

Abstract
A variety of bioinspired materials have been successfully synthesized to mimic the sophisticated structures or functions of biological systems. However, it is still challenging to develop materials with multiple functions that can be performed synergistically or sequentially. The purpose of this work was to demonstrate a novel bioinspired hydrogel that can interact with cancer cells, functionally similar to Drosera in catching and killing prey. This hydrogel had two layers with the top one functionalized with oligonucleotide aptamers and the bottom one functionalized with double-stranded DNA. The results show that the top hydrogel layer was able to catch target cells with high efficiency and specificity, and that the bottom hydrogel layer could sequester doxorubicin (Dox) for sustained drug release. Importantly, the released Dox could kill 90% of the cells after 1-h residence of the cells on the hydrogel. After the cell release, this bifunctional hydrogel could be regenerated for continuous cell catching and killing. Therefore, the data presented in this study has successfully demonstrated the potential of developing a material system with the functions of attracting, catching and killing diseased cells (e.g., circulating tumor cells) or even invading microorganisms (e.g., bacteria).
Development of materials for mimicking the sophisticated structures or functions of biological systems not only creates opportunities to gain deeper understanding of biological mechanisms1,2,3,4,5,6, but also holds the potential for discovering new applications7,8,9,10,11,12,13,14,15,16,17,18,19. For instance, the water-repellent surface structure of the lotus leaf has inspired the development of lotus-leaf-like nanostructured materials7,8; and the autonomic healing function of bone after fracture has inspired the formulation of composites that are able to release healing agents upon crack instrusion17,18,19. These bioinspired materials have been under rigorous investigation for various promising applications such as synthesis of super-hydrophobic materials and self-healing concrete. However, successful efforts have been primarily focused on the development of materials with the ability to mimic the singular functions of biological systems. It is still challenging to develop bioinspired materials with multiple functions that can be performed synergistically or sequentially.
The purpose of this work was to explore a hydrogel with the potential of mimicking the functions of Drosera in catching and killing prey (Fig. 1A). Drosera is a genus of carnivorous plants, whose leaves have tentacles covered with adhesive secretions20,21,22,23. When flying prey touch the leaves, they will be captured by the sticky tentacles24,25,26. In addition to presenting an adhesive surface to the environment, this plant releases digestive enzymes such as esterase, peroxidase, and protease on its leaves27. These enzymes can further digest and kill the captured prey. Since an unhealthy human body may have circulating diseased cells (e.g., cancer cells) or invading microorganisms (e.g., bacteria)28,29,30,31,32, the development of a bioinspired material with the functions of catching targets and releasing toxic drugs to destroy the targets may lead to potential biomedical applications. Thus, we were inspired to develop a novel hydrogel with the target-catching and drug-releasing functions.
Figure 1. Conceptual illustration of Drosera-bioinspired hydrogel.

(A) Photos of Drosera before and after the prey catch. (B) Schematic illustration of the bilayer hydrogel made of acrydite-DNA, acrylamide and bisacrylamide. ID1 and ID2 are chemically incorporated into the top and bottom hydrogel layers during free radical polymerization, respectively. ID1 hybridizes with CD1 on the top layer for catching target cells; and ID2 hybridizes with CD2 to sequester drug (Doxorubicin) in the bottom hydrogel. (C) Schematic illustration of cell catch and drug release for killing target cells on the hydrogel.
This bifunctional hydrogel was made of two hydrogel layers (Fig. 1B). Both layers were functionalized with oligonucleotides. The top layer hydrogel was functionalized with nucleic acid aptamers. Nucleic acid aptamers are single-stranded oligonucleotides that can be, in principle, selected from oligonucleotide libraries with high affinities and specificities against any target molecules or cells33,34,35. Since aptamers are tolerant of harsh chemical conditions, they can be immobilized to a substrate without losing their binding capabilities36,37,38,39. Thus, aptamers were used to functionalize the top layer hydrogel for catching target cells via aptamer-mediated cell recognition. The chemical incorporation of aptamers into the hydrogel was evaluated with fluorescence imaging. The aptamer-mediated cell catch was studied by measurement of the kinetics of cell binding onto the top layer hydrogel. The bottom layer hydrogel was chemically functionalized with a double-stranded DNA that was used as an affinity site for sequestration of small toxic drugs. Thus, the drugs in the hydrogel could be locally released in a sustained manner. Drug sequestration and release were characterized with fluorescence spectroscopy. The function of the released drugs in killing the cells was examined with the cell viability assays.
Results and Discussion
Synthesis and characterization of the bilayer hydrogel
The bifunctional hydrogel was synthesized with two layers via two-step free radical polymerization (Figure S1). The top layer was ~25 μm thick and the bottom one was ~200 μm thick (Fig. 2A). During free radical polymerization, the immobilizing DNAs (IDs) including ID1 and ID2 were chemically incorporated into the top and bottom layers, respectively. To demonstrate the success of chemical incorporation of IDs into the hydrogel and intermolecular hybridization between the IDs and their complementary DNAs (CDs), the hydrogels were treated with FAM-CD1 and Cy5.5-CD2, washed thoroughly and examined under fluorescence microscopy. The hydrogels prepared with ID1 and/or ID2 without acrydite exhibited negligible fluorescence signals of Cy5.5 and/or FAM (Fig. 2B). In contrast, the hydrogels prepared with acrydite-modified ID1 and/or ID2 exhibited strong fluorophore signals. These results demonstrate that acrydite-modified ID1 and ID2 could be chemically incorporated into the two layers via free radical polymerization, and that IDs and CDs formed stable complexes via sequence-specific intermolecular hybridization. An in-depth observation of the bilayer hydrogel with confocal microscopy confirmed the incorporation and hybridization of the oligonucleotides (Fig. 2C). The two hydrogel layers played two different roles: the top one was used to present the hybridized aptamer (i.e., the ID1-CD1 complex) for cell catch and the bottom one was used to accommodate the drug intercalating double-stranded DNA (i.e., the ID2-CD2 complex) for Dox loading and release.
Figure 2. Characterization of the hydrogel.

(A) SEM image of the hydrogel. Scale bar: 50 μm. (B) Fluorescence images of the hydrogel after treated with FAM-CD1 and Cy5.5-CD2. The green fluorescence was emitted by FAM-CD1 from the top layer hydrogel and the red fluorescence was emitted by Cy5.5-CD2 from the bottom layer hydrogel. All of the hydrogel samples were washed thoroughly before fluorescence imaging. Scale bar: 100 μm. (C) Confocal images of the hydrogel treated with FAM-CD1 and Cy5.5-CD2. The three demonsional image was generated by FV10-ASW version 3.0 software. Scale bar: 100 μm.
Examination of cell catch on the hydrogel
To catch target cells in a biologically relevant environment, the hybridized aptamer needs to have stability. Thus, the hybridized aptamer was examined using gel electrophoresis after it was incubated in the cell culture medium for 1 h. The gel electrophoresis image shows that the fluorescence intensity of the hybridized aptamer incubated in PBS and the cell culture medium was virtually the same (Fig. 3A), demonstrating that the aptamer complex exhibited stability during the 1-h incubation in the cell culture medium. The model aptamer used in this work was originally selected via a cell-SELEX procedure and it can bind the CCRF-CEM cell line with high specificity40. We incubated the CCRF-CEM cells with the hydrogel displaying he hybridized aptamer to test whether the hydrogel could catch the cells. Approximately 1,200 CCRF-CEM cells per mm2 were observed on the CD1 functionalized hydrogel whereas only ~10 control cells per mm2 was observed on the hydrogel (Fig. 3B). Thus, the data show that the top layer hydrogel with the hybridized aptamer has the ability to induce cell-type-specific catch in the biologically relevant environment.
Figure 3. Examination of specific cell catch.
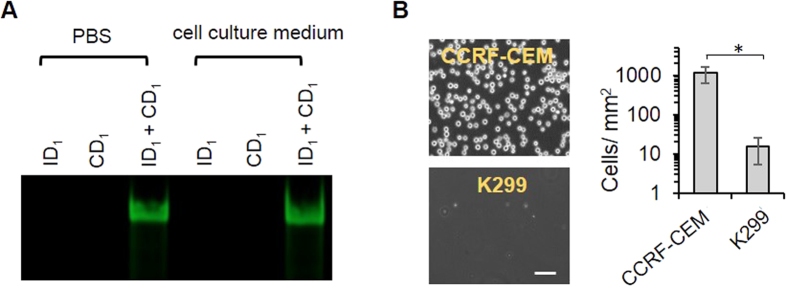
(A) Gel image showing ID1-CD1 hybridization in two different solutions. (B) Imaging and quantitative analysis of cell catch on the hydrogels. The CCRF-CEM (target) cells and K299 (control) cells were incubated on the hydrogel surface for 1 h. Quantitative analysis was performed by the examination of three areas randomly chosen from each image. Cell numbers were analyzed by ImageJ. Scale bar: 50 μm. *P < 0.05; n = 3.
While other affinity ligands such as antibodies and peptides may be applied to functionalize the top layer hydrogel to acquire the cell-catching function41,42, we chose to use nucleic acid aptamers to develop the top hydrogel layer for several reasons. Nucleic acid aptamers are selected in an in vitro environment43,44. They have high binding affinities and specificities against their targets owing to the rigorous procedure of aptamer selection. They are small in size and can be facilely synthesized using standard chemical methods. They are also tolerant of harsh chemical and physical conditions and suitable for chemical incorporation into a material45. These advantages distinguish nucleic acid aptamers from other ligands in the functionalization of the hydrogel.
A number of elegant material systems have been successfully developed with great potential to capture circulating tumor cells from blood for cell separation or cancer diagnosis46,47,48. However, most of them are made of hard materials presenting a rigid interface between the materials and the cells. By contrast, hydrogel is a soft material with many properties similar to human tissues. In addition, it is more flexible to conjugate affinity ligands to hydrogels or optimize hydrogels for controlled drug release in comparison to hard materials. Thus, hydrogels were used in this work to catch cancer cells and control drug release (next section).
Examination of drug loading and release
To study the function of the bottom layer hydrogel in drug loading and release, Dox was used as a model drug. With the increase of the concentration of the ID2-CD2 complex, the fluorescence intensity of the Dox solution was decreased (Fig. 4A). The fluorescence intensity was decreased by 50% and 94% when the molar ratio was increased to ~0.07 and 0.5, respectively. Since the intercalation of Dox into double-stranded DNA leads to the quenching of Dox fluorescence49, the result indicates that Dox would be effectively loaded into the DNA-functionalized hydrogel. To directly illustrate this point, we incubated the hydrogel in the Dox solution for 24 h and subsequently transferred it into DPBS for the release test. The native hydrogel (without DNA) released 65.5% Dox during the first 1 h (Fig. 4B). The cumulative Dox release reached 86.8% and the release reached the plateau approximately after 4 h. In contrast, 19.8% and 36.0% of Dox was released from the dsDNA-functionalized hydrogel at 1 and 4 h, respectively. These data show that the incorporation of the ID2-CD2 complex was able to significantly reduce the initial burst release. Moreover, the release of Dox from the ID2-CD2 complex-functionalized hydrogel exhibited a pseudo-linear release profile after 4 h. Specifically, the concentrations of Dox in the release media were higher than or close to 1 μM during the entire 144 h test and were above 2 μM within the first 48 h (Fig. 4C). Since the function of the bottom layer hydrogel depends on whether the released Dox could kill the cells on the top layer hydrogel, we examined the relationship of cell viability versus the concentration of Dox. The results show that the viability of the cells was decreased to 36.7 and 4.9% after the 1-h exposure to 1 and 2 μM of Dox, respectively (Fig. 4D). Increasing the exposure time of cells to Dox could significantly decrease the cell viability in the low Dox concentration groups, e.g., 1 μM.
Figure 4. Sustained Dox release from the hydrogel.

(A) Characterization of Dox (5 μM) sequestration by dsDNA using fluorescence spectroscopy. The labeling numbers from the top to the bottom indicate the molar ratios of DNA: Dox. The quantitative relationship between Dox and dsDNA is shown by the peak emission intensity at 592 nm versus the molar ratio. (B) Kinetics of cumulative Dox released from the hydrogel with or without the chemically conjugated dsDNA. (C) Concentrations of Dox released from the hydrogel with the chemically conjugated dsDNA at each sampling time points. (D) Effect of the Dox concentration on the viability of CCRF-CEM cells that were treated by Dox for either 1 h or 2 h.
In this work, we applied the ID2-CD2 complex to functionalize the bottom layer hydrogel to achieve the sustained Dox release. Other methods may be used to optimize and improve the bottom layer hydrogel for a longer period of drug release. For instance, biodegradable microparticles with the ability to control the release of drugs for weeks or months have been used to functionalize hydrogels50,51,52. Such a method may be applied to develop the bottom hydrogel. The bottom hydrogel may also be functionalized with enzymes that can covert prodrugs into drugs for a long-term sustained release53. In addition to using hydrogels to control drug release, a variety of other polymeric systems have been studied for long-term sustained drug release54,55. Those sustained release systems can, in principle, be used to replace the bottom hydrogel for the optimization of the sustained drug release.
Examination of cell catch and killing on the bifunctional hydrogel
After demonstrating the cell-catching and drug-releasing functions of each hydrogel layer, we examined cell behavior on the aptamer-functionalized drug-loaded hydrogel. The number of cells increased linearly during the first half hour and gradually reached a plateau between 0.5 and 1.5 h (Fig. 5A). Moreover, the results show that the presence or absence of Dox did not affect the outcome of cell catch, suggesting that the presence of drugs did not affect the cell-binding functionality of the hybridized aptamer. In addition to the examination of cell catch during the first 1.5 h, we examined the cell release kinetics. With the increase of the incubation time, the ID1-CD1 complex was gradually degraded (Fig. 5B). As a result, the attached cells were released from the hydrogel surface. The density of the cells on the hydrogel was decreased to 50.1 and 99.8% after 8 and 24 h incubation in the presence of serum (Fig. 5C). The results also show that the presence or absence of Dox did not affect the kinetics of cell release (Fig. 5C).
Figure 5. Cell catch and release kinetics on the hydrogel.

(A) Kinetics of CCRF-CEM cell catch on hydrogels in the presence or absence of Dox. Scale bar: 100 μm. (B) Degradation of dsDNA in the culture medium. The electrophoretic gel image was analyzed with the software of Maestro imaging system to generate the profile of dsDNA degradation versus time. (C) Kinetics of CCRF-CEM cell release from the hydrogels in the presence or absence of Dox. For the cell release study, the starting 0 h indicates 1 h after cell catch. Scale bar: 50 μm.
Since the kinetic profiles of cell-hydrogel interactions show that the cell density reached a plateau at 1 h, we harvested the cells from the hydrogel for examining cellular Dox uptake using both fluorescence microscopy and flow cytometry. The fluorescence images show that the cells on the hydrogel exhibited a strong fluorescence signal from Dox (Fig. 6A). The imaging result was confirmed by the flow cytometry (Fig. 6B). Thus, the data show that the cells were able to take up a significant amount of Dox released from the bottom layer hydrogel within the first 1 h before the onset of cell release.
Figure 6. Examinations of drug uptake and cell viability.

(A) Fluorescence images of CCRF-CEM cells after 1-h residence on the Dox-releasing hydrogel. The cells were harvested and analyzed after 1-h residence on the hydrogel. Scale bar: 20 μm. (B) Flow cytometry histogram. The CCRF-CEM cells were harvested and analyzed after 1-h residence on the hydrogel. (C) Examination of cell viability via the cell staining assay. The harvested CCRF-CEM cells were cultured in the cell culture medium for 48 h and then stained with Hoechst 33342 and Calcein AM. Hoechst 33342 was used to stain the nuclei of cells; Calcein AM was used to stain live cells. Scale bar: 20 μm. (D) Analysis of cell viability with the MTS assay. *P < 0.05; n = 3.
After the examination of Dox uptake by the cells, the effectiveness of Dox-mediated cell killing was investigated via the cell viability assays. As shown in the fluorescence images, the cells without Dox treatment could maintain the same bioactivity and morphology as those intact cells (i.e., cells incubated in normal cell culture medium) (Fig. 6C). In contrast, most of the cells harvested from the Dox-releasing hydrogel died with irregular shapes. The quantitative data showed that the viability of these cells was decreased to less than 10% (Fig. 6D), suggesting that the short duration of drug uptake was sufficient to kill the majority of the cells. To illustrate the effect of the residence time on drug uptake, we harvested the cells from the hydrogel at the different time points and characterized their Dox uptake via flow cytometry. The results show that the amount of Dox uptake increased with the time (Figure S2A). The 12-h exposure of the cells to Dox resulted in cellular fragmentation into membrane-bound apoptotic bodies, which is a hallmark of cell apoptosis and death (Figure S2B).
Evaluation of the sustainability of catching and killing cells
After catching prey, the tentacles of Drosera can return to their original position to continuously catch and kill new prey. Since aptamer degradation occurred during the procedure of cell release (Fig. 5), the hydrogel may lose the ability to catch target cells without fresh aptamers displayed on the top hydrogel layer. To regenerate the surface with fresh aptamers for a new batch of cell catch, we used a chemically modified ID1 for the functionalization of the top hydrogel layer. One of the non-bridging oxygen in each phosphate was replaced by sulfur (Fig. 7A). The image shows that ID1 degradation was undetectable during a 7-day degradation test (Fig. 7B), indicating that the sulfurization of the backbone stabilized ID1 in the serum. Thus, chemically modified ID1 on the top layer hydrogel was able to hybridize with fresh aptamers after the original ones were degraded (Fig. 7C). Resultantly, the cell-catching function of the hydrogel can be regenerated. Indeed, the regenerated hydrogel was able to sustainably catch and kill cells (Fig. 8). In addition to the replacement of the non-bridging oxygen with sulfur, there is a diverse array of other chemical methods available to stabilize oligonucleotides against nuclease degradation56,57. Those methods can also be used to optimize the chemical modification of ID1.
Figure 7. Examination of the stability of phosphorothiolated DNA.

(A) Chemical structure of ID1 on the top layer hydrogel. The backbone of ID1 has the phosphorothioate bond resistant to nuclease degradation. The 3′ ends was modified with the inverted T. (B) Gel electrophoresis image demonstrating the resistance of ID1 against nuclease degradation during the 7-day degradation test. After ID1 was degraded in the cell culture medium for different days, CD1 was added into ID1 solutions before gel electrophoresis. (C) Gel image of the ID1-CD1 duplex before and after the incubation in the cell culture medium in a two-round test. The duplex was incubated in the cell culture medium for 24 h. This procedure was repeated after the duplex solution was added with fresh CD1.
Figure 8. Evaluation of sustained cell catch and killing.

(A) Schematic illustration of alternate aptamer display and nuclease degradation on the top layer hydrogel. ID1 is nondegradable owing to the full modification of its backbone whereas unmodified CD1 is degradable. (B) Microscopic images and quantitative analysis of CCRF-CEM cell catch and release in a two-round test. Scale bar: 50 μm. (C) Analysis of cell viability. *P < 0.05; n = 3.
The major focus of the current work was to develop a bioinspired hydrogel with multiple functions. However, one may inquire whether this material can be used in the human body for catching and killing diseased cells or invading microorganisms. We envision that this multifunctional material holds potential of treating human diseases for the following reasons. First, in nature, circulating cancer cells can be captured onto the endothelium surface during circulation via molecular recognition58. Similarly, invading bacteria in the circulation can bind to implant surfaces59. These biological effects suggest that a material with the function of cell recognition would be able to catch target cells or microorganisms in an in vivo setting. Second, the human body is virtually a closed system for circulation. Thus, the multifunctional material has multiple opportunities to catch the circulating cells in a given period of time. This differs from in vitro diagnostic tools (e.g., microfluidic devices for separation of circulating cancer cells) that usually need to capture target cells during the first pass of the cells over the surface of the affinity material. Third, while high shear stress is not favorable for cell catch on a surface, there are many locations (e.g., veins or lymphatic vessels) in the human body where the shear stress of the circulation is low. Fourth, the multifunctional material can be designed in diverse forms such as a depot or a conduit to satisfy the different requirements of implantation in appropriate locations. Certainly, more work needs to be performed in the future to demonstrate the feasibility of using this multifunctional bioinspired material in vivo.
Conclusion
In summary, we synthesized a novel bioinspired hydrogel functionally similar to Drosera in catching and killing prey. This hydrogel is able to catch cancer cells effectively and release drugs locally to kill the cells. In addition, it can repetitively display the cell-catching aptamer on the surface. As a result, the sustained drug release from this bioinspired hydrogel can continuously kill cancer cells. The functions of this material will be further studied in an in vivo setting. Future work may also be performed to integrate the controlled chemokine release function into the hydrogel for attracting cancer cells. Thus, the Drosera-bioinspired hydrogel will have the ability to attract, catch, and kill target diseased cells or even invading microorganisms for biomedical applications.
Methods
Chemical reagents
Doxorubicin, 3-(trimethoxysilyl) propyl methacrylate (TMSPM), and magnesium chloride (MgCl2) solution (1.0 M) were obtained from Sigma Aldrich (Louis, MO). Dulbecco’s phosphate buffered saline (DPBS), SYBR-Safe, Calcein AM, and Hochest 33342 were purchased from Invitrogen (Carlsbad, CA). RPMI-1640 was purchased from ATCC (Manassas, VA). Fetal bovine serum (FBS) and the penicillin/streptomycin solution were purchased from Hyclone (Logan, UT). The CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) kit was from Promega (Madison, WI). Nucleic acid oligonucleotides (Table 1 in supporting information) were synthesized by Integrated DNA Technologies (Coralville, IA). Other chemicals were purchased from Fisher Scientific (Suwanee, GA).
Synthesis of the bilayer hydrogels
The bilayer hydrogels were synthesized on the surface of silanized glass slides with a dimension of 7.5 × 7.5 mm2. The silanization of glass slides was performed according to the method described previously35. In brief, the NaOH-treated glass slides were reacted with the TMSPM solution for 5 min and subsequently washed with ethanol and dried in the air. To synthesize the bottom hydrogel, a pregel solution was prepared with ammonium persulfate (APS, 0.5 μL, 10% w/v), N,N,N’,N’-tetramethylenediamine (TEMED, 0.5 μL, 10% v/v), acrylamide solution (19 μL, 10% w/v) and acrydite-modified ID1 (125 μM). This pregel solution was transferred onto the surface of silanized glass and covered with coverslip. After the hydrogel was cured for 2 h, the coverslip was gently removed from the cured hydrogel. The hydrogel on the silanized glass square was washed in PBS for 1 h to remove the unreacted molecules. The top layer hydrogel was synthesized on the bottom hydrogel using a similar procedure. The pregel solution containing acrylamide solution (4 μL, 10% w/v), acrydite-modified ID2 (100 μM), APS (0.25 μL, 10% w/v), and TEMED (0.25 μL, 10% v/v) was sandwiched between the bottom hydrogel and a piece of supporting naked glass slide. After cured for 2 h, the bilayer hydrogel was flipped off the supporting glass slide and washed thoroughly with PBS.
Gel electrophoresis
Gel electrophoresis was used to examine intermolecular hybridization and degradation kinetics. To test intermolecular hybridization, ID1, ID2 were incubated with complementary sequences at a molar ratio of 1:1 in PBS for 1 h at room temperature. To study the degradation of oligonucleotides, the oligonucleotides either in a single-stranded or hybridized form were incubated in the culture medium (RPMI 1640 medium, 10% FBS, and 100 IU/mL penicillin-streptomycin) at 37 °C. At predetermined time points, 25 μL aliquot of the mixture was collected and stored at −20 °C. The solution of single-stranded oligonucleotides was mixed with the complementary sequences prior to the analysis. To examine the functionality of ID1 in repeatedly hybridizing CD1, a solution containing ID1 (0.8 μM) and CD1 (0.8 μM) was incubated in the cell culture medium at 37 °C. At 1 h and 24 h, 25 μL of the solution was collected and stored at −20 °C. The solution collected at 24 h was also mixed with the fresh CD1 solution (0.8 μM). The samples were collected at 1 h and 24 h. The collected samples were loaded into a 10% polyacrylamide gel. The electrophoresis was run in the TBE buffer (89 mM tris-borate, 89 mM boric acid, and 2 mM EDTA; pH 8.2) at 140 V for 40 min using a Mini-PROTEAN Tetra Cell system (Bio-Rad, Hercules, CA). The gel was stained in SYBR-Safe solution for 30 min and imaged with a green fluorescence filter using a CRI Maestro EX System (Woburn, MA). The images were analyzed using the software provided by the supplier.
SEM imaging of the hydrogels
The bilayer hydrogel was treated with a series of ethanol solutions (50%, 60%, 70%, 80%, 90%, and 100%) for gradient dehydration. After the dehydration, the hydrogels were dried in air and coated with gold. The cross-section of the hydrogels were imaged under a Hitachi S-3500N scanning electron microscope (Hitachi, Ltd, Tokyo, Japan). SEM imaging was acquired at an accelerating voltage of 20 kV.
Fluorescence imaging of the hydrogels
The hydrogels prepared with ID1 and ID2 (with or without acrydite) were incubated with 20 μL Cy5.5-CD2 solution (125 μM) for 2 h. After incubated in PBS for 1 h washing, the hydrogels were incubated with 20 μL FAM-CD1 (20 μM) for 1 h. The hydrogels were washed thoroughly in PBS to remove the free fluorophore-labeled DNA molecules. The hydrogels were imaged under the Olympus FV1000 laser scanning confocal microscope (Olympus America Inc., Melville, NY) with a 20X objective. Blue Argon (488 nm) and red HeNe (633 nm) lasers were used to catch the signals of Cy5.5 and FAM, respectively. The hydrogel was scanned from the top where fluorescence appeared to the bottom until fluorescence disappeared. The images was analyzed using FV10-ASW version 3.0 software. In addition to the in-depth observation of the fluorescence signals, the hydrogels were also imaged under a fluorescence microscope (Olympus IX73, Melville, NY) with a 10X objective to examine the incorporation of ID1 and ID2. The fluorescence signals were recorded by using two sets of fluorescence filter cubes at 480 ± 20/535 ± 25 nm (Ex./Em.) and 620 ± 30/700 ± 40 nm (Ex./Em.).
Dox loading and release
The hydrogels were incubated with the CD2 solution of 20 μL (125 μM in DPBS) at room temperature for 2 h and washed with PBS thoroughly. The hydrogels were incubated with 40 μL of DPBS containing 5 mM MgCl2 and Dox (500 μM) at 4 °C for 24 h. To examine Dox release, the Dox-loaded hydrogels were incubated in 500 μL of DPBS with a shaking speed of 70 rpm at 37 °C. At predetermined time points, the supernatants were collected and replaced with 500 μL of fresh DPBS. The collected supernatants were transferred into a fluorescence 96-well plate (Perkin Elmer, Waltham, MA) and measured by a Tecan F200 Pro Micro-plate reader (Tecan US Inc., San Jose, CA) at 592 nm.
Cell culture
CCRF-CEM cell (CCL-119, human T lymphocytic leukemia cell line) was obtained from ATCC (Manassas, VA); K299 (Karpas 299, human T cell lymphoma cell line) was purchased from Sigma-Aldrich. The cells were cultured in the cell culture medium in an incubator at 37 °C in a 5% CO2 atmosphere.
Examination of the interactions between cells and the hydrogels
The hydrogel was washed in cell culture medium for 2 h and then incubated with 20 μL of CD1 solution (20 μM in cell culture medium) at 37 °C for 1 h for the immobilization of CD1 on the top layer hydrogel. The hydrogel was washed with 500 μL of culture medium for 10 min to remove free CD1 and then incubated with 500 μL of cell suspension (3 × 105 cells in the cell culture medium supplemented with 5 mM MgCl2) in a 48-well plate at 37 °C for 1 h. The unbound cells were removed after the 48-well plate was gently shaken at 70 rpm for 1 min. Five hundred microliters of fresh cell culture medium was added into the plate and the hydrogel was incubated at 37 °C in the CO2 incubator for 24 h. At predetermined time points, the hydrogel was examined under the microscope. Three areas of the hydrogel surface were randomly selected for imaging. The cells in each area were enumerated using ImageJ. To examine the sustainability of using the hydrogel to catch cells, the whole procedure of immobilizing CD1 and capturing cells was repeated after the first round of cell catch and release.
Examination of Dox uptake by cells
At predetermined time points after cell attachment onto the hydrogel, the cells were collected from hydrogel surface. After washing with 1 mL of DPBS twice, the collected cells were imaged using the fluorescence microscope with a red fluorescence filter (535 ± 25/610 ± 40 nm, Ex./Em.) and a 40X objective. The collected cells were also examined with a Coulter FC500 flow cytometer (Beckman Coulter, Miami, FL). A total of 5,000 events were counted for each group. The data were analyzed with FlowJo 7.0.
SEM imaging of the cells on the hydrogel
The cells on the hydrogel surface were fixed in glutaraldehyde solution (3% in PBS) for 3 h. After rinsing with PBS, the samples were dehydrated in a series of ethanol solutions (50%, 60%, 70%, 80%, 90%, and 100%). Subsequently, the samples were dehydrated in Hexamethyldisilazane (HMDS) solutions (33%, 66%, and 100%) and dried in air overnight. The cells were coated with a gold layer. The SEM imaging was operated under a Hitachi S-3500N scanning electron microscope (Hitachi, Ltd, Tokyo, Japan) at an accelerating voltage of 15.0 kV.
Analysis of cell viability
The cells were incubated in 8-well chamber slide with an initial concentration of 2.5 × 105 cells/mL in the culture medium for 48 h and were then stained in the mixture of Hochest 33342 (2.5 μg/mL) and Calcein AM (1 μM) at 37 °C for 30 min. After the replacement of the staining medium with DPBS, the cells were imaged under the fluorescent microscope with two sets of fluorescence filter cubes (350 ± 25/460 ± 25 nm (Ex./Em.) and 480 ± 20/535 ± 25 nm (Ex./Em.) and a 40X objective. Quantitative analysis of the cell viability was performed using a CellTiter MTS cell assay kit. In brief, CCRF-CEM cells were collected from the hydrogel surface, seeded into 96-well plate with a population of 2.5 × 104 cells in 100 μL of cell culture medium and cultured in CO2 incubator for 48 h. The CellTiter reagent (20 μL) was added to each well and incubated with the cells at 37 °C for 3 h. The absorbance at 490 nm was recorded using the Tecan Micro-plate reader. The absorbance in each group was normalized to the average absorbance of the group with intact cells to present the viability.
Statistical analysis
Student’s t-test (two-sided) was used to determine the significance of results. P-value < 0.05 was considered as significant. Error bars in all figures represent standard deviations.
Additional Information
How to cite this article: Li, S. et al. A Drosera-bioinspired hydrogel for catching and killing cancer cells. Sci. Rep. 5, 14297; doi: 10.1038/srep14297 (2015).
Supplementary Material
Acknowledgments
This work was supported in part by the U.S. National Science Foundation (1340173, 1322332) and the Penn State Start-Up Fund. Shihui Li and Niancao Chen were supported in part by the China Scholarship Council. Mr. Avery Chan and Mr. Enric Bringués are acknowledged for taking the photos of Drosera. Mr. Kyle Plasterer and the Husk Institute Microscopy Facility (University Park, PA) are acknowledged for technical support in using the microscopes.
Footnotes
Author Contributions S.L. designed and performed experiments, analyzed data and wrote the paper; N.C., E.G. and X.Z. performed experiments; D.C. analyzed data; and Y.W. conceived the concept, designed experiments, analyzed data and wrote the paper.
References
- Barthlott W. & Neinhuis C. Purity of the sacred lotus, or escape from contamination in biological surfaces. Planta 202, 1–8 (1997). [Google Scholar]
- Autumn K. et al. Adhesive force of a single gecko foot-hair. Nature 405, 681–685 (2000). [DOI] [PubMed] [Google Scholar]
- Autumn K. et al. Evidence for van der Waals adhesion in gecko setae. Proc. Natl. Acad. Sci. USA 99, 12252–12256 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn H. F. & Federle W. Insect aquaplaning: Nepenthes pitcher plants capture prey with the peristome, a fully wettable water-lubricated anisotropic surface. Proc. Natl. Acad. Sci. USA 101, 14138–14143 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woesz A. et al. Micromechanical properties of biological silica in skeletons of deep-sea sponges. J. Mater. Res. 21, 2068–2078 (2006). [Google Scholar]
- Vukusic P. & Sambles J. R. Photonic structures in biology. Nature 424, 852–855 (2003). [DOI] [PubMed] [Google Scholar]
- Jiang L., Zhao Y. & Zhai J. A lotus-leaf-like superhydrophobic surface: a porous microsphere/nanofiber composite film prepared by electrohydrodynamics. Angew. Chem. Int. Ed. 43, 4338–4341 (2004). [DOI] [PubMed] [Google Scholar]
- Cheng Q. et al. Janus interface materials: superhydrophobic air/solid interface and superoleophobic water/solid interface inspired by a lotus leaf. Soft Matter 7, 5948 (2011). [Google Scholar]
- Wilson P. W. et al. Inhibition of ice nucleation by slippery liquid-infused porous surfaces (SLIPS). Phys. Chem. Chem. Phys. 15, 581–585 (2013). [DOI] [PubMed] [Google Scholar]
- Wong T.-S. et al. Bioinspired self-repairing slippery surfaces with pressure-stable omniphobicity. Nature 477, 443–447 (2011). [DOI] [PubMed] [Google Scholar]
- Kim P. et al. Liquid-infused nanostructured surfaces with extreme anti-ice and anti-frost performance. ACS Nano 6, 6569–6577 (2012). [DOI] [PubMed] [Google Scholar]
- Epstein A. K., Wong T.-S., Belisle R. A., Boggs E. M. & Aizenberg J. Liquid-infused structured surfaces with exceptional anti-biofouling performance. Proc. Natl. Acad. Sci. USA 109, 13182–13187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geim A. K. et al. Microfabricated adhesive mimicking gecko foot-hair. Nat. Mater. 2, 461–463 (2003). [DOI] [PubMed] [Google Scholar]
- King D. R., Bartlett M. D., Gilman C. A., Irschick D. J. & Crosby A. J. Creating gecko-like adhesives for ‘real world’ surfaces. Adv. Mater. 26, 4345–4351 (2014). [DOI] [PubMed] [Google Scholar]
- Lee U. J., Bush B., Maboudian R. & Fearing R. S. Gecko-inspired combined lamellar and nanofibrillar array for adhesion on nonplanar surface. Langmuir 25, 12449–12453 (2009). [DOI] [PubMed] [Google Scholar]
- Sethi S., Ge L., Ci L., Ajayan P. M. & Dhinojwala A. Gecko-inspired carbon nanotube-based self-cleaning adhesives. Nano Lett. 8, 822–825 (2008). [DOI] [PubMed] [Google Scholar]
- White S. R. et al. Autonomic healing of polymer composites. Nature 409, 794–797 (2001). [DOI] [PubMed] [Google Scholar]
- Brown E., Sottos N. R. & White S. R. Fracture testing of a self-healing polymer composite. Exp. Mech. 42, 372–379 (2002). [Google Scholar]
- Rule J. D., Brown E. N., Sottos N. R., White S. R. & Moore J. S. Wax-protected catalyst microspheres for efficient self-healing materials. Adv. Mater. 17, 205–208 (2005). [Google Scholar]
- Darwin C. Insectivorous Plants (Appleton, London, 1875). [Google Scholar]
- Albert V. A., Williams S. E., Chase M. W. & Chaset M. W. Carnivorous plants: phylogeny and structural evolution. Science 257, 1491–1495 (1992). [DOI] [PubMed] [Google Scholar]
- Ellison A. M. & Gotelli N. J. Evolutionary ecology of carnivorous plants. Trends Ecol. Evol. 16, 623–629 (2001). [Google Scholar]
- Ragetli H. W. J., Weintraub M. & Lo E. Characteristics of Drosera tentacles. I. anatomical and cytological detail. Can. J. Bot. 50, 159–168 (1972). [Google Scholar]
- Hartmeyer R. H. Summary of comparative examination of Drosera tentacles. Carniv. Pl. Newslett. 39, 101–114 (2010). [Google Scholar]
- Gilchrist A. J. & Juniper B. E. An excitable membrane in the stalked glands of Drosera capensis L. Planta 119, 143–147 (1974). [DOI] [PubMed] [Google Scholar]
- Chandler G. E. & Anderson J. W. Uptake and metabolism of insect metabolites by leaves and tentacles of Drosera species. New Phytol. 77, 625–634 (1976). [Google Scholar]
- Clancy G. & Coffey D. Acid phosphatase and protease release by the insectivorous plant Drosera rotundifolial. Can. J. Bot. 55, 480–488 (1977). [Google Scholar]
- Williams S. C. P. Circulating tumor cells. Proc. Natl. Acad. Sci. USA 110, 4861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B., Peterse J. L. & van ’t Veer L. J. Breast cancer metastasis: markers and models. Nat. Rev. Cancer 5, 591–602 (2005). [DOI] [PubMed] [Google Scholar]
- Hayes D. F. & Smerage J. B. Circulating tumor cells. Prog. Mol. Biol. Transl. Sci. 95, 95–112 (2010). [DOI] [PubMed] [Google Scholar]
- Meng S. et al. Circulating tumor cells in patients with breast cancer dormancy. Clin. Cancer Res. 10, 8152–8162 (2004). [DOI] [PubMed] [Google Scholar]
- Levine M. M., Kaper J. B., Black R. E. & Clements M. L. New knowledge on pathogenesis of bacterial enteric infections as applied to vaccine development. Microbiol. Rev. 47, 510–550 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann T. & Patel D. J. Adaptive recognition by nucleic acid aptamers. Science 287, 820–825 (2000). [DOI] [PubMed] [Google Scholar]
- Keefe A. D., Pai S. & Ellington A. Aptamers as therapeutics. Nat. Rev. Drug Discovery 9, 537–550 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Chen N., Li S., Battig M. R. & Wang Y. Programmable hydrogels for controlled cell catch and release using hybridized aptamers and complementary sequences. J. Am. Chem. Soc. 134, 15716–15719 (2012). [DOI] [PubMed] [Google Scholar]
- Potyrailo R. A., Conrad R. C., Ellington A. D. & Hieftje G. M. Adapting selected nucleic acid ligands (aptamers) to biosensors. Anal. Chem. 70, 3419–3425 (1998). [DOI] [PubMed] [Google Scholar]
- Balamurugan S., Obubuafo A., Soper S. A. & Spivak D. A. Surface immobilization methods for aptamer diagnostic applications. Anal. Bioanal. Chem. 390, 1009–1021 (2008). [DOI] [PubMed] [Google Scholar]
- Chen N., Zhang Z., Soontornworajit B., Zhou J. & Wang Y. Cell adhesion on an artificial extracellular matrix using aptamer-functionalized PEG hydrogels. Biomaterials 33, 1353–1362 (2012). [DOI] [PubMed] [Google Scholar]
- Liu J. Oligonucleotide-functionalized hydrogels as stimuli responsive materials and biosensors. Soft Matter 7, 6757 (2011). [Google Scholar]
- Shangguan D. et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc. Natl. Acad. Sci. USA 103, 11838–11843 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisic M., Iyer R. K. & Murthy S. K. Micro- and nanotechnology in cell separation. Int. J. Nanomed. 1, 3–14 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung J. H., Gajjar K. A., Saric J., Eddington D. T. & Hong S. Dendrimer-mediated multivalent binding for the enhanced capture of tumor cells. Angew. Chem. Int. Ed. 50, 11769–11772 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. S. & Szostak J. W. In vitro selection of functional nucleic acid. Annu. Rev. Biochem. 68, 611–647 (1999). [DOI] [PubMed] [Google Scholar]
- Stoltenburg R., Reinemann C. & Strehlitz B. SELEX–a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 24, 381–403 (2007). [DOI] [PubMed] [Google Scholar]
- Juliano R. L., Ming X. & Nakagawa O. The chemistry and biology of oligonucleotide conjugates. Acc. Chem. Res. 45, 1067–1076 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagrath S. et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. et al. Highly efficient capture of circulating tumor cells by using nanostructured silicon substrates with integrated chaotic micromixers. Angew. Chem., Int. Ed. Engl. 50, 3084–3088 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. et al. Aptamer-based microfluidic device for enrichment, sorting, and detection of multiple cancer cells. Anal. Chem. 81, 7436–7442 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasiuk J., Frézard F., Garnier-Suillerot A. & Gattegno L. Anthracycline incorporation in human lymphocytes. Kinetics of uptake and nuclear concentration. Biochim. Biophys. Acta. 1013, 109–117 (1989). [DOI] [PubMed] [Google Scholar]
- Galeska I. et al. Controlled release of dexamethasone from PLGA microspheres embedded within polyacid-containing PVA hydrogels. AAPS J. 7, e231–240 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsioris K. et al. Fabrication of silk microneedles for controlled-release drug delivery. Adv. Func. Mater. 22, 330–335 (2012). [Google Scholar]
- Demuth P. C., Garcia-Beltran W. F., Ai-Ling M. L., Hammond P. T. & Irvine D. J. Composite dissolving microneedles for coordinated control of antigen and adjuvant delivery kinetics in transcutaneous vaccination. Adv. Func. Mater. 23, 161–172 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varghese O. P., Sun W. & Ossipov D. A. In situ cross-linkable high molecular weight hyaluronan-bisphosphonate conjugate for localized delivery and cell-specific targeting: a hydrogel linked prodrug approach. J. Am. Chem. Soc. 4, 8781–8783 (2009). [DOI] [PubMed] [Google Scholar]
- De Las Heras Alarcon C., Pennadam S. & Alexander C. Stimuli responsive polymers for biomedical applications. Chem. Soc. Rev. 34, 276–285 (2005). [DOI] [PubMed] [Google Scholar]
- Duncan R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discovery 2, 347–360 (2003). [DOI] [PubMed] [Google Scholar]
- Johnston J. F. Nuclease resistance and antisense activity of modified oligonucleotides targeted to Ha-ras. J. Biol. Chem. 271, 14533–14540 (1996). [DOI] [PubMed] [Google Scholar]
- Miller P. Oligonucleoside Methylphosphonates as Antisense Reagents. Nature Biotechnol. 9, 358–362 (1991). [DOI] [PubMed] [Google Scholar]
- Orr F. W., Wang H. H., Lafrenie R. M., Scherbarth S. & Nance D. M. Interactions between cancer cells and the endothelium in metastasis. J. Pathol. 190, 310–329 (2000). [DOI] [PubMed] [Google Scholar]
- Popat K. C., Eltgroth M., Latempa T. J., Grimes C. A. & Desai T. A. Decreased Staphylococcus epidermis adhesion and increased osteoblast functionality on antibiotic-loaded titania nanotubes. Biomaterials 28, 4880–4888 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.