

Abstract
Ranbp1, a Ran GTPase-binding protein implicated in nuclear/cytoplasmic trafficking, is included within the DiGeorge/22q11.2 Deletion Syndrome (22q11.2 DS) critical region associated with behavioral impairments including autism and schizophrenia. Ranbp1 is highly expressed in the developing forebrain ventricular/subventricular zone but has no known obligate function during brain development. We assessed the role of Ranbp1 in a targeted mouse mutant. Ranbp1−/− mice are not recovered live at birth, and over 60% of Ranbp1−/− embryos are exencephalic. Non-exencephalic Ranbp1−/− embryos are microcephalic, and proliferation of cortical progenitors is altered. At E10.5, radial progenitors divide more slowly in the Ranpb1−/− dorsal pallium. At E14.5, basal, but not apical/radial glial progenitors, are compromised in the cortex. In both E10.5 apical and E14.5 basal progenitors, M phase of the cell cycle appears selectively retarded by loss of Ranpb1 function. Ranbp1−/−-dependent proliferative deficits substantially diminish the frequency of layer 2/3, but not layer 5/6 cortical projection neurons. Ranbp1−/− cortical phenotypes parallel less severe alterations in LgDel mice that carry a deletion parallel to many (but not all) 22q11.2 DS patients. Thus, Ranbp1 emerges as a microcephaly gene within the 22q11.2 deleted region that may contribute to altered cortical precursor proliferation and neurogenesis associated with broader 22q11.2 deletion.
Keywords: basal progenitor, cell cycle, cerebral cortex, cortical development, intermediate progenitor
Introduction
DiGeorge/22q11.2 Deletion Syndrome (22q11.2 DS) is a copy number variant genetic syndrome strongly associated with disorders of cortical connectivity including autism, attention deficit disorder, and schizophrenia (Fine et al. 2005; Philip and Bassett 2011; Baker and Vorstman 2012; Schneider et al. 2014). The behavioral disorders associated with 22q11.2 DS likely reflect neural circuit anomalies that result from disrupted cortical development; however, little is known about the function of individual 22q11.2 genes in the developing cortex. 22q11.2 gene-mediated disruptions could occur during early patterning of the telencephalon (Maynard et al. 2013; Karpinski et al. 2014), or generation and migration of cortical projection neurons and interneurons (Meechan et al. 2009, 2012; Schaer et al. 2009; Baker et al. 2011). We asked whether Ranbp1, a 22q11.2 gene whose expression is enhanced in the developing cerebral cortex (Maynard et al. 2002, 2003), is critical for optimal cortical development and is thus a candidate gene for 22q11.2 DS-associated cortical circuit disruption.
Ranbp1 seems a likely regulator of early central and peripheral nervous system development. This binding partner of Ran, a small Ras-like GTPase essential for nuclear/cytoplasmic transport (Freitas and Cunha 2009), is 1 of a small subset of 22q11.2 genes apparently expressed in neural progenitors during early CNS patterning and differentiation (Maynard et al. 2003; Meechan et al. 2009). Ranbp1 is first strongly expressed in neural crest populations that are critical for early forebrain patterning (LaMantia 1999; Siegenthaler et al. 2009), and subsequently in the forebrain ventricular and subventricular zones. In the fetal mouse as well as fetal human brain, Ranbp1 is maximally expressed during the time of peak forebrain neurogenesis (Meechan et al. 2006). Accordingly, Ranbp1 is a compelling candidate for disruptions of cortical development that accompany 22q11.2 DS. We therefore asked whether Ranbp1 function was necessary for key steps in forebrain or cortical development by analyzing phenotypes that result from complete Ranbp1 loss of function.
Ranpb1 may influence multiple cellular mechanisms critical for forebrain differentiation. Ranbp1 regulates the multi-protein Ran complex, thereby modulating a key effector of nuclear/cytoplasmic shuttling (Plafker and Macara 2000; Kehlenbach et al. 2001; Freitas and Cunha 2009). Ranbp1 can also induce primary cilia formation (Fan et al. 2011), alter cell-cycle progression (Battistoni et al. 1997; Di Fiore et al. 2003), modulate re-formation of the nuclear envelope following mitosis (Ciciarello et al. 2010), and regulate signaling in response to axonal injury (Yudin et al. 2008). We found that Ranbp1 is indispensable for normal fetal morphogenesis and viability. Furthermore, Ranbp1 has an obligate function in regulation of cortical progenitor proliferation most likely via modulation of mitosis, particularly in rapidly dividing precursor populations, leading first to diminished cortical size and then diminished neurogenesis in cortical layers 2 and 3, but not layers 5 and 6. Thus, Ranpb1 emerges as a microcephaly gene, and as a viable candidate for influencing cortical circuit development relevant for behavioral disorders associated with 22q11.2 DS.
Materials and Methods
Generation and Characterization of Ranbp1 Mutant Mouse Line
Ranbp1 mutant mice were generated from Line S17-6E1 Soriano ROSAFARY gene-trap ES-cells (Chen and Soriano 2003) by the UC Davis Mutant Mouse Regional Resource Center. The hygromycin cassette in the gene-trap construct was excised by crossing to a Rosa26-FLP1 line, and the resulting line backcrossed for >8 generations to C57/Bl6N (Charles River Laboratories) before analysis. Detailed methodology is included in Supplementary Material.
Mouse husbandry, embryo preparation, and tissue collection were performed as described in previous studies (Meechan et al. 2009; Maynard et al. 2013). A detailed qPCR methodology and a list of qPCR primers used in this study are included as Supplementary Material.
Immunofluorescent and Western Blot Analysis
Embryos were dissected in ice-cold PBS, immersion-fixed overnight in 4% paraformaldehyde in PBS, cryosectioned, and stained as described previously (Maynard et al. 2003; Meechan et al. 2009). Western blot analysis was performed as described previously (Maynard et al. 2008). Primary antibodies used in this study are included as Supplementary Table 2.
BrdU Labeling and Cell Counts
For birth-dating and proliferation analysis, BrdU (50 mg/kg body weight) was injected IP into timed pregnant mice. Embryos were harvested at E10.5 1 h after labeling with BrdU, at E14.5, either 1 or 16 h after labeling with BrdU, or at E17.5, after labeling with BrdU at E14.5. Standard BrdU immunolabeling techniques were used after sodium citrate steam treatment (antigen retrieval). Only heavily labeled cells were scored as BrdU positive. Specimens were imaged as photomontages on either a Zeiss 710 confocal microscope or a Leica DM6000 epifluorescence microscope with automated tiling stages. After imaging, sections were coded and counted blind. For E10.5 embryos, sections for analysis were chosen within the anterior telencephalon, at the level of the olfactory epithelium, and cells were counted along the complete length of the telencephalic vesicle. To account for size differences between individual embryos, the perimeter of the telencephalic vesicle was measured for each section, and cell counts were normalized to this length (i.e., cells/mm). For E14.5 embryos, cells were counted from coronal sections taken at the level of mid-ganglionic eminence approximately at the level of the eye, whereas E17.5 embryos were analyzed at the level of the anterior commissure. These regions have similar morphology in both null mutant and wild-type embryos despite overall changes in cortical size. For cell counting, 150-um- or 300-um-wide counting boxes were superimposed onto micrographs at described locations as noted. We used two-tailed Student's t-tests to evaluate significance of mean cell number differences between pairs of Ranbp1 null mutant and wild-type samples, and two-way ANOVA to compare cell distributions when multiple bins were assessed.
Results
A Mutant Allele of Ranbp1 Results in Complete Loss of Function
We used a mouse carrying a “gene-trap” insertion of a βGeo reporter into the coding sequence of the Ranpb1 locus (Chen and Soriano 2003) to recover a null mutation and evaluate its obligate functions during early brain development. We mapped the insertion site of the ROSAFARY reporter to the 98th base of exon 2 (Fig. 1A), which prevents expression of all but the initial 44 N-terminal amino acids of Ranbp1. Due to the atypical insertion of the reporter into the exon (rather than into an intron as designed), the βGeo reporter is almost undetectable by X-gal staining and acts as a poor indicator for Ranbp1 expression. Using an antibody to the N-terminus of Ranbp1 (including the 44 amino acid fragment), we did not detect expression of the residual protein in homozygous Ranbp1 mutant embryos, either by immunofluorescent staining (Fig. 1B) or western blotting (Fig. 1C). Any sub-threshold level of the residual 44-amino acid peptide that may remain is likely nonfunctional. It lacks most of the presumptive Ran-binding domain and the requisite nuclear export sequence (Richards et al. 1996) and does not mediate the same cell-cycle disruption (abnormal chromatin condensation; Battistoni et al. 1997) caused by full-length Ranbp1 when overexpressed in HEK-293 cells (data not shown). Accordingly, this mutation can be considered an effective null, referred to as Ranbp1−/−.
Figure 1.

A mutant allele of Ranbp1 disrupts early brain development. (A) Overview of Ranbp1 mutant allele, detailing insertion of transgene into Exon 2. (B) Immunostain of E10.5 WT and Ranbp1−/− neuroepithelium, showing undetectable levels of protein expression (red) in null embryo. Right side of images show overlaid nuclear DAPI staining (blue). (C) Analysis of expression levels of RANBP1 protein in E13.5 cortex obtained from WT (+/+) and heterozygous (+/−) or homozygous Ranbp1 mutant embryos. Reduced levels are observed in heterozygote samples, whereas no detectable band was observed in homozygous samples. The same blot was simultaneously probed for beta-Actin as a loading control. (D) Reduced expression levels of Ranbp1 transcript are observed in E13.5 cortical samples from heterozygous Ranbp1 mutant embryos, similar to levels observed in LgDel embryos. No reduction was observed in heterozygous Ranbp1 embryos for a chromosomally adjacent transcript, Trmt2a. (D). (E–H) Examination of E10.5 embryos shows that Ranbp1−/− embryos are dysmorphic, including a shortened telencephalic vesicle (bracket) in the non-exencephalic cohort (F), and open anterior neural tube in the exencephalic cohort (G,G′ is an alternate view of same embryo). (H–J) E14.5 Ranbp1−/− embryos have visible anomalies including aberrant eye position (arrows) and frequent eye anomalies (insets). (K–P) Cortex of Ranbp1−/− embryos is distinctively smaller at E14.5, as shown in dorsal (K,L) and lateral (M,N) views, and in hematoxylin and eosin-stained sagittal sections (O,P). (Q–S) Quantification of cortical thinning at E14.5, obtained by measuring thickness of the cortical mantle in 10 WT and 10 Ranbp1−/− cortices, at 6 evenly spaced landmarks across the mantle. P < 0.001 by two-way ANOVA. (T–V) Quantification of 22q11.2 transcript mRNA by quantitative RT-qPCR. (T) Estimation of expression levels in E14 cortex, measured by ▵CT relative to Gapdh expression (lower numbers indicate higher expression levels). (U,V) Expression of 22q11.2 transcripts in cortex of heterozygous Ranbp1 mutant cortex (U) and null mutant cortex (V). Asterisk in (U,V) indicates that expression for a transcript is significant, assessed by one-way ANOVA with Dunnett's post hoc test. Scale bars = 1 mm, except B = 10 µm; R,S = 200 µm.
We next confirmed that this genomic lesion specifically and uniquely disrupts Ranbp1 transcription. We compared Ranbp1 mRNA levels in E13.5 cortex from Ranbp1+/− mutants to that in the cortex of E13.5 LgDel embryos who also are heterozygous null for Ranbp1 as well as 27 other 22q11.2 orthologues (Meechan et al. 2006, 2009). Ranbp1 message was diminished by ∼50% in Ranbp1+/− cortex, as expected (52%, P < 0.001), similar to that observed in LgDel embryos heterozygously deleted for the entire 22q11.2 orthologous “minimal critical region” (Fig. 1D; see also Fig. 1U). Furthermore, the Trmt2a locus (also known as Htf9c), which is immediately 5′ to Ranbp1 on mmChr 16 (the murine orthologue of hChr 22 as well as 21) and shares the same bicistronic promoter, was expressed in the Ranbp1 heterozygote cortex at levels similar to wild type (WT), rather than LgDel levels (P > 0.9). Together these results confirm that this complete loss-of-expression mutation is targeted specifically to the Ranbp1 locus and can be used to identify obligate functions of Ranpb1 during embryonic development.
Ranbp1 Mutation Leads to Gross Morphological Defects with Variable Penetrance
We evaluated the gross morphology of Ranbp1−/− embryos at 2 developmental stages: E10.5, when initial patterning is complete throughout the embryo and the neural tube is fully closed, and E14.5, when brain regionalization and organogenesis have progressed substantially. At E10.5, homozygous mutation of Ranbp1 profoundly disrupts morphogenesis. E10.5 Ranbp1−/− embryos appear at the expected frequency (Table 1); however, they are highly—but variably—dysmorphic (Fig. 1E–G). All exencephalic (11/11) and most non-exencephalic (6/7) E10.5 Ranbp1−/− embryos have hypoplastic first and second branchial arches (Fig. 1F–G). Approximately 60% of mutant embryos are exencephalic from E10.5 onward (Table 2). In most exencephalic E10.5 Ranbp1−/− embryos (11/14), neural tube closure is disrupted from the rhombencephalon forward, and the spinal cord is spared (Fig. 1G,G′). The non-exencephalic cohort of E10.5 Ranbp1−/− embryos is less dysmorphic than their exencephalic counterparts, but not completely normal. These embryos are smaller, the branchial arches are dysmorphic, and the forebrain vesicle is shortened in the anterior–posterior axis (Fig. 1F).
Table 1.
Ratio of Ranbp1 mutant recovery by age of analysis
| Age | +/+ | +/− | −/− | P (χ2) |
|---|---|---|---|---|
| E10.5 | 87 | 143 | 75 | 0.34 |
| E14.5 | 74 | 125 | 41 | 0.009 |
| P7–10 | 49 | 83 | 0 | >0.0001 |
Table 2.
Observed exencephalic and non-exencephalic Ranbp1 null embryos
| Exencephalic (% of total) | Non-exencephalic (% of total) | % exencephaly in nulls | |
|---|---|---|---|
| E10.5 | 53 (17%) | 22 (7.2%) | 70% |
| E14.5 | 25 (10%) | 16 (6.7%) | 61% |
| E17.5 | 12 (13%) | 7 (7.9%) | 63% |
E14.5 Ranbp1−/− embryos remain dysmorphic (Fig. 1H–J). Both non-exencephalic and exencephalic embryos are smaller than their WT counterparts. In non-exencephalic mutants, the head appears smaller (Fig. 1I). Anomalies are also apparent in the developing eyes. Their position is shifted (Fig. 1H–J, arrows), and microphthalmia or severe colobomas (incomplete iris closure) are present in 21/22 eyes of exencephalic Ranbp1−/− embryos, and 13/26 eyes of non-exencephalic null embryos (insets, Fig. 1H–J). Gross dissection of the E14.5 brain of non-exencephalic embryos shows that it is reduced in size (Fig. 1K–N), and sagittal sections confirm that the cortex appears thinned in non-exencephalic null mutants (Fig. 1O,P). Other telencephalic structures, such as the olfactory bulbs and ganglionic eminences, also appear to be smaller. We quantified this apparent change in E14.5 cortical thickness in a cohort of non-exencephalic Ranbp1−/− and WT embryos, at multiple locations along the ventrolateral to dorsomedial extent of the hemisphere (Fig. 1Q,R). The cortical mantle in E14.5 Ranpb1−/− embryos is reduced between 11 and 23% of WT (P < 0.001 by two-way ANOVA), with the greatest reduction observed near the midpoint of the ventrolateral to dorsomedial axis (Fig. 1S). Complete loss of Ranbp1 is not compatible with postnatal survival. We did not recover any Ranbp1−/− pups at 7–10 days after birth (49 WT, 83 +/−, 0−/−; P < 0.001 by χ2; Table 1). A significant portion of this failure in postnatal survival can be accounted for by the 61–70% incidence of exencephaly; however, we have yet to identify a non-exencephalic Ranbp1−/− embryo that survives postnatally. Our data also suggest modestly reduced viability of heterozygous progeny in Ranbp1−/+ × Ranpb1−/+ matings (+/+ : +/− ratio of 1.11 : 1.89), as well as for Ranbp1+/− × WT C57/Bl6N matings (110 WT, 93+/−); however, these changes do not reach statistical significance.
To further confirm the specificity of our Ranbp1 mutation, to better establish a potential contribution of Ranpb1 to cortical development, and to evaluate the consequences of its mutation for expression of other 22q11.2 genes in the developing cortex, we quantified mRNA levels of Ranpb1 and additional murine orthologues from the 22q11.2 minimal critical region in microdissected samples of the E14.5 cortical mantle of WT, Ranbp1+/−, and non-exencephalic Ranbp1−/− embryos. Ranbp1 is 1 of the most robustly expressed genes in WT E14.5 cortex (Fig. 1T). As expected, heterozygous deletion diminishes Ranpb1 expression in the E14.5 cortex by ∼50%; however, it does not significantly alter expression levels of any other 22q11.2 gene in the cortex (Fig. 1U). In homozygous mutants, Ranpb1 mRNA is undetectable (Fig. 1V). In addition, homozygous deletion modestly disrupts the expression of several other 22q11.2 genes (Fig. 1S), including Tbx1 (P = 0.0005), Arvcf (P = 0.016), Trmt2a (P = 0.0004), Slc25a1 (P = 0.008), and Dgcr2 (P = 0.02). These expression changes may be a direct consequence of the loss of Ranbp1, or secondary to cellular changes that lead to cortical thinning in the null embryo. Of the 5 statistically significant expression changes observed in the E14.5 Ranbp1−/− cortex, 4 are seen for genes expressed at substantial levels. The fifth, Tbx1, is not robustly expressed in the cortex at midgestation. Indeed, we have reported previously that Tbx1 is not readily detectable by in situ hybridization in the cortex at this age (Meechan et al. 2009), consistent with the very high detection threshold (+12.6 CT) in our current qPCR analysis. Thus, the change in Tbx1 expression in the Ranpb1−/− cortex likely reflects detection threshold rather than a meaningful expression change. Thus, our expression analysis of the cortex from WT, Ranpb1+/− and Ranbp1−/− embryos shows that Ranbp1 is robustly expressed in the normal cortex, confirms the specificity of our mutation for the Ranpb1 locus, including in the developing cortical mantle, and suggests that full loss of function of Ranbp1 can modulate—but not eliminate—expression levels of additional 22q11.2 genes.
Neural Crest is Not Significantly Altered by Homozygous Loss of Ranbp1
Craniofacial rudiments, especially the branchial arches whose development depends critically upon the integrity of the cranial neural crest, are smaller and dysmorphic in Ranpb1−/− embryos. Previous studies have demonstrated that cranial neural crest cell populations are also essential for forebrain induction (LaMantia et al. 1993) including establishing or maintaining intrinsic forebrain Shh and Fgf8 “organizer” domains (Tucker et al. 2008) as well as maintaining telencephalic neural progenitors (Siegenthaler et al. 2009). Disrupted neural crest differentiation can also phenocopy key aspects of 22q11.2 DS (Scambler 2000). Accordingly, we evaluated the integrity of the cranial neural crest in E10.5 Ranpb1−/− embryos to determine whether branchial arch and forebrain hypotrophy reflects altered neural crest-mediated morphogenetic interactions. Since many changes in the exencephalic Ranbp1−/− brain are likely secondary to the open neural tube, possibly reflecting the consequences of direct contact with extraembryonic fluids (Lehtinen et al. 2011), we focused exclusively on the non-exencephalic Ranpb1−/− cohort for this and all subsequent analyses.
In E10.5 WT embryos, neural crest-derived cells are a primary constituent of the mesenchyme between the telencephalic neuroepithelium and the overlying cranial surface epithelium. These cells secrete a fibronectin-rich basal lamina, and we saw no apparent differences in the distribution of fibronectin between WT and Ranpb1−/− embryos (Fig. 2A,B). Cranial crest also robustly express Crabp1, a key binding protein for retinoic acid (RA), which is produced by the crest-derived cranial mesenchyme (LaMantia et al. 2000; Bhasin et al. 2003). Ranpb1−/− and WT embryos have similar Crabp1 labeling in the cranial mesenchyme, particularly in the substantial population of mesenchymal cells interposed between the olfactory placode and ventral forebrain (Fig. 2C,D). Finally, we assessed the integrity of the neural crest-derived presumptive meningeal cells adjacent to the dorsal telencephalon, also a source of RA (Siegenthaler et al. 2009). These cells, robustly labeled by the neural crest marker P75, appear similar in E10.5 Ranbp1−/− and WT embryos (Fig. 2E,F). Thus, it is unlikely that a significant failure of migration or initial differentiation of the neural crest underlies craniofacial or forebrain morphogenetic anomalies in Ranbp1−/− embryos.
Figure 2.

Patterning of neural crest and dorsal-ventral markers appear normal in Ranbp1−/− embryos. (A,B) Distribution of craniofacial mesenchyme, as indicated by fibronectin staining, appears normal in Ranbp1−/−, as does neural crest-derived mesenchyme in both the frontonasal process (labeled by CRABP1, C,D) and in the presumptive meninges, adjacent to the dorsal telencephalon (labeled by P75, E,F). (G,H) Expression of dorsal (Pax6) and ventral (Nkx2.1) markers is appropriately localized in E10.5 embryos. (I,J) Both the ventromedial (MGE, Nkx2.1/Ascl1 double-labeled) and lateral (LGE, Ascl1) compartments of the subpallium are present in Ranbp1−/− mutants at E14.5. Scale bars = 250 µm, except C,D = 100 µm and E,F = 25 µm.
Forebrain Patterning is Not Significantly Disrupted by Ranbp1 Mutation
Cerebral cortical size is significantly smaller in Ranbp1−/− mutants; however, major changes of extrinsic patterning of the nascent forebrain due to neural crest anomalies do not seem likely to account for these changes. Thus, we assessed several aspects of intrinsic forebrain patterning in the non-exencephalic cohort to determine whether local morphogenetic mechanisms are responsible for cortical hypotrophy in Ranbp1−/− mutants. In E10.5 Ranbp1−/− embryos, Pax6, a marker of the dorsal pallium which will form the neocortex (Yun et al. 2001; O'Leary et al. 2007) and Nkx2.1, a marker of the ventral telencephalon/subpallium which gives rise to the ganglionic eminences (Kimura et al. 1996; Sussel et al. 1999) remain segregated as in the WT embryo (Fig. 2G,H). The ventral Nkx2.1 domain appears somewhat smaller in the mutants; however, it is difficult to determine whether this is an effect of disrupted patterning, or a consequence of the smaller size of the telencephalon. We next asked whether the medial and lateral ganglionic eminences (MGE/LGE) form normally in the Ranbp1−/− ventral telencephalon. We analyzed the expression domains of Nkx2.1, restricted to the MGE, and Ascl1, which labels the MGE and LGE in the E14.5 telencephalon. Although the E14.5 telencephalon is smaller, the MGE and LGE are morphologically and molecularly distinct in Ranbp1−/− embryos (Fig. 2I,J). Thus, despite its smaller size, it appears that some fundamental aspects of intrinsic forebrain dorsoventral patterning are intact in Ranbp1−/− mice.
Neuroepithelial Apical-Basal Polarity is Maintained in Ranbp1 Mutant Mice
Pallial neuroepithelial cell polarity is critical for proliferation, migration, and differentiation of cortical progenitors and their projection neuron progeny (Chenn and Walsh 2002; Zechner et al. 2003; Kwan et al. 2012; Evsyukova et al. 2013). Thus, we next asked whether essential aspects of neuroepithelial polarity were compromised in the Ranbp1−/− E10.5 dorsal pallium. We first evaluated the morphology of radial glial cells, which become forebrain stem cells as well as migratory substrates (Malatesta et al. 2008; Kriegstein and Alvarez-Buylla 2009). In both WT and Ranpb1−/− E10.5 embryos, nascent radial glial progenitors have long, basally oriented processes (i.e., toward the pial surface), with end-feet that contact the basal lamina (Fig. 3A,B). We next assessed the integrity of the apical domain by evaluating β-catenin, which is concentrated at apical tight junctions. We saw no consistent differences in apical β-catenin labeling between WT and Ranbp1−/− mutants (Fig. 3C,D). We also examined the apical localization of the polarity marker atypical protein kinase C (aPKC). Ranbp1−/− mutants have the same basic distribution of aPKC as WT embryos (Fig. 3E,F). Together, these results indicate that essential aspects of polarity of the cortical neuroepithelium and its constituent radial glial stem cells are not substantially disrupted by the loss of Ranbp1 function. Based upon in vitro observations (Fan et al. 2011), disrupted Ranbp1 function may compromise an additional aspect of epithelial polarity: the generation of primary apical cilia that distinguish all neural progenitor and stem cells (Bay and Caspary 2012; Higginbotham et al. 2013). Thus, we evaluated cilia frequency in the pallial neuroepithelium of E10.5 embryos, using Arl13b (Caspary et al. 2007) as a marker (Fig. 3G,H). We did not see a statistically significant difference in cilia frequency in the E10.5 telencephalon (WT = 728 ± 100 cilia/mm; Ranbp1−/− = 607.6 ± 144 cilia/mm; n = 5 WT, 5 null; P = 0.51); however, the average number is lower in Ranbp1−/− embryos. Together, these results show that several key aspects of neuroepithelial polarity often associated with disrupted cortical neurogenesis are relatively undisturbed in Ranbp1−/− embryos.
Figure 3.
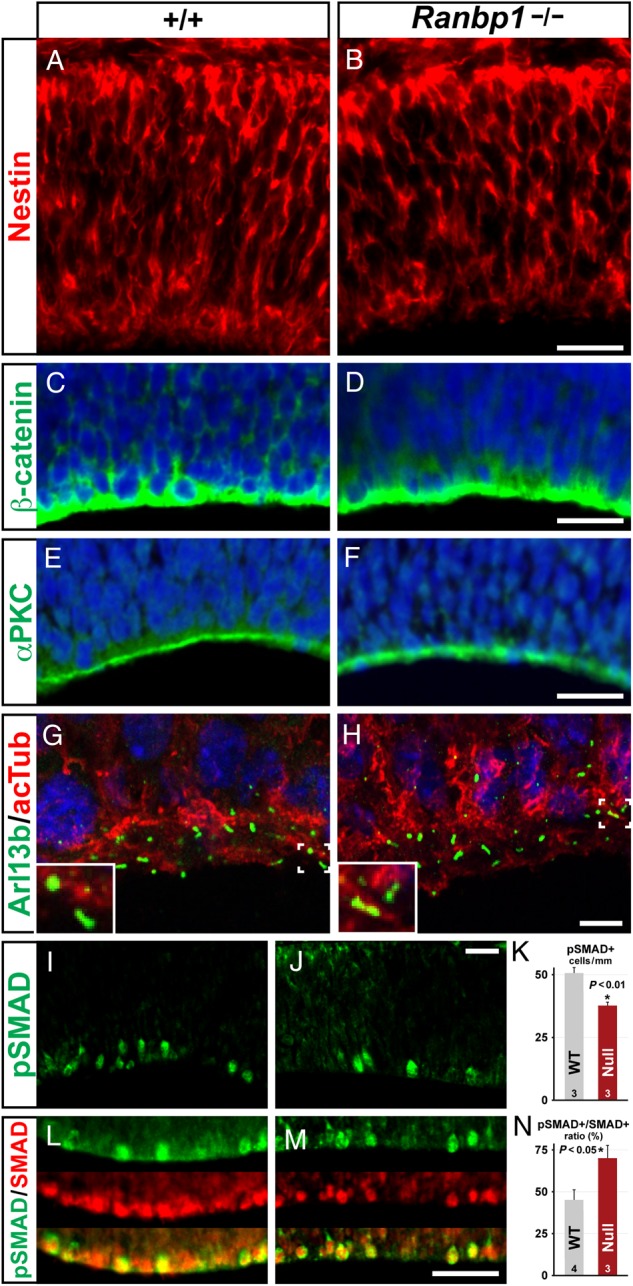
Markers of neuroepithelial polarity are maintained in the E10.5 Ranbp1−/− telencephalon. (A,B) Nestin-labeled progenitors are present and have apparently normal morphology, and the apical markers β-catenin (C,D) and αPKC (E,F) appear normal in Ranbp1−/− embryos. (G,H) Arl13B-labeled primary cilia are present and appear grossly normal in Ranbp1−/− embryos. (I–N) SMAD signaling appears disrupted in apical neuroepithelial cells: fewer phosphorylated SMAD 1/5/8 (pSMAD) cells are evident (I–K), although there are also fewer cells expressing either phosphorylated or unphosphorylated SMAD1, leading to an increase in the pSMAD+/SMAD+ ratio in the apical population (L–N). Scale bars: A–F = 50 µm, G–H = 5 µm, and I,J,L,M = 25 µm. Inset in G,H is ×2.5 magnification. Number of embryos assessed for each data point and P-values are noted on each graph.
Other aspects of signaling at the cortical apical/ventricular surface may be compromised by loss of Ranbp1 function. We noted apparent gross-phenotypic similarities between Ranbp1 and Noggin null mutations. Both result in similar proportions of exencephalic embryos, as well as reduced telencephalic vesicle size in the non-exencephalic cohort (McMahon et al. 1998). This suggested that signaling via TGFβ-family members, particularly BMPs and related signaling intermediates known to act on forebrain progenitors (Gross et al. 1996; Mehler et al. 2000), might be compromised by Ranbp1 loss of function. Thus, we asked whether Ranbp1 mutation disrupts BMP/SMAD signaling during early telencephalic development, possibly by disrupting nuclear import/export of activated SMADs (Nicolas et al. 2004). The SMADs are downstream effectors of signaling via TGFβ-family proteins; SMADs 1/5/8 are primary targets of embryonic BMP signaling (Feng and Derynck 2005). BMPs (and possibly other TGFβ-family signals) influence early cortical patterning (Shimogori et al. 2004), are present in embryonic CSF (Lehtinen et al. 2011), and are a likely signal to telencephalic progenitors at the ventricular surface, presumably via SMAD phosphorylation to influence neuronal or radial glial fates (Li and Grumet 2007). Increased and ectopic activation of BMP signaling in the telencephalon accompanies exencephaly or microcephaly in Nog−/− embryos; therefore, we asked whether BMP signaling was similarly disrupted in the Ranbp1−/− by quantifying the frequency of phospho-SMAD 1/5/8 labeled telencephalic ventricular zone cells. Contrary to predictions, based upon the phenotypically similar Nog−/− mutant, we found diminished frequency of pSMAD-labeled cells in the dorsal pallium of E10.5 Ranbp1−/− embryos (P < 0.01 by T-test; Fig. 3I–K), as well as an increased proportion of pSMAD/SMAD (P < 0.05 by Mann–Whitney U-test; Fig. 3L–N). Apparently, the phenotypic similarity between the Ranbp1 and Noggin null embryos is not due to increased BMP/SMAD signaling. Nevertheless, additional aspects of TGFβ signaling at the apical/ventricular surface, independent of radial glial polarity or cilia-based transduction, are disrupted by Ranbp1 loss of function.
Proliferation of Forebrain Progenitors is Altered by the Loss of Ranbp1
Our analysis of the embryonic Ranbp1−/− forebrain did not reveal dramatic changes in neuroepithelial patterning or polarity; nevertheless, the cortex is visibly smaller at E10.5 (see Fig. 1F) and measurably thinner by E14.5 (see Fig. 1S,T). Ranbp1 modulates mitotic progression and proliferation in cultured cells following over-expression or siRNA depletion (Battistoni et al. 1997; Di Fiore et al. 2003; Ciciarello et al. 2010). Thus, we asked whether reduced cortical thickness is a consequence of disrupted early cortical progenitor proliferation, perhaps due to aberrant M phase kinetics—reflecting essential contributions of Ranbp1 to spindle formation and nuclear envelope assembly/disassembly (Di Fiore et al. 2003; Tedeschi et al. 2007; Ciciarello et al. 2010; Hwang et al. 2011)—in the E10.5 pallial neuroepithelium.
We compared proliferative progenitors in WT and non-exencephalic E10.5 Ranpb1−/− embryos using 3 markers: BrdU, which after a 1-h pulse labels cells in S phase; Ki67, which is expressed at high levels in the nuclei of cells in early M phase, and PH3 to identify cells in M phase. At E10.5, cortical progenitors (radial glia) divide rapidly (Misson et al. 1988; Noctor et al. 2002). We did not see a significant decrease in BrdU incorporation in the Ranpb1−/− pallial neuroepithelium (Fig. 4A–C). In contrast, there was a significant decrease in E10.5 mitotic progenitors based upon diminished frequency of nuclear Ki67 (73% ± 2% SEM of WT, P = 0.0005; Fig. 4D–F) and PH3-labeled cells (83% ± 6% SEM of WT, P = 0.03; Fig. 4G–I). These changes in apparent mitotic efficiency are not accompanied by a detectable increase in cell death, which is equivalent in WT and Ranpb1−/− cortices (Fig. 4J–N). Thus, it is unlikely that cell death accounts for reduced cortical size. Instead, these data indicate that the diminished size of the dorsal pallium in E10.5 Ranbp1−/− embryos reflects disrupted entry and completion of the M phase of the cell cycle of rapidly dividing cortical radial glial progenitors in the E10.5 neuroepithelium.
Figure 4.

Proliferation, but not cell death or early neurogenesis, is disrupted in the Ranbp1−/− telencephalon. (A–I) Proliferation of E10.5 neuroepithelial cells was assessed in WT (+/+) and null (−/−) embryos by assessing BrdU incorporation 1 h after a BrdU injection (A–C), by Ki67 (G2/M phase) expression (D–F), and by PH3 (M phase) expression (G–I). (J–N) Cell death was assessed in E10.5 neuroepithelium (J–K) and E14 cortex (L,M) by assessing cells expressing activated (cleaved) caspase. (O–Q) Early neurogenesis was assessed by quantifying E10.5 neuroepithelial cells expressing the neuronal marker TuJ1. Scale bars = 50 µm, except O,P = 25 µm.
Premature exit from the cell cycle would increase the frequency/density of cortical plate neurons, deplete the pool of proliferative stem and precursor cells, diminish the frequency of M phase progenitors, and reduce overall cortical size and thickness. To determine whether this occurs in Ranpb1−/− embryos, we assessed initial neurogenesis in the forebrain neuroepithelium at E10.5 based upon increased frequency of cells expressing the early neuronal marker TuJ1 (Fig. 4O–Q). This approach has been used to identify premature cell-cycle exit caused by other factors including pharmacological disruption of cell-cycle progression (Calegari and Huttner 2003). We saw no evidence of premature cell-cycle exit or increased neuron frequency in E10.5 Ranbp1−/− embryos. Indeed, we saw a non-significant trend toward reduced frequency of TuJ1 cells (Fig. 4Q). Apparently, reduced cortical size in the Ranbp1 mutant is not due to premature cell-cycle exit and enhanced neurogenesis at E10.5.
Loss of Ranbp1 Disrupts Cortical Progenitor Proliferation
Once cortical neurogenesis begins, apical progenitors in the ventricular zone (VZ) become distinct from basal progenitors in the subventricular zone (SVZ): apical progenitors divide substantially more slowly, whereas basal progenitors divide more rapidly (shortened S/G2/M phase; Arai et al. 2011). We asked whether the expression of Ranbp1 in the E14.5 cortex might vary, identifying particular sub-classes of VZ or SVZ progenitors or perhaps early postmitotic neurons (Gleeson et al. 1999; Englund et al. 2005). Ranbp1 can be seen in apical progenitors in the VZ (labeled by Pax6, Fig. 5A), basal progenitors of the SVZ (labeled by Tbr2, Fig. 5B), and postmitotic neurons (labeled by Dcx, Fig. 5C). Nevertheless, levels of Ranbp1 immunoreactivity vary substantially across the cortical mantle at E14.5 (Fig. 5D). Ranbp1 is attenuated in Pax6-labeled presumed apical progenitors in the VZ (Fig. 5E), robust in Tbr2-labeled presumed basal progenitors in the SVZ (Fig. 5F), and substantially diminished in Dcx-labeled nascent cortical neurons in the IZ and cortical plate (Fig. 5G,H). A similar high level of Ranbp1 expression is noted in the SVZ of prenatal human cortical samples in microarray studies (BrainSpan Atlas of the Developing Human Brain, http://www.brainspan.org/lcm/gene/5870) Accordingly, Ranbp1 is present in and could potentially influence cortical neurogenesis during all stages of development; however, the most robust Ranbp1 labeling is seen in Tbr2-expressing, presumed basal progenitors in the SVZ.
Figure 5.

Neurogenesis is disrupted in the Ranbp1−/− cortex. (A–H) Relative expression of Ranbp1 was assessed in E14.5 cortex by double labeling with markers for distinct progenitor subpopulations, including Pax6-expressing apical progenitors (A), Tbr2-expressing basal progenitors (C), as well as Doublecortin (Dcx)-positive intermediate zone (IZ) and cortical plate (CP) neuronal precursor cells, and immature neurons (C). Ranbp1 is expressed in all cortical layers (D), although high-magnification confocal micrographs indicate that levels are more robust in VZ (E) and SVZ (F), relative to IZ and CP. (I–L) Thickness of the TuJ1-expressing neuronal population is reduced in the Ranbp1 mutant, as measured at 6 evenly distributed locations across the cortical mantle (white lines, I). Quantification of the thickness of the TuJ1 layer shows reduction in Ranbp1−/− cortex (K; P < 0.001 by two-way ANOVA), whereas the thickness of the underlying VZ/SVZ layer is unchanged (L,P = 0.18). (M–O) Quantification of early generated neurons in 3 evenly distributed 150-µm-wide counting boxes (dashed lines, M) shows a reduction in the number of Ctip2-expressing neurons in the Ranbp1−/− cortex (O; P < 0.001 by two-way ANOVA). Scale bar for A–D = 50 µm; 5 µm for E-H, and 200 µm for I,J and M,N. Inset for I,J is ×2 magnification; ×3 magnification for M,N.
Based upon potential activity of Ranbp1 in E14.5 cortical progenitors and early postmitotic neurons, we assessed the distribution of postmitotic neurons in the E14.5 cortex to determine whether neurogenesis or differentiation was disrupted. TuJ1-labeled early differentiating neurons appear substantially diminished in the CP/IZ in Ranbp1−/− embryo (Fig. 5I,J). To confirm this impression, we measured the thickness of the CP/IZ defined by TuJ1 across the entire pallial surface and found that the CP/IZ is significantly thinner in E14.5 Ranbp1−/− cortex (P > 0.001; Fig. 5K). In contrast, the thickness of the VZ/SVZ, defined as the zone where TuJ1 expression is absent or minimal, is not substantially reduced (P = 0.16; Fig. 5L). To more precisely quantify newly generated cortical neurons, we next determined the frequency of Ctip2-labeled cells, an early-generated projection neuron subpopulation found primarily in layers 5/6 and present in sufficient numbers at E14.5 to be accurately quantified (Fig. 5M–O). We found significantly fewer Ctip2-labeled cells across the ventrolateral to anteromedial extent of the E14.5 Ranbp1−/− cortex (P < 0.001; Fig. 5O). Together, these results indicate that, following an initial change in M phase kinetics at E10.5, subsequent cortical neurogenesis is slowed or diminished in Ranpb1−/− mutants.
Ranbp1 is Necessary for Optimal Proliferation of Basal, But Not Apical Progenitors
The apparent disruption of cortical neurogenesis in E14.5 Ranbp1−/− embryos could reflect the combined consequences of a smaller pool of E10.5 radial glial stem cells due to altered M phase kinetics and continued slowed radial glial proliferation, or aberrant basal-progenitor proliferation. Thus, we examined the frequency and proliferative capacity of these 2 distinct cortical progenitor populations in at E14.5. We assessed multiple phases of the cell cycle in Tbr2-negative cells (Tbr2−; presumed radial glia) in the VZ and SVZ and Tbr2-positive cells (Tbr2+; presumed basal progenitors) in the SVZ. Once again, we used multiple markers, including Cyclin D1, whose nuclear expression distinguishes G1, acute BrdU (1 h before collecting fetuses) to identify cells in S-phase, and PH3 to assess M phase.
The frequency of Tbr2− presumed apical progenitors in the VZ/SVZ does not differ between Ranpb1−/− and WT (Fig. 6A–C). Similarly, there is no significant difference in Tbr2+-presumed basal-progenitor cells in the Ranbp1−/− cortex. Both observations are consistent with the lack of significant change in VZ/SVZ thickness in the E14.5 Ranbp1−/− cortex (see Fig. 5G). Nevertheless, cortical progenitor proliferative capacity is selectively disrupted: VZ apical progenitors are spared, and SVZ basal progenitors are compromised. G1 integrity in Ranpb1−/− apical progenitors, based upon frequency of nuclear Cyclin D expression in Tbr2− VZ/SVZ cells, is unaltered (Ranbp1−/−: 85% ± 8% SEM of WT, P = 0.27; Fig. 6D–F). In contrast, the frequency of Cyclin D nuclear labeling is significantly reduced for Ranbp1−/− basal progenitors (73% ± 4% SEM of WT, P = 0.024). We found no significant differences in the number of S-phase apical progenitors, identified as BrdU+/Tbr2− VZ/SVZ cells (94% ± 9% SEM of WT, P = 0.66; Fig. 6F). In contrast, there were significantly fewer BrdU+/Tbr2+ basal progenitors in the Ranbp1−/− SVZ (74% ± 8% SEM of WT, P < 0.006; Fig. 6F, right). Finally, we did not detect a statistically significant change in PH3-labeled Ranbp1−/− apical precursors (84% ± 4% SEM of WT, P = 0.08; Fig. 6J–L); in contrast, there is a substantial, statistically significant change in the number of PH3-labeled mitotic basal Tbr2+ Ranbp1−/− basal progenitors (56% ± 8% SEM of WT, P = 0.04). Apparently several phases of the cell cycle—most dramatically M phase—are disrupted in basal, but not apical E14.5 cortical progenitors.
Figure 6.

Proliferation of basal progenitors is disrupted in the Ranbp1−/− cortex. (A–C) Tbr2-positive basal progenitors are present in similar numbers in both WT (A) and Ranbp1−/− (B) cortex, as assessed in a 150-µm counting box in a medial position (equivalent to Position 2 in Fig. 5I) of the E14.5 cortex, as are non-Tbr2-expressing (DAPI+/Tbr2−) cortical cells (C). Blue overlay in A,B shows nuclear DAPI staining. (D–L) Immunofluorescent double-labeling was used to quantify Tbr2-expressing (Tbr2+) basal progenitors and Tbr2-negative (Tbr2−, primarily apical progenitors) in the VZ/SVZ of E14.5 cortex that express the G1-phase marker Cyclin D1 (Ccnd1, D–F), S phase incorporation of BrdU from a 1-h administration of BrdU (G–I), or express the M phase marker PH3 (J–L). Significance of each independent pair was assessed by Student's t test for the data shown in A–L. (M) Assessment of the cell-cycle phase of proliferating cells was assessed by examining PH3-labeled condensed chromatin in cells with 2 identifiable γ-tubulin-labeled spindle poles; examples of each cell-cycle phase are shown at bottom. No significant difference was observed in cell-cycle phase by Fisher's exact test (P = 0.15, top). (N) No significant difference was observed in the angle of the spindle poles (measured relative to the ventricular surface) for all cells (P = 0.48, n = 46 WT, 43 Ranbp1−/− cells assayed from 3 embryos of each genotype), although a trend was observed when only the small number of anaphase/telophase cells are considered (P = 0.054, n = 5 WT, 4 Ranbp1−/− cells). All scale bars = 25 µm.
The decrease in these 3 measures of mitotic activity in VZ/SVZ progenitors suggests that the loss of Ranbp1 at E14.5 retards cell-cycle progression, especially in M phase, and primarily in rapidly dividing basal progenitors. We therefore asked whether additional aspects of mitosis might be disrupted in Ranbp1−/− mitotic progenitors. Previous reports suggest that Ranbp1 is required for optimal chromosome segregation (Guarguaglini et al. 2000; Tedeschi et al. 2007; Hwang et al. 2011). Accordingly, we examined mitotic cells for spindle pole formation and apparent mitotic cleavage plane orientation in mitotic figures in the cortical VZ/SVZ (Fig. 6M,N). By examining PH3-labeled cells with identifiable spindle poles (labeled with γ-tubulin) with high-resolution confocal microscopy, we identified cells in each stage of mitosis (metaphase, anaphase, telophase; Fig. 6M, bottom). We found no statistically significant difference in the frequency of each mitotic phase among the PH3-labeled VZ/SVZ mitotic population (P = 0.14 by Fisher's Exact test), and we saw no evidence of any chromosomal anomalies (e.g., lagging chromosomes or cytokinesis asymmetries) in any examined cell, although relatively few anaphase and telophase cells were present in either WT or Ranbp1−/− cortical samples. Furthermore, we saw no significant difference in the mitotic cleavage angle of the apical population (Fig. 6N) as distinguished by the angle of the spindle poles relative to the ventricular surface. Thus, major disruptions in mitotic spindle formation or chromosome segregation do not accompany the change in proliferative activity in Ranbp1 VZ/SVZ precursors.
Homozygous deletion of Ranbp1 shares a key phenotype with the LgDel mouse model of 22q11.2 DS: disrupted basal-progenitor proliferation (Meechan et al. 2009). Thus, we asked whether heterozygous deletion of Ranbp1 disrupts basal-progenitor proliferation. We found no significant difference in basal-progenitor proliferation (BrdU+/Tbr2+ cells) in E14.5 Ranbp1+/− cortex (95% ± 6% SEM of WT of WT, n = 14, P = 0.54; data not shown). Thus, diminished dosage of Ranbp1 is apparently not the sole contributor to disrupted basal-progenitor proliferation and related phenotypes in the LgDel mouse, which models the minimal (1.5 Mb) 22q11.2 deletion (Carlson et al. 1997; Michaelovsky et al. 2012). Heterozygous deletion of Ranbp1 in the context of broader 22q11.2 deletion likely acts in concert with diminished dosage of other genes within and beyond the minimal critical 22q11.2 deleted region to modify basal-progenitor proliferation in the LgDel mouse model of 22q11.2 DS (Meechan et al. 2009).
Loss of Ranbp1 Disrupts Exit of Basal Progenitor Progeny from the Subventricular Zone
To determine the significance of reduced basal-progenitor proliferation in the E14.5 Ranbp1−/− cortex, we examined their presumed early neural progeny labeled by BrdU on E13.5, 16 h prior to collecting embryos. We quantified 2 distinct populations, BrdU+/Tbr2+ cells in the SVZ and presumed postmitotic neurons (BrdU+/Tbr2−) in the SVZ/IZ (Fig. 7A,B). We found a significant increase in BrdU+/Tbr2+ cells in the Ranpb1−/− SVZ (145% of WT, P = 0.023; Fig. 7C, left), in sharp contrast to decreased Tbr2+/BrdU+ SVZ following a 1 h of BrdU pulse (see Fig. 6G–I). In addition, fewer likely postmitotic neurons (Tbr2−/BrdU+ cells in the IZ) are generated (67% of WT, P = 0.013; Fig. 7C, right). Ranpb1 loss of function does not appear to cause early cell-cycle exit; instead, it likely prolongs cell division, perhaps through retarding M phase, and thereby diminishes cortical neurogenesis. We also asked whether selective disruption of proliferative kinetics in basal progenitors influences migration of postmitotic neuroblasts into the cortical plate in Ranpb1−/− embryos. In E14.5 WT cortex, newly born neurons (BrdU+/Tbr2− after 16 h of BrdU) are seen primarily in the IZ; few Tbr2+/BrdU+ cells remain in the SVZ (Fig. 7A, right). In contrast, newly born neurons in the E14.5 Ranbp1−/− cortex are clustered along the upper boundary of the SVZ (Fig. 7B). Apparently, altered proliferative kinetics in the Ranbp1−/− cortex compromises migratory capacity, and perhaps acquisition of neuronal characteristics (suggested by maintenance of Tbr2 expression) in postmitotic, presumed cortical neurons.
Figure 7.

Genesis of upper-layer neurons is disrupted in the Ranbp1−/− cortex. (A–C) Fate of proliferative cells was determined by harvesting embryos at E14.5, 16 h after a maternal injection of BrdU. Distribution of single-labeled BrdU+/Tbr2− (red) and double-labeled BrdU+/Tbr2+ (yellow) cells is illustrated on the right side of the WT (A) and Ranbp1−/− (B) panels, as established by measuring and plotting the laminar (Y-axis) position of each single- and double-labeled cell from 4 embryos of each genotype. (C) Quantification of double-labeled (Tbr2+/BrdU+) and single-labeled (Tbr2−/BrdU+) populations in cortex 16 h after BrdU injection. (D–L) Upper layer neurons are preferentially disrupted in the Ranbp1−/− cortex, as assessed in E17.5 cortex, following BrdU injection at E14.5. (D–F) Assessment of Satb2 (green) and Ctip2 (red) in E17.5 WT (D) and Ranbp1−/− (E) cortex. (F) Quantification of upper-layer Satb2+ cells (above the Ctip2-expressing cells) and lower-layer Ctip2+ cells as assessed in a 150-µm counting box in a medial position of the cortex. (G–I) Quantification of Cux1+ cells (green) and Ctip2+ (red) cells, assessed using similar methodology to R–T. (J–L) Assessment of fate of E14.5 BrdU-labeled neuronal cells in WT (J) and Ranbp1−/− (K) cortex at E17.5. Tissue was triple-labeled for upper-layer (Cux1, green) and lower-layer neurons (Ctip2, blue), as well as for BrdU (red). For clarity, each panel is separated to show Ctip2/BrdU labeling (J,K) on the left and Cux1/BrdU labeling (J′,K′) on the right. (L) Fate of the E14.5 BrdU-labeled cohort was quantified by counting strongly labeled BrdU+ cells coexpressing either Ctip2 or Cux1. Significance of each independent pair was assessed by Student's t test. All scale bars = 25 µm.
Reduced Basal-Progenitor Proliferation Leads to Loss of Layer 2/3 Projection Neurons
The disruption of cortical progenitor proliferation in Ranbp1−/− mutants suggests several possible scenarios for subsequent cortical differentiation. Disruption of radial glial/neuroepithelial stem cell division at E10.5 might yield a smaller cortex where the relative thickness of lamina and overall organization does not differ substantially from WT. Alternately, the change in cortical size may reflect a combination of fewer radial glia due to E10.5 disruption, followed by disrupted genesis of cortical neurons in all layers, due to slowed proliferation of basal progenitors. If this was the case, and if basal progenitors contribute substantially to all layers, there would be a proportionate loss of all laminar cohorts across a thinner cortex. Finally, if basal progenitors are biased toward genesis of laminar subsets, there should be a disproportionate reduction of specific laminar subpopulations. To resolve these possibilities, we evaluated projection neuron identities and laminar organization in the cortex of E17.5 Ranbp1−/− and WT fetuses.
In the E17.5 Ranbp1−/− cortex, we found reduced frequency of Satb2-labeled cells (56% of WT; P = 0.01; Fig. 7D–F) thought to represent commissural neurons (Alcamo et al. 2008) found predominantly (but not exclusively) in layer 2/3, and Cux1-labeled cells (70% of WT, P = 0.02, Fig. 7G–I) that include a broader population of layer 2/3 projection neurons (Cubelos et al. 2010). In contrast, layer 5/6 projection neuron frequency, identified by Ctip2, is not significantly reduced in the E17.5 Ranpb1−/− cortex (Fig. 7D–F). Apparently, E10.5 as well as E14.5 proliferative changes result in relative preservation of the layer 5/6 cohort of projection neurons at the expense of layer 2/3 projection neurons. To confirm this impression, we evaluated E14.5-generated layer 5/6 versus layer 2/3 neurons. We found no change in the frequency of E14.5 BrdU/Ctip2-labeled layer 5/6 neurons in E17.5 WT versus Ranpb1−/− cortex; however, there was a substantial decrease in BrdU/Cux1-labeled layer 2/3 neurons (Fig. 7J–L). Thus, even at the midpoint of cortical neurogenesis, when both infra and supragranular layer neurons are generated, layer 5/6 projection neurons are produced at normal levels whereas layer 2/3 projection neurons decline in concert with altered basal-progenitor proliferation caused by Ranbp1 loss of function. Accordingly, the regulation of rapid basal-progenitor division, apparently during M phase by Ranbp1, at least from E14.5 onward, is critical for the genesis of layer 2/3 neurons.
Discussion
Ranbp1, the mouse orthologue of 1 of 32 protein-encoding genes in the human 22q11.2 DS minimal critical deleted region, has essential functions in craniofacial, eye and cortical development. Exencephaly in 61–70% of Ranbp1−/− embryos suggests that Ranbp1 is crucial for early cranial/anterior neural tube morphogenesis, perhaps in the context of additional polymorphic loci, maternal, or environmental factors. Ranbp1−/− embryos without exencephaly have milder craniofacial and ocular anomalies, and their brains—especially their cerebral cortices—are smaller. We focused on cortical development because candidate 22q11.2 genes for cortical differentiation phenotypes in the LgDel as well as Df1 mouse models of 22q11.2 DS (Meechan et al. 2009, 2012, 2015; Fenelon et al. 2013; Ellegood et al. 2014) have not yet been identified. Ranbp1 loss-of-function alters cortical growth by first compromising early neuroepithelial/radial glial progenitors as they divide rapidly to establish the rudimentary dorsal pallium. Subsequently, Ranbp1 loss-of-function disrupts proliferation of rapidly dividing basal progenitors, while sparing more slowly dividing apical/radial glial progenitors. This results in reduced generation of layer 2/3 projection neurons, without significant changes in layer 5/6 projection neurons. Throughout cortical development, M phase of the cell cycle seems to be most significantly modified by Ranpb1 loss-of-function. The diminished layer 2/3 projection neuron phenotype in Ranbp1−/− embryos parallels decreased layer 2/3 projection neuron frequency in the 22q11.2 DS LgDel mouse model (Meechan et al. 2009, 2015). We suggest that diminished dosage of Ranbp1, in concert with that of other 22q11.2 genes, contributes to this phenotype, and thus to disruption of cortical circuit development that compromises behavior due to 22q11.2 deletion.
A Novel Mouse Mutant for Studying Ranbp1 Function
We generated a new loss-of-function allele for Ranbp1 that disrupts the second exon of the Ranbp1 locus and does not yield detectable amounts of Ranbp1 mRNA or Ranbp1 protein. Expression of adjacent genes in the 22q11.2 orthologous region, including Trmt2a (Htf9c) that shares a bidirectional promoter with Ranbp1, is not disrupted by this Ranbp1 mutation. The core phenotypes in our Ranbp1 mutant have remained stable after numerous outcrosses to commercially obtained C57Bl6-N stock (outcrossed at least 8 generations by the vendor). Thus, it is unlikely that independent segregation of any unlinked modifier gene accounts for the core Ranbp1 phenotypes. A brief report using a different targeted Ranbp1 mutation (Nagai et al. 2011) on a mixed background (hybrid C57Bl6:S129 strain) identified a small number (25% of the expected Mendelian ratio) of homozygous mutants that survive birth and into maturity. These mutant mice are small, apparently microcephalic, and infertile. Analysis of mutant embryos or fetuses—presumably including the 75% of Ranbp1−/− that are not recovered live—is not reported for this line. We did not recover viable postnatal mice across multiple litters from our Ranbp1−/− line. Thus, the Ranbp1 mutant allele reported by Nagai et al. may be modified by other factors—most likely the mixed background used to generate the mutants for analysis. In addition, selective husbandry—culling of WT littermates shortly after birth to stabilize the small Ranbp1−/− pups, or the use of foster mothers—may facilitate survival and study of a small proportion of mutant mice that are born live. Sensitivity to background strain has been observed for several other mutations associated with partially penetrant exencephaly, including Noggin (McMahon et al. 1998) and Gli3 (Theil et al. 1999). In these cases background strain variation modulates the frequency of exencephaly and modestly modulates the severity of other phenotypes. Thus the discrepancy in postnatal survival of the 2 Ranbp1 alleles likely reflects differences in background strain, as well as focus on prenatal development in our study versus postnatal viability in the previous report.
Ranbp1 is A Novel Microcephaly Gene
Ranbp1 loss of function in non-exencephalic mutant embryos results in a significantly smaller head, forebrain and cerebral cortex. Ranbp1 regulation of brain size may contribute to a fairly penetrant phenotype in 22q11.2 DS: reduced brain volume in up to 50% of 22q11.2 DS patients (Bearden et al. 2007; Jalbrzikowski et al. 2013). It is unlikely that Ranbp1 alone accounts for this 22q11.2 DS phenotype; Ranbp1+/− embryos do not have noticeably smaller heads or brains. Instead, Ranbp1 may have dosage-dependent interactions with other cell-cycle regulators in the minimal critical region that are expressed in the VZ/SVZ during cortical neurogenesis including Hira, Cdc45l, and Trmt2a (Maynard et al. 2003; Meechan et al. 2006; Meechan et al. 2009). In addition there may be an additive effect for diminished dosage of Ranbp1 and mitochondrial 22q11.2 genes that subsequently influence neuronal growth (Maynard et al. 2008). One additional 22q11.2 DS gene—Tbx1—that contributes to cardiovascular dysmorphogenesis and cranial nerve anomalies in 22q11.2 DS (Merscher et al. 2001; Karpinski et al. 2014) is not likely a direct contributor to microcephaly due to altered cortical precursor proliferation. It is not expressed at significant levels in the VZ/SVZ (Meechan et al. 2009), and cortical phenotypes have not been noted in the extensive literature on Tbx1-mediated cardiac and craniofacial development. Additional genetic modifiers may influence the severity of the Ranbp1 null cortical phenotype, or its contribution to related 22q11.2 deletion phenotypes. Interaction with other non-genetic factors is also possible. Ranbp1 expression can be enhanced in response to NMDA receptor signaling (Chao et al. 2012). In the context of 22q11.2 deletion, diminished dosage of Ranbp1 may alter normal NMDA receptor-mediated responses, or exacerbate the consequences of another mutation that disrupts NMDA signaling.
Most primary microcephaly disorders for which single genes have been identified reflect disruption of the centriole, centrosome, and spindle apparatus (Megraw et al. 2011; Gilmore and Walsh 2013). Our data, and previously published observations in cell culture (Guarguaglini et al. 2000; Di Fiore et al. 2003; Tedeschi et al. 2007), implicate Ranbp1 in M phase regulation; however, its specific role is not yet established. If Ranbp1 mediates rapid disassembly and reassembly of the nuclear envelope during mitosis (Ciciarello et al. 2010; Hwang et al. 2011), then loss of function may prolong the time necessary to divide and reestablish the nucleus. Such changes may secondarily compromise centriole integrity, cell polarity, spindle assembly, and disassembly. We note, however, that we did not find a significant change in number or distribution of primary cilia—anchored by the centriole—at the cortical ventricular surface in Ranpb1−/− mutants, nor did we find any substantial disruption of cleavage orientation or chromosome segregation. Ranbp1, therefore, is a novel microcephaly gene that likely disrupts mitotic capacity via its influence on nuclear envelope assembly/ disassembly and may act in concert with other 22q11.2 genes to cause diminished brain size in 22q11.2 DS patients.
Ranpb1 Modulates Rapidly Dividing Cortical Progenitors
We found that Ranbp1 does not act selectively in a specific cortical precursor class, but rather in cortical precursors that are in a specific state: rapid cell division/M-phase. Thus, as neuroepithelial/radial glial progenitors shift from rapid to slower rates of division between E10.5 and E14.5 (Caviness et al. 1999), and as rapidly dividing (shortened S/G2/M) basal progenitors emerge by E14.5 (Arai et al. 2011), there is a shift in precursor populations compromised by Ranpb1 loss of function from apical to basal progenitors. The most severe proliferative deficit for E10.5 apical and E14.5 basal progenitors is seen for M phase, consistent with the suggested function of Ranbp1 in regulation of mitosis. Basal progenitors also showed significant reductions in G1 and S phase; however, this may be a consequence of slowed reentry of daughter cells into the cell cycle due to M phase changes. This is consistent with reports suggesting Ranbp1 modulates other aspects of cell-cycle progression, including entry into S phase (Battistoni et al. 1997; Guarguaglini et al. 2000; Ciciarello et al. 2010). Together, these data indicate that Ranbp1 function is associated with the state of rapid proliferation and division—especially the kinetics of M phase—rather than a particular cortical precursor class.
Many previously described genetic forms of microcephaly have been associated with specific disruption of apical progenitor/radial glial division, which and can result in precocious neurogenesis at the expense of the apical progenitor pool, thus limiting cortical expansion (Chae and Walsh 2007). We did not find premature neurogenesis in E10.5 Ranbp1−/− mutants. In addition, there is no evidence of enhanced cell death in the E10.5 Ranpb1−/− cortex. The altered proportions of pSMAD versus SMAD VZ cells suggest that E10.5 Ranbp1−/−apical precursors retain signaling responses characteristic of neurogenic radial glial progenitors (Li and Grumet 2007), but perhaps activate signaling more slowly. These changes may reflect the central role of Ranbp1 in modulating nuclear export (Fung and Chook 2014). Signaling via Bmp, Shh, and Wnt family members relies at least in part on regulated nuclear import/export of activator proteins, and our data indicates that at least 1 of these pathways, that via SMAD1/5/8, is altered in the Ranbp1−/− cortex. In addition, miRNA processing relies on facilitated nuclear import/export (Iwasaki et al. 2013). It is intriguing to speculate that Ranbp1 interacts with another 22q11.2 gene, Dgcr8, which regulates miRNA processing and apparently contributes to cortical phenotypes in 22q11.2 DS mouse models (Fenelon et al. 2011, 2013; Chun et al. 2014). The available evidence, however, suggests that Dgcr8 does not likely compromise basal-progenitor proliferation in the Ranbp1−/−or LgDel cortex. Our preliminary observations suggest that Dgcr8 protein is undetectable by immunofluorescent staining in the VZ/SVZ (data not shown), similar to its mRNA expression profile in prenatal human cortex (BrainSpan, http://www.brainspan.org/lcm/gene/33778); however, Dgcr8 is detected in the cortical plate. Thus, in the context of 22q11.2 deletion, diminished dosage of Ranbp1 likely modifies basal progenitors independent of Dgcr8. Key interactions between Ranbp1 and other 22q11.2 genes remain to be defined.
Ranpb1 Regulates Genesis of Layer 2/3 Projection Neurons
Ranbp1 is apparently essential for the genesis of appropriate numbers of layer 2/3 cortical projection neurons. Diminished frequency of layer 2/3 projection neurons, without similar changes in layer 5/6, is the most quantitatively substantial cortical phenotype in the Ranpb1−/− fetus. This loss of upper-layer neurons may reflect the reduced proliferation of basal progenitors. It has been suggested that this population, at least from E14.5 onward, primarily produces layer 2/3 cells (Tarabykin et al. 2001; Cubelos et al. 2008). We found that Ranbp1 loss of function biases cortical neurogenesis toward production of layer 5/6 cells, either at the expense of cells that would otherwise be specified as layer 2/3 projection neurons, or because layer 5/6 neurons are generated from more slowly dividing progenitors. Several mechanisms might account for this change. First, the genesis of layer 2/3 versus layer 5/6 neurons might depend upon numbers of apical or basal progenitor divisions. If this is the case, the early slowing of apical progenitors, followed by slow basal progenitors in Ranbp1−/− embryos would yield cortical progenitors capable of generating layer 5/6 cells at a time when layer 2/3 cells would normally be produced. Second, the altered proliferative kinetics of basal progenitors, which may be biased (but not specified) to produce layer 2/3 neurons may selectively prevent postmitotic progeny from responding to key signals that establish or maintain layer 2/3 identities; therefore, daughter cells acquire or retain anomalous layer 5/6 identities. Finally, the failure of cell migration in the Ranpb1−/− cortex may disrupt essential cell–cell interactions facilitated by normal cortical neuronal migration, leading to retention or inappropriate acquisition of layer 5/6 identities by later generated neurons that arise from proliferatively retarded basal progenitors.
Ranbp1 loss of function may selectively disrupt molecularly distinct subsets of apical and basal progenitors with characteristic proliferative properties due to variation in Ranbp1 levels in subsets of VZ/SVZ cells. Recent studies suggest significant molecular heterogeneity within apical and basal-progenitor classes. Accordingly, the apparent selectivity of Ranbp1 may be due to its relative activity, based upon expression level or post-translational modification state, in subsets of Cux2-expressing radial glia that may be biased to produce layer 2/3 neurons (Franco et al. 2012; Franco and Muller 2013). Similarly, subsets of apical progenitors that express high levels of Fezf2 may also preferentially generate upper-layer neurons (Guo et al. 2013). Ranbp1 loss of function might specifically compromise these apical cells thereby further amplifying disrupted upper-layer neurogenesis. Alternatively, loss of Ranbp1 activity may have selective effects due to differences in cell-cycle kinetics or other molecular differences independent of characteristic molecular and cell-fate distinctions between apical and basal progenitors. Further experiments using conditional approaches to inactivate Ranbp1 in specific apical and basal subpopulations, or in precursors in distinct proliferative states, would clarify this issue.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/.
Funding
This work was supported by a National Institute of Child Health and Human Development grant (HD042182 to A.S.L.), and by a Simons Foundation grant (SFARI 306796) to A.S.L. Confocal microscopy was supported by National Institutes of Health grants from the National Center for Research Resources (S10RR025565) and National Institute of Child Health and Human Development (P30HD40677).
Notes
Conflict of Interest: None declared.
Supplementary Material
References
- Alcamo EA, Chirivella L, Dautzenberg M, Dobreva G, Farinas I, Grosschedl R, McConnell SK. 2008. Satb2 regulates callosal projection neuron identity in the developing cerebral cortex. Neuron. 57:364–377. [DOI] [PubMed] [Google Scholar]
- Arai Y, Pulvers JN, Haffner C, Schilling B, Nusslein I, Calegari F, Huttner WB. 2011. Neural stem and progenitor cells shorten S-phase on commitment to neuron production. Nat Commun. 2:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Chaddock CA, Baldeweg T, Skuse D. 2011. Neuroanatomy in adolescents and young adults with 22q11 deletion syndrome: comparison to an IQ-matched group. Neuroimage. 55:491–499. [DOI] [PubMed] [Google Scholar]
- Baker K, Vorstman JA. 2012. Is there a core neuropsychiatric phenotype in 22q11.2 deletion syndrome? Curr Opin Neurol. 25:131–137. [DOI] [PubMed] [Google Scholar]
- Battistoni A, Guarguaglini G, Degrassi F, Pittoggi C, Palena A, Di Matteo G, Pisano C, Cundari E, Lavia P. 1997. Deregulated expression of the RanBP1 gene alters cell cycle progression in murine fibroblasts. J Cell Sci. 110(Pt 19):2345–2357. [DOI] [PubMed] [Google Scholar]
- Bay SN, Caspary T. 2012. What are those cilia doing in the neural tube? Cilia. 1:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden CE, van Erp TG, Dutton RA, Tran H, Zimmermann L, Sun D, Geaga JA, Simon TJ, Glahn DC, Cannon TD, et al. 2007. Mapping cortical thickness in children with 22q11.2 deletions. Cereb Cortex. 17:1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhasin N, Maynard TM, Gallagher PA, LaMantia AS. 2003. Mesenchymal/epithelial regulation of retinoic acid signaling in the olfactory placode. Dev Biol (NY 1985). 261:82–98. [DOI] [PubMed] [Google Scholar]
- Calegari F, Huttner WB. 2003. An inhibition of cyclin-dependent kinases that lengthens, but does not arrest, neuroepithelial cell cycle induces premature neurogenesis. J Cell Sci. 116:4947–4955. [DOI] [PubMed] [Google Scholar]
- Carlson C, Sirotkin H, Pandita R, Goldberg R, McKie J, Wadey R, Patanjali SR, Weissman SM, Anyane-Yeboa K, Warburton D, et al. 1997. Molecular definition of 22q11 deletions in 151 velo-cardio-facial syndrome patients. Am J Hum Genet. 61:620–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. 2007. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 12:767–778. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T, Nowakowski RS. 1999. The G1 restriction point as critical regulator of neocortical neuronogenesis. Neurochem Res. 24:497–506. [DOI] [PubMed] [Google Scholar]
- Chae TH, Walsh CA. 2007. Genes that control the size of the cerebral cortex. Novartis Found Symp. 288:79–90. Discussion 91–78. [DOI] [PubMed] [Google Scholar]
- Chao HW, Lai YT, Lu YL, Lin CL, Mai W, Huang YS. 2012. NMDAR signaling facilitates the IPO5-mediated nuclear import of CPEB3. Nucleic Acids Res. 40:8484–8498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WV, Soriano P. 2003. Gene trap mutagenesis in embryonic stem cells. Methods Enzymol. 365:367–386. [PubMed] [Google Scholar]
- Chenn A, Walsh CA. 2002. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 297:365–369. [DOI] [PubMed] [Google Scholar]
- Chun S, Westmoreland JJ, Bayazitov IT, Eddins D, Pani AK, Smeyne RJ, Yu J, Blundon JA, Zakharenko SS. 2014. Specific disruption of thalamic inputs to the auditory cortex in schizophrenia models. Science. 344:1178–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciarello M, Roscioli E, Di Fiore B, Di Francesco L, Sobrero F, Bernard D, Mangiacasale R, Harel A, Schinina ME, Lavia P. 2010. Nuclear reformation after mitosis requires downregulation of the Ran GTPase effector RanBP1 in mammalian cells. Chromosoma. 119:651–668. [DOI] [PubMed] [Google Scholar]
- Cubelos B, Sebastian-Serrano A, Beccari L, Calcagnotto ME, Cisneros E, Kim S, Dopazo A, Alvarez-Dolado M, Redondo JM, Bovolenta P, et al. 2010. Cux1 and Cux2 regulate dendritic branching, spine morphology, and synapses of the upper layer neurons of the cortex. Neuron. 66:523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubelos B, Sebastian-Serrano A, Kim S, Moreno-Ortiz C, Redondo JM, Walsh CA, Nieto M. 2008. Cux-2 controls the proliferation of neuronal intermediate precursors of the cortical subventricular zone. Cereb Cortex. 18:1758–1770. [DOI] [PubMed] [Google Scholar]
- Di Fiore B, Ciciarello M, Mangiacasale R, Palena A, Tassin AM, Cundari E, Lavia P. 2003. Mammalian RanBP1 regulates centrosome cohesion during mitosis. J Cell Sci. 116:3399–3411. [DOI] [PubMed] [Google Scholar]
- Ellegood J, Markx S, Lerch JP, Steadman PE, Genc C, Provenzano F, Kushner SA, Henkelman RM, Karayiorgou M, Gogos JA. 2014. Neuroanatomical phenotypes in a mouse model of the 22q11.2 microdeletion. Mol Psychiatry. 19:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. 2005. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 25:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evsyukova I, Plestant C, Anton ES. 2013. Integrative mechanisms of oriented neuronal migration in the developing brain. Annu Rev Cell Dev Biol. 29:299–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan S, Whiteman EL, Hurd TW, McIntyre JC, Dishinger JF, Liu CJ, Martens JR, Verhey KJ, Sajjan U, Margolis B. 2011. Induction of ran GTP drives ciliogenesis. Mol Biol Cell. 22:4539–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenelon K, Mukai J, Xu B, Hsu PK, Drew LJ, Karayiorgou M, Fischbach GD, Macdermott AB, Gogos JA. 2011. Deficiency of Dgcr8, a gene disrupted by the 22q11.2 microdeletion, results in altered short-term plasticity in the prefrontal cortex. Proc Natl Acad Sci USA. 108:4447–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenelon K, Xu B, Lai CS, Mukai J, Markx S, Stark KL, Hsu PK, Gan WB, Fischbach GD, MacDermott AB, et al. 2013. The pattern of cortical dysfunction in a mouse model of a schizophrenia-related microdeletion. J Neurosci. 33:14825–14839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng XH, Derynck R. 2005. Specificity and versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev Biol. 21:659–693. [DOI] [PubMed] [Google Scholar]
- Fine SE, Weissman A, Gerdes M, Pinto-Martin J, Zackai EH, McDonald-McGinn DM, Emanuel BS. 2005. Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J Autism Dev Disord. 35:461–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Gil-Sanz C, Martinez-Garay I, Espinosa A, Harkins-Perry SR, Ramos C, Muller U. 2012. Fate-restricted neural progenitors in the mammalian cerebral cortex. Science. 337:746–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco SJ, Muller U. 2013. Shaping our minds: stem and progenitor cell diversity in the mammalian neocortex. Neuron. 77:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas N, Cunha C. 2009. Mechanisms and signals for the nuclear import of proteins. Curr Genomics. 10:550–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung HY, Chook YM. 2014. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol. 27:52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore EC, Walsh CA. 2013. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip Rev Develop Biol. 2:461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA. 1999. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 23:257–271. [DOI] [PubMed] [Google Scholar]
- Gross RE, Mehler MF, Mabie PC, Zang Z, Santschi L, Kessler JA. 1996. Bone morphogenetic proteins promote astroglial lineage commitment by mammalian subventricular zone progenitor cells. Neuron. 17:595–606. [DOI] [PubMed] [Google Scholar]
- Guarguaglini G, Renzi L, D'Ottavio F, Di Fiore B, Casenghi M, Cundari E, Lavia P. 2000. Regulated ran-binding protein 1 activity is required for organization and function of the mitotic spindle in mammalian cells in vivo. Cell Growth Differ. 11:455–465. [PubMed] [Google Scholar]
- Guo C, Eckler MJ, McKenna WL, McKinsey GL, Rubenstein JL, Chen B. 2013. Fezf2 expression identifies a multipotent progenitor for neocortical projection neurons, astrocytes, and oligodendrocytes. Neuron. 80:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham H, Guo J, Yokota Y, Umberger NL, Su CY, Li J, Verma N, Hirt J, Ghukasyan V, Caspary T, et al. 2013. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat Neurosci. 16:1000–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HI, Ji JH, Jang YJ. 2011. Phosphorylation of Ran-binding protein-1 by Polo-like kinase-1 is required for interaction with Ran and early mitotic progression. J Biol Chem. 286:33012–33020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki YW, Kiga K, Kayo H, Fukuda-Yuzawa Y, Weise J, Inada T, Tomita M, Ishihama Y, Fukao T. 2013. Global microRNA elevation by inducible Exportin 5 regulates cell cycle entry. RNA. 19:490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalbrzikowski M, Jonas R, Senturk D, Patel A, Chow C, Green MF, Bearden CE. 2013. Structural abnormalities in cortical volume, thickness, and surface area in 22q11.2 microdeletion syndrome: relationship with psychotic symptoms. NeuroImage Clin. 3:405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinski BA, Maynard TM, Fralish MS, Nuwayhid S, Zohn I, Moody SA, Lamantia AS. 2014. Dysphagia and disrupted cranial nerve development in a mouse model of DiGeorge/22q11 deletion syndrome. Dis Models Mech. 7:245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehlenbach RH, Assheuer R, Kehlenbach A, Becker J, Gerace L. 2001. Stimulation of nuclear export and inhibition of nuclear import by a Ran mutant deficient in binding to Ran-binding protein 1. J Biol Chem. 276:14524–14531. [DOI] [PubMed] [Google Scholar]
- Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ. 1996. The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev. 10:60–69. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. 2009. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 32:149–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Sestan N, Anton ES. 2012. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development. 139:1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMantia AS. 1999. Forebrain induction, retinoic acid, and vulnerability to schizophrenia: insights from molecular and genetic analysis in developing mice. Biol Psychiatry. 46:19–30. [DOI] [PubMed] [Google Scholar]
- LaMantia AS, Bhasin N, Rhodes K, Heemskerk J. 2000. Mesenchymal/epithelial induction mediates olfactory pathway formation. Neuron. 28:411–425. [DOI] [PubMed] [Google Scholar]
- LaMantia AS, Colbert MC, Linney E. 1993. Retinoic acid induction and regional differentiation prefigure olfactory pathway formation in the mammalian forebrain. Neuron. 10:1035–1048. [DOI] [PubMed] [Google Scholar]
- Lehtinen MK, Zappaterra MW, Chen X, Yang YJ, Hill AD, Lun M, Maynard T, Gonzalez D, Kim S, Ye P, et al. 2011. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron. 69:893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Grumet M. 2007. BMP and LIF signaling coordinately regulate lineage restriction of radial glia in the developing forebrain. Glia. 55:24–35. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Appolloni I, Calzolari F. 2008. Radial glia and neural stem cells. Cell Tissue Res. 331:165–178. [DOI] [PubMed] [Google Scholar]
- Maynard TM, Gopalakrishna D, Meechan DW, Paronett EM, Newbern JM, LaMantia AS. 2013. 22q11 Gene dosage establishes an adaptive range for sonic hedgehog and retinoic acid signaling during early development. Hum Mol Genet. 22:300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Haskell GT, Bhasin N, Lee JM, Gassman AA, Lieberman JA, LaMantia AS. 2002. RanBP1, a velocardiofacial/DiGeorge syndrome candidate gene, is expressed at sites of mesenchymal/epithelial induction. Mech Dev. 111:177–180. [DOI] [PubMed] [Google Scholar]
- Maynard TM, Haskell GT, Peters AZ, Sikich L, Lieberman JA, LaMantia AS. 2003. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proc Natl Acad Sci USA. 100:14433–14438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard TM, Meechan DW, Dudevoir ML, Gopalakrishna D, Peters AZ, Heindel CC, Sugimoto TJ, Wu Y, Lieberman JA, Lamantia AS. 2008. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol Cell Neurosci. 39:439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon JA, Takada S, Zimmerman LB, Fan CM, Harland RM, McMahon AP. 1998. Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 12:1438–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Maynard TM, Wu Y, Gopalakrishna D, Lieberman JA, LaMantia AS. 2006. Gene dosage in the developing and adult brain in a mouse model of 22q11 deletion syndrome. Mol Cell Neurosci. 33:412–428. [DOI] [PubMed] [Google Scholar]
- Meechan DW, Rutz HL, Fralish MS, Maynard TM, Rothblat LA, Lamantia AS. 2015. Cognitive ability is associated with altered medial frontal cortical circuits in the LgDel mouse model of 22q11.2DS. Cereb Cortex. 25:1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, LaMantia AS. 2012. Cxcr4 regulation of interneuron migration is disrupted in 22q11.2 deletion syndrome. Proc Natl Acad Sci USA. 109:18601–18606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meechan DW, Tucker ES, Maynard TM, LaMantia AS. 2009. Diminished dosage of 22q11 genes disrupts neurogenesis and cortical development in a mouse model of 22q11 deletion/DiGeorge syndrome. Proc Natl Acad Sci USA. 106:16434–16445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megraw TL, Sharkey JT, Nowakowski RS. 2011. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 21:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehler MF, Mabie PC, Zhu G, Gokhan S, Kessler JA. 2000. Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Dev Neurosci. 22:74–85. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. 2001. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 104:619–629. [DOI] [PubMed] [Google Scholar]
- Michaelovsky E, Frisch A, Carmel M, Patya M, Zarchi O, Green T, Basel-Vanagaite L, Weizman A, Gothelf D. 2012. Genotype-phenotype correlation in 22q11.2 deletion syndrome. BMC Med Genetics. 13:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misson JP, Edwards MA, Yamamoto M, Caviness VS., Jr 1988. Mitotic cycling of radial glial cells of the fetal murine cerebral wall: a combined autoradiographic and immunohistochemical study. Brain Res. 466:183–190. [DOI] [PubMed] [Google Scholar]
- Nagai M, Moriyama T, Mehmood R, Tokuhiro K, Ikawa M, Okabe M, Tanaka H, Yoneda Y. 2011. Mice lacking Ran binding protein 1 are viable and show male infertility. FEBS Lett. 585:791–796. [DOI] [PubMed] [Google Scholar]
- Nicolas FJ, De Bosscher K, Schmierer B, Hill CS. 2004. Analysis of Smad nucleocytoplasmic shuttling in living cells. J Cell Sci. 117:4113–4125. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, Kriegstein AR. 2002. Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci. 22:3161–3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary DD, Chou SJ, Sahara S. 2007. Area patterning of the mammalian cortex. Neuron. 56:252–269. [DOI] [PubMed] [Google Scholar]
- Philip N, Bassett A. 2011. Cognitive, behavioural and psychiatric phenotype in 22q11.2 deletion syndrome. Behav Genet. 41:403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plafker K, Macara IG. 2000. Facilitated nucleocytoplasmic shuttling of the Ran binding protein RanBP1. Mol Cell Biol. 20:3510–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards SA, Lounsbury KM, Carey KL, Macara IG. 1996. A nuclear export signal is essential for the cytosolic localization of the Ran binding protein, RanBP1. J Cell Biol. 134:1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scambler PJ. 2000. The 22q11 deletion syndromes. Hum Mol Genet. 9:2421–2426. [DOI] [PubMed] [Google Scholar]
- Schaer M, Debbane M, Bach Cuadra M, Ottet MC, Glaser B, Thiran JP, Eliez S. 2009. Deviant trajectories of cortical maturation in 22q11.2 deletion syndrome (22q11DS): a cross-sectional and longitudinal study. Schizophr Res. 115:182–190. [DOI] [PubMed] [Google Scholar]
- Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree MB, Owen M, Murphy KC, Niarchou M, Kates WR, et al. for the International Consortium on B, Behavior in 22q11.2 Deletion S. 2014. Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the international consortium on brain and behavior in 22q11.2 deletion syndrome. Am J Psychiatry. 171:627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimogori T, Banuchi V, Ng HY, Strauss JB, Grove EA. 2004. Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development. 131:5639–5647. [DOI] [PubMed] [Google Scholar]
- Siegenthaler JA, Ashique AM, Zarbalis K, Patterson KP, Hecht JH, Kane MA, Folias AE, Choe Y, May SR, Kume T, et al. 2009. Retinoic acid from the meninges regulates cortical neuron generation. Cell. 139:597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. 1999. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 126:3359–3370. [DOI] [PubMed] [Google Scholar]
- Tarabykin V, Stoykova A, Usman N, Gruss P. 2001. Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development. 128:1983–1993. [DOI] [PubMed] [Google Scholar]
- Tedeschi A, Ciciarello M, Mangiacasale R, Roscioli E, Rensen WM, Lavia P. 2007. RANBP1 localizes a subset of mitotic regulatory factors on spindle microtubules and regulates chromosome segregation in human cells. J Cell Sci. 120:3748–3761. [DOI] [PubMed] [Google Scholar]
- Theil T, Alvarez-Bolado G, Walter A, Ruther U. 1999. Gli3 is required for Emx gene expression during dorsal telencephalon development. Development. 126:3561–3571. [DOI] [PubMed] [Google Scholar]
- Tucker ES, Segall S, Gopalakrishna D, Wu Y, Vernon M, Polleux F, Lamantia AS. 2008. Molecular specification and patterning of progenitor cells in the lateral and medial ganglionic eminences. J Neurosci. 28:9504–9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yudin D, Hanz S, Yoo S, Iavnilovitch E, Willis D, Gradus T, Vuppalanchi D, Segal-Ruder Y, Ben-Yaakov K, Hieda M, et al. 2008. Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron. 59:241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun K, Potter S, Rubenstein JL. 2001. Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development. 128:193–205. [DOI] [PubMed] [Google Scholar]
- Zechner D, Fujita Y, Hulsken J, Muller T, Walther I, Taketo MM, Crenshaw EB, 3rd, Birchmeier W, Birchmeier C. 2003. beta-Catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev Biol (NY 1985). 258:406–418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.