkB.T1 mice compared with wild-type littermates (0.37 ± 0.03, n = 10 cells from 3 mice, vs. 0.36 ± 0.03, n = 9 cells from 3 mice; P = 0.56; Fig. 4F). Together, these data show that the deficit in network activity observed in broadly expressing TrkB.T1 mice cannot be explained by a problem in postsynaptic NMDA receptor expression or by altered intrinsic firing properties of pyramidal neurons (see Discussion).
Neurons in Broadly Expressing TrkB.T1 Mice Receive Less Inhibitory Input
We next analyzed whether inhibitory synapses onto pyramidal neurons were also differentially affected by expression of TrkB.T1 in most or sparse pyramidal neurons in V1. We therefore assessed inhibitory input to layer II/III pyramidal neurons in V1 by whole-cell recordings in TrkB.T1-broad mice and littermate controls (Fig. 5A). We found that mIPSC frequencies were strongly decreased in TrkB.T1-broad mice. The average distribution of time intervals between events was increased in TrkB.T1-broad mice (474 ± 57 ms, n = 6 cells from 5 mice) compared with the controls (315 ± 30 ms, n = 18 cells from 11 mice, P = 0.02) (Fig. 5B, cumulative distributions of the mIPSC interevent intervals are shown in Fig. 5D). Miniature IPSCs in TrkB.T1-broad mice showed no significant difference in amplitude compared with controls (26.8 ± 2.9 pA, n = 6 cells from 5 mice, vs. 26 ± 1.51 pA, respectively, n = 18 cells from 11 mice, P = 0.72; Fig. 5C, cumulative distribution of the mIPSC amplitudes are shown in Fig. 5E). These findings indicate that either the number of inhibitory synapses onto TrkB.T1-expressing neurons or the probability of presynaptic vesicle release of the inhibitory inputs was reduced. Because spontaneous excitatory input to inhibitory neurons is unaltered in TrkB.T1-broad mice, this suggests that cortical pyramidal neurons in these mice receive less inhibitory input.
Figure 5.

Inhibitory input to TrkB.T1-EGFP neurons shows a decrease in frequency without any change in amplitude. (A) Representative mIPSCs in TrkB.T1-broad mice and littermate controls. (B and D) Averages and cumulative distributions of interevent intervals in TrkB.T1-broad mice (n = 6 cells) and littermate controls (n = 18) show a significant difference (P = 0.021). (C and E) Averages and cumulative distributions of amplitudes of mIPSCs in TrkB.T1-broad mice and littermate controls. There is no significant difference between the 2 groups (P = 0.72).
Inhibitory Input of Sparse TrkB.T1-Expressing Neurons Is Unaltered
To understand whether this reduction in inhibitory input was regulated cell autonomously or depended on changes in neuronal activity at the network level, we also analyzed inhibitory synaptic input in sparse TrkB.T1-expressing neurons. To this end, mIPSCs were recorded in whole-cell configuration in TrkB.T1-sparse mice or in EGFP-sparse control mice (Fig. 6A). We found that there was no change in the interevent intervals (567 ± 48 ms, n = 7 cells from 5 mice vs. 598 ± 64 ms, n = 9 cells from 8 mice; P = 0.54; Fig. 6B), nor in mIPSC amplitudes (31.5 ± 1.64 pA, n = 7 cells from 5 mice vs. 31.4 ± 1.3 pA, n = 9 cells from 7 mice, P = 0.96) in TrkB.T1-expressing pyramidal neurons and their EGFP-expressing counterparts (Fig. 6C). We conclude that inhibitory synaptic inputs in sparse TrkB.T1-expressing neurons are unaffected. This implies that the decrease in inhibitory inputs observed in pyramidal neurons in TrkB.T1-broad mice is not mediated through a cell-autonomous mechanism, but that it is more likely a homeostatic compensation for reduced network activity.
Figure 6.

No change in inhibitory inputs to sparsely expressing TrkB.T1 neurons compared with controls sparsely expressing EGFP. (A) Representative mIPSCs in individual EGFP-expressing neurons compared with TrkB.T1-expressing neurons. (B) Interevent intervals of mIPSCs in individual TrkB.T1-expressing neurons (n = 4 cells) compared with EGFP-expressing neurons (n = 9) is not changed (P = 0.54). (C) Amplitudes in the same groups of neurons are also similar (P = 0.96).
Numbers of Perisomatic Boutons of Parvalbumin-Expressing Interneurons Are Only Reduced if TrkB.T1 Is Expressed in Most Pyramidal Neurons
The observation that the mIPSC frequency was reduced in pyramidal cells of TrkB.T1-broad mice could be explained by a reduction of inhibitory synapse numbers, which should be detectable by morphological analyses. We therefore quantified PV-positive inhibitory boutons surrounding pyramidal neurons in TrkB.T1-broad mice and control animals. We found that the number of boutons per cell was 25% lower in TrkB.T1-broad mice (9.6 ± 0.32, n = 110 cells from 3 mice) when compared with their littermate controls (12.83 ± 0.41, n = 114 cells from 3 mice P = 0.041; Fig. 7B). This suggests that the number of perisomatic inhibitory synapses is reduced when TrkB.T1 is expressed in most pyramidal neurons in V1.
Figure 7.
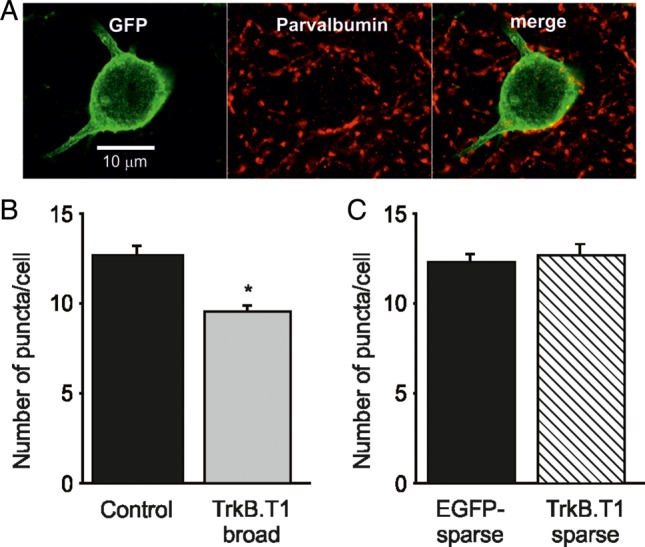
Perisomatic parvalbumin boutons are decreased only around broadly expressing TrkB.T1-EGFP neurons. (A) A representative image showing parvalbumin (PV) puncta around a cell body stained for GFP. (B) Number of PV-expressing boutons around cell bodies of TrkB.T1-broad mice (110 cells, 1056 boutons) compared with their littermate controls (114 cells, 1463 boutons) is significantly decreased (P = 0.041). (C) Numbers of PV-expressing boutons in sparse TrkB.T1 (31 cells, 393 boutons) or EGFP-expressing neurons (44 cells, 541 boutons) are similar (P = 0.69).
Because individual pyramidal neurons expressing the TrkB.T1 transgene in TrkB.T1-sparse mice did not show any difference in mIPSC frequency or amplitude, we expected that these cells would not show this reduction in inhibitory synapse numbers. To confirm this, sections of V1 of TrkB.T1-sparse mice and EGFP-sparse control mice were stained for GFP and PV (Fig. 7A). We found that the number of puncta per cell was indeed not changed in TrkB.T1-sparse mice (12.67 ± 0.60, n = 31 cells from 3 mice) compared with the EGFP-sparse controls (12.30 ± 0.45, n = 44 cells from 6 mice, P = 0.69; Fig. 7C). We conclude that inhibitory perisomatic boutons formed by PV-expressing interneurons are reduced only when most pyramidal neurons in V1 express the TrkBT1 transgene.
Discussion
In this study, we show that expression of the dominant-negative BDNF receptor TrkB.T1 in the majority of pyramidal neurons in adult V1 has strikingly different effects on cortical wiring than expression of the same transgene in a sparse neuron population. Broad expression does not lead to the reduction of dendritic spine numbers and synaptic connections that occurs when sparse neurons express the transgene. Very recently, it was observed that reduced expression of neuroligin-1 (Kwon et al. 2012) or BDNF (English et al. 2012) in individual neurons in the developing cortex is also more detrimental to spine density than gene modification in all neurons. Together, these studies establish that relative differences in the levels of these synaptic signaling molecules are more important than their absolute levels for attracting presynaptic partners, and that this mechanism contributes to competition between postsynaptic neurons for presynaptic inputs.
An important conclusion from our study is that the competition of postsynaptic neurons for synaptic input in the cortex also occurs in adulthood. Earlier studies in hippocampal cultures have shown similar processes during network development (Burrone et al. 2002; Hartman et al. 2006). However, these studies suggested that this form of synaptic competition only occurs during early development, and that it is absent when synapse formation has already occurred. In our study, the expression of TrkB.T1 started at an age at which synapse formation and neurite outgrowth have reached mature levels, excluding developmental effects of defective BDNF signaling on neuronal morphology (Bibel and Barde 2000). Therefore, we conclude that in the visual cortex competition for postsynaptic partners is not limited to development but continues into adulthood. Interestingly, the apparent discrepancy of our findings with hippocampal culture experiments may represent a difference between cortex and hippocampus, as we have shown in an earlier study that extensive spine loss also does not occur in individual TrkB.T1-expressing CA1 neurons in vivo (Chakravarthy et al. 2006).
To our knowledge, it is unknown whether inhibitory neurons compete with excitatory neurons for presynaptic partners. Our finding that fast-spiking interneurons (which are mainly PV positive) in TrkB.T1-broad mice show an increased mEPSC frequency suggests that they may. These interneurons do not express the TrkB.T1 transgene. Therefore, it could be that if most neighboring excitatory neurons express this transgene, interneurons become more attractive postsynaptic partners. However, we cannot rule out other explanations for this observation.
Although spine densities and mEPSC frequencies and amplitudes in broad TrkB.T1-broad mice are normal, we previously found that excitatory synaptic transmission is hampered in these mice. Spontaneous and visually evoked activity in V1 of TrkB.T1-broad mice is reduced in vivo and evoked excitatory currents are reduced in cortical slices (Heimel et al. 2010). Such differences between evoked and miniature excitatory currents have been observed before and can be mediated by various underlying mechanisms (Fredj and Burrone 2009; Weber et al. 2010; Hua et al. 2011; Sara et al. 2011; Kavalali et al. 2011; Ramirez and Kavalali 2012; Raingo et al. 2012; Ramirez et al. 2012). The cause of the reduced synaptic efficacy in TrkB.T1-broad mice is not clear. It could be that the increase in excitatory inputs to interneurons leads to a stronger recruitment of feed-forward inhibition, which in turn could shunt excitatory synaptic transmission. However, in our view, this is an unlikely explanation, as we observed the weaker excitatory synaptic responses within 2–3 ms after stimulation, which is too early for disynaptic inhibitory input to be recruited (Heimel et al. 2010). Our results also exclude a change in excitability or postsynaptic NMDA receptor expression as a possible explanation (Madara and Levine 2008). Future experiments will thus need to determine which other mechanism may underlie the reduction in evoked excitatory currents in TrkB.T1-broad mice.
Apart from the different effects of sparse and broad expression of TrkB.T1 on excitatory synapses, we found that inhibitory synapses onto TrkB.T1 expressing cells are also affected differentially in these 2 mouse lines, but in the opposite way. Morphological analysis and mIPSC recordings showed that inhibitory inputs onto pyramidal neurons were reduced when TrkB.T1 is expressed broadly in V1. Inhibitory inputs onto sparse neurons expressing the transgene remain unchanged, however. This means that the reduction of inhibitory inputs in TrkB.T1-broad mice cannot be mediated through cell-autonomous downregulation of inhibitory synapses by TrkB.T1-expressing pyramidal cells. The loss of inhibitory synapses also cannot be explained by defective TrkB signaling in interneurons themselves (Rutherford et al. 1997, 1998; Swanwick et al. 2006) as they do not express the transgene in TrkB.T1-broad mice. The loss of inhibitory innervation must thus depend on changes in network activity of cortical neurons.
Possibly, the decreased number of inhibitory synapses is a homeostatic response to the previously observed reduction of excitatory synaptic efficacy and population activity in TrkB-T1-broad mice in vivo (Heimel et al. 2010). This would be in line with previous studies showing changes in inhibitory synaptic transmission as a homeostatic scaling mechanism (Kilman et al. 2002; Knott et al. 2002; Karmarkar and Buonomano 2006; Bartley et al. 2008; Gandhi et al. 2008; Maffei and Turrigiano 2008; Maffei et al. 2010; Keck et al. 2011; Yang et al. 2011). An interesting hypothesis about how inhibitory neurons may normalize network activity is that they normalize their own activity by adjusting their output. In this scenario, reduced activity of few excitatory neurons does not affect population activity, and the output of PV interneurons will thus not be adjusted. If many excitatory neurons are less active, however, PV interneurons receive less input and reduce the number of inhibitory synapses they form. Because PV interneurons are driven by the pooled activity of surrounding neurons (Kerlin et al. 2010; Hofer et al. 2011) and form powerful inhibitory synapses onto the cell bodies and proximal dendrites of their postsynaptic targets they are in an excellent position to control network activity through this mechanism. The observation that in the TrkB.T1-broad mice this homeostatic mechanism does not manage to completely normalize cortical activity could be due to the increase in excitatory synapses that we observe in fast-spiking interneurons of these mice. This may cause these interneurons to “overestimate” the amount of network activity and consequently reduce their output insufficiently.
In conclusion, we demonstrate that adjustments of both inhibition and excitation only occur when TrkB/BDNF signaling is reduced in many neurons in the cortex, effectively normalizing overall cortical connectivity and activity while maintaining differences in postsynaptic drive. We find that competition occurs between neighboring excitatory neurons and possibly also between excitatory and inhibitory neurons. Finally, we show that if network activity is compromised, homeostatic mechanisms such as reducing synaptic inhibition are in place to restore activity to normal levels. These forms of homeostatic plasticity differ strongly from those that regulate excitatory or inhibitory input at a cell-autonomous level, as cell-autonomous mechanisms would result in the disappearance of differences in postsynaptic drive between neurons. The fact that the homeostatic mechanisms we describe here only become apparent when many neurons are challenged may explain why synaptic deficits observed in neuronal cultures, caused by genetic targeting of low numbers of neurons, can sometimes not be observed in transgenic or knockout mouse models in which many or all neurons are targeted (Okamura et al. 2004; Bozdagi et al. 2010). Finally, we show that both synaptic competition and adjustment of inhibitory innervation remain active in the adult cortex. This may have important consequences for our understanding of the pathogenesis of neuronal diseases as the continued activity of both processes can mask deficits in synaptic transmission over prolonged periods of time during disease progression.
Funding
This work was funded by a grant from AgentschapNL to the Neuro-Basic PharmaPhenomics consortium and by a grant from the Netherlands Organization for Scientific Research to R.M. (NWO 863.12.006). Funding to pay the Open Access publication charges for this article was provided by The Netherlands Organization for Scientific Research.
Notes
The authors thank Nargess Saiepour and Dr Alexander Heimel for data analysis, Drs Alexander Heimel, Helmut Kessels, and Christian Lohmann for helpful discussions and critical reading of the manuscript, Dr Chris Pool and Joop van Heerikhuize for assistance with image analyses and Emma Ruimschotel for technical assistance. Conflict of Interest: None declared.
References
- Aarts E, Verhage M, Veenvliet JV, Dolan CV, van der Sluis S. 2014. A solution to dependency: using multilevel analysis to accommodate nested data. Nat Neurosci. 17:491–496. [DOI] [PubMed] [Google Scholar]
- Bartley AF, Huang ZJ, Huber KM, Gibson JR. 2008. Differential activity-dependent, homeostatic plasticity of two neocortical inhibitory circuits. J Neurophysiol. 100:1983–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benediktsson AM, Schachtele SJ, Green SH, Dailey ME. 2005. Ballistic labeling and dynamic imaging of astrocytes in organotypic hippocampal slice cultures. J Neurosci Methods. 141:41–53. [DOI] [PubMed] [Google Scholar]
- Bibel M, Barde YA. 2000. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 14:2919–2937. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Wang XB, Nikitczuk JS, Anderson TR, Bloss EB, Radice GL, Zhou Q, Benson DL, Huntley GW. 2010. Persistence of coordinated long-term potentiation and dendritic spine enlargement at mature hippocampal CA1 synapses requires N-cadherin. J Neurosci. 30:9984–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, von Saint PF, Svoboda K. 2006. Interdigitated paralemniscal and lemniscal pathways in the mouse barrel cortex. PLoS Biol. 4:e382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN. 2002. Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature. 420:414–418. [DOI] [PubMed] [Google Scholar]
- Chakravarthy S, Keck T, Roelandse M, Hartman R, Jeromin A, Perry S, Hofer SB, Mrsic-Flogel T, Levelt CN. 2008. Cre-dependent expression of multiple transgenes in isolated neurons of the adult forebrain. PLoS One. 3:e3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy S, Saiepour MH, Bence M, Perry S, Hartman R, Couey JJ, Mansvelder HD, Levelt CN. 2006. Postsynaptic TrkB signaling has distinct roles in spine maintenance in adult visual cortex and hippocampus. Proc Natl Acad Sci USA. 103:1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English CN, Vigers AJ, Jones KR. 2012. Genetic evidence that brain-derived neurotrophic factor mediates competitive interactions between individual cortical neurons. Proc Natl Acad Sci USA. 109:19456–19461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredj NB, Burrone J. 2009. A resting pool of vesicles is responsible for spontaneous vesicle fusion at the synapse. Nat Neurosci. 12:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel MY, Bear MF. 2004. How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron. 44:917–923. [DOI] [PubMed] [Google Scholar]
- Gandhi SP, Yanagawa Y, Stryker MP. 2008. Delayed plasticity of inhibitory neurons in developing visual cortex. Proc Natl Acad Sci USA. 105:16797–16802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman KN, Pal SK, Burrone J, Murthy VN. 2006. Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci. 9:642–649. [DOI] [PubMed] [Google Scholar]
- Heimel JA, Saiepour MH, Chakravarthy S, Hermans JM, Levelt CN. 2010. Contrast gain control and cortical TrkB signaling shape visual acuity. Nat Neurosci. 13:642–648. [DOI] [PubMed] [Google Scholar]
- Hofer SB, Ko H, Pichler B, Vogelstein J, Ros H, Zeng H, Lein E, Lesica NA, Mrsic-Flogel TD. 2011. Differential connectivity and response dynamics of excitatory and inhibitory neurons in visual cortex. Nat Neurosci. 14:1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Z, Leal-Ortiz S, Foss SM, Waites CL, Garner CC, Voglmaier SM, Edwards RH. 2011. v-SNARE composition distinguishes synaptic vesicle pools. Neuron. 71:474–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubel DH, Wiesel TN, LeVay S. 1977. Plasticity of ocular dominance columns in monkey striate cortex. Philos Trans R Soc Lond B Biol Sci. 278:377–409. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Hanover JL, England PM, Stryker MP. 2008. TrkB kinase is required for recovery, but not loss, of cortical responses following monocular deprivation. Nat Neurosci. 11:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. 2008. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 58:673–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Xie Y, An JJ, Stryker MP, Xu B. 2012. Dendritic BDNF synthesis is required for late-phase spine maturation and recovery of cortical responses following sensory deprivation. J Neurosci. 32:4790–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmarkar UR, Buonomano DV. 2006. Different forms of homeostatic plasticity are engaged with distinct temporal profiles. Eur J Neurosci. 23:1575–1584. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Chung C, Khvotchev M, Leitz J, Nosyreva E, Raingo J, Ramirez DM. 2011. Spontaneous neurotransmission: an independent pathway for neuronal signaling? Physiology (Bethesda). 26:45–53. [DOI] [PubMed] [Google Scholar]
- Keck T, Scheuss V, Jacobsen RI, Wierenga CJ, Eysel UT, Bonhoeffer T, Hubener M. 2011. Loss of sensory input causes rapid structural changes of inhibitory neurons in adult mouse visual cortex. Neuron. 71:869–882. [DOI] [PubMed] [Google Scholar]
- Kerlin AM, Andermann ML, Berezovskii VK, Reid RC. 2010. Broadly tuned response properties of diverse inhibitory neuron subtypes in mouse visual cortex. Neuron. 67:858–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilman V, van Rossum MC, Turrigiano GG. 2002. Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J Neurosci. 22:1328–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GW, Quairiaux C, Genoud C, Welker E. 2002. Formation of dendritic spines with GABAergic synapses induced by whisker stimulation in adult mice. Neuron. 34:265–273. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Kozorovitskiy Y, Oh WJ, Peixoto RT, Akhtar N, Saulnier JL, Gu C, Sabatini BL. 2012. Neuroligin-1-dependent competition regulates cortical synaptogenesis and synapse number. Nat Neurosci. 15:1667–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Nevian T, Sandler M, Polsky A, Schiller J. 2009. Synaptic integration in tuft dendrites of layer 5 pyramidal neurons: a new unifying principle. Science. 325:756–760. [DOI] [PubMed] [Google Scholar]
- Madara JC, Levine ES. 2008. Presynaptic and postsynaptic NMDA receptors mediate distinct effects of brain-derived neurotrophic factor on synaptic transmission. J Neurophysiol. 100:3175–3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Lambo ME, Turrigiano GG. 2010. Critical period for inhibitory plasticity in rodent binocular V1. J Neurosci. 30:3304–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Turrigiano GG. 2008. Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci. 28:4377–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrsic-Flogel TD, Hofer SB, Ohki K, Reid RC, Bonhoeffer T, Hubener M. 2007. Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron. 54:961–972. [DOI] [PubMed] [Google Scholar]
- Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Colman DR, Miki N. 2004. Cadherin activity is required for activity-induced spine remodeling. J Cell Biol. 167:961–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingo J, Khvotchev M, Liu P, Darios F, Li YC, Ramirez DM, Adachi M, Lemieux P, Toth K, Davletov B, et al. 2012. VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat Neurosci. 15:738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Kavalali ET. 2012. The role of non-canonical SNAREs in synaptic vesicle recycling. Cell Logist. 2:20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Khvotchev M, Trauterman B, Kavalali ET. 2012. Vti1a identifies a vesicle pool that preferentially recycles at rest and maintains spontaneous neurotransmission. Neuron. 73:121–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, DeWan A, Lauer HM, Turrigiano GG. 1997. Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J Neurosci. 17:4527–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford LC, Nelson SB, Turrigiano GG. 1998. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 21:521–530. [DOI] [PubMed] [Google Scholar]
- Sara Y, Bal M, Adachi M, Monteggia LM, Kavalali ET. 2011. Use-dependent AMPA receptor block reveals segregation of spontaneous and evoked glutamatergic neurotransmission. J Neurosci. 31:5378–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NB, Frenkel MY, Philpot BD, Nakazawa K, Tonegawa S, Bear MF. 2003. NMDA receptor-dependent ocular dominance plasticity in adult visual cortex. Neuron. 38:977–985. [DOI] [PubMed] [Google Scholar]
- Swanwick CC, Murthy NR, Kapur J. 2006. Activity-dependent scaling of GABAergic synapse strength is regulated by brain-derived neurotrophic factor. Mol Cell Neurosci. 31:481–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JP, Reim K, Sorensen JB. 2010. Opposing functions of two sub-domains of the SNARE-complex in neurotransmission. EMBO J. 29:2477–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Weiner BD, Zhang LS, Cho SJ, Bao S. 2011. Homeostatic plasticity drives tinnitus perception in an animal model. Proc Natl Acad Sci USA. 108:14974–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]