

Abstract
Halophilic Martelella strain AD-3, isolated from highly saline petroleum-contaminated soil, can efficiently degrade polycyclic aromatic hydrocarbons (PAHs), such as phenanthrene and anthracene, in 3–5% salinity. Gentisic acid is a key intermediate in the microbial degradation of PAH compounds. However, there is little information on PAH degradation by moderately halophilic bacteria. In this study, a 1,077-bp long gene encoding gentisate 1,2-dioxygenase (GDO) from a halophilic Martelella strain AD-3 was cloned, sequenced, and expressed in Escherichia coli. The recombinant enzyme GDO was purified and characterized in detail. By using the 18O isotope experiment and LC-MS analysis, the sources of the two oxygen atoms added onto maleylpyruvate were identified as H2O and O2, respectively. The Km and kcat values for gentisic acid were determined to be 26.64 μM and 161.29 s−1, respectively. In addition, optimal GDO activity was observed at 30 °C, pH 7.0, and at 12% salinity. Site-directed mutagenesis demonstrated the importance of four highly conserved His residues at positions 155, 157, 167, and 169 for enzyme activity. This finding provides new insights into mechanism and variety of gentisate 1,2-dioxygenase for PAH degradation in high saline conditions.
Gentisate (2,5-dihydroxybenzoate) is a key intermediate metabolite in the aerobic degradation of a large number of monocyclic and polycyclic aromatic hydrocarbons (PAHs) by microorganisms. In the gentisate pathway, gentisate is cleaved by gentisate 1,2-dioxygenase (GDO), to form maleylpyruvate1. Maleylpyruvate is isomerized to fumarylpyruvate and then hydrolyzed to TCA cycle intermediates, such as fumarate and pyruvate.
Until now, GDOs have been purified and characterized from Ralstonia (Pseudomonas) sp. U21, Klebsiella pneumoniae M5a11, Corynebacterium glutamicum ATCC 130322, Rhodococcus opacus R73, Sphingomonas sp. RW54, Xanthobacter polyaromaticivorans5, Pseudomonas alcaligenes NCIB 9867 (xlnE, xbzE)6,7, Haloarcula sp. D18, Haloferax sp. D12278,9, Pseudomonas alcaligenes NCIMB 986710, and Silicibacter (Ruegeria) pomeroyi DSS-311. However, to our knowledge, there is relatively little information about GDO from moderately halophilic bacteria. Comparative studies of the characterization of different gentisate dioxygenases from moderately halophilic bacteria will contribute to the comprehension of the limitations of degrading of PAHs.
Martelella sp. strain AD-3, a moderate halophilic bacterium, was isolated from highly saline petroleum-contaminated soil in Shandong province, China12. It is highly effective in degrading many PAHs, such as naphthalene, anthracene, and phenanthrene, under broad salinities (0.1–15%) and varying pH (6.0–10.0)13. Salicylic acid was also utilized by Martelella sp. strain AD-3. At the same time, significantly, high activity of GDO was observed13. Furthermore, genes related to degrading gentisic acid has been annotated in the genome sequence of strain AD-314.
In this study, we cloned, expressed, and characterized the gene encoding gentisate 1,2-dioxygenase from Martelella AD-3. Moreover, through site-directed mutagenesis, we determined that four His residues in GDO from strain AD-3 are important for its enzymatic activity. Finally, the source of the two oxygen atoms added to the maleylpyruvate was also analyzed.
Results
Gene cloning, identification, and amino acid sequence analysis
The 1,077-bp gentisate 1,2-dioxygenase gene gdo, was found by mining the genome sequence of strain AD-314. Amino acid sequence alignment of GDO from strain AD-3 with other related proteins was performed with the Vector NTI program. Compared with other known sequences, the highest identity (70.8%) was found with Rhodococcus opacus CIR2 (RnoH, AB186916)3, and 60.4% identity with Corynebacterium glutamicum ATCC 130322. Amino acid sequence of the AD-3 shared 30.9% and 29.1% sequence identities with two amino acid sequences from Pseudomonas alcaligenes NCIB 98677. Compared with other two Haloferax strains (Haloferax sp. D1 and Haloferax sp. D1227)8, strain AD-3 had 24.9% and 24.2% identity, respectively. A phylogenetic tree was constructed with GDO proteins from 15 other strains and demonstrated that the protein from strain AD-3 is most closely related to Rhodococcus opacus R73 and Rhodococcus jostii RHA115 (Fig. 1B).
Figure 1. Amino acid sequence analysis results.

(A) Neighbor-joining tree of the predicted GDO amino acid sequence. (B) Excerpts of GDO sequences used for multiple sequence alignment performed by Vector NTI program. Aligned sequences are from halophilic Martelella sp. AD-3 (WP_024706076), Corynebacterium glutamicum ATCC 13032 (NP_602217.1), Rhodococcus opacus R7 (ABH01038.1), Rhodococcus jostii RHA1 (ABG93677.1), Bradyrhizobium japonicum (NP_766750), Pseudomonas alcaligenes NCIB 9867 (xlnE) (AAD49427.1), Xanthobacter polyaromaticivorans (BAC98955.1), Klebsiella pneumoniae M5a1 (WP_004870415), Pseudomonas aeruginosa UCBPP-PA14 (ZP00135722), Sphingomonas sp. RW5 (CAA12267.1), Haloarcula sp. D1 (AAQ79814.1), Haloferax sp. D1227 (AAQ62856.1), Pseudomonas alcaligenes NCIB 9867 (hbzE) (ABD64513.1), Ralstonia (Pseudomonas) sp. U2 (AAD12619.1), Silicibacter (Ruegeria) pomeroyi DSS-3 (AAV97252.1). The positions of the four highly conserved His residues are indicated in the figures.
Expression of gentisate 1,2-dioxygenase in E. coli BL21 (DE3)
E. coli strain BL21(DE3) cells containing the pET28a-gdo plasmid, were cultured for 8 h in the presence of IPTG (1 mM) in LB medium at 0, 16, or 30 °C. The most suitable temperature for expression was at 30 °C (Fig. 2A). Protein GDO was purified, and obtained at 12 mg from 1 L LB medium. SDS-PAGE showed the purified protein to be approximately 38 kDa in size (Fig. 2A). Analysis of the eluted fraction by Native-PAGE produced a prominent band of ~120 kDa (Fig. 2B). When stored at −20 °C for 98 h, enzymatic activity was lost.
Figure 2. Full wavelength scanning results.
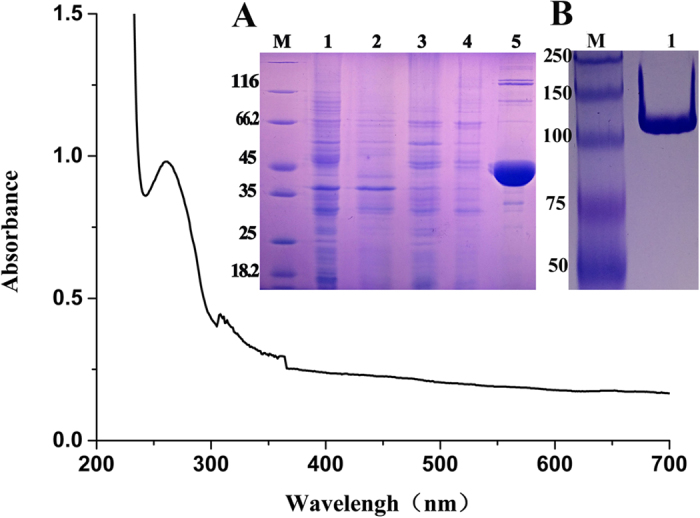
(A) SDS-PAGE analysis of expressed GDO in BL21 (DE3) on a 12.5% gel. Lane M, protein molecular weight marker (MBI); Lane 1, uninduced cells; Lane 2, cell culture induced with 1 mM IPTG; Lane 3, the supernatant of the induced cells; Lane 4, the precipitate of the induced cells; Lane 5, purified recombinant His6-GDO protein; (B) NATIVE-PAGE of purified GDO-His6. Lane M, protein molecular weight marker (MBI); Lane 1, 5 μl purified GDO-His6.
Characteristic of the purified GDO
Effects of pH on GDO activity was measured 5 min after adding GDO to reactions at different pHs. Buffers with pH 4.0–7.0, 7.0–9.0, and 9.0–11.0 were adjusted accordingly with citric acid-sodium citrate, Tris-HCl, and sodium carbonate-sodium bicarbonate buffers, respectively. Enzyme activity of the purified GDO was observed between pH 6.0–8.0 with optimal activity at pH 7.0 (Fig. 3A). Then, effect of temperature on GDO activity was measured at pH 7.0 at temperature ranging from 15–60 °C, maximum enzyme activity was observed at 30 °C (Fig. 3B). Effect of salinity on GDO activity was measured at pH 7.0, 30 °C and salinity ranging from 0 to 16%, optimal activity for the purified GDO was observed at 30 °C, pH 7.0, and salinity at 12% (Fig. 3C). The effects of metal salts at 0.25 mM (Fe2+, Cu2+, Mn2+, Ca2+, Ni2+, Mg2+, Zn2+, and Co2+) were assessed, indicating that Fe2+, Cu2+, Mn2+ can activate the enzyme, while Ca2+, Ni2+, Mg2+, and Zn2+ have no impact on GDO activity. Moreover, the addition of 5 μl Co2+ did inhibit enzyme activity to 53% (Fig. 3D).
Figure 3. Characterization of GDO.

(A) pH optimization of GDO. GDO has optimal activity at pH 7.0. (B) Temperature sensitivity of GDO enzyme activity; optimal activity was obtained at 30 °C. (C) Salinity-dependence of GDO activity; maximum enzyme activity was observed at 12%. (D) Effects of metal salts on enzyme activity; control, without metal salt. Ni, Ni2+; Co, Co2+; Ca, Ca2+; Cu, Cu2+; Mn, Mn2+; Zn, Zn2+; Mg, Mg2+; K, K+; Fe, Fe2+.
Enzyme kinetics of GDO
First, full wavelength scanning of GDO was performed. GDO showed a characteristic absorption peak at 279 nm, which is not the characteristic absorption peak FAD or NADH. Secondly, the result of enzyme kinetics showed that the absorbance at 330 nm steadily increased with time in 1 s, without any transition (Fig. 4). Finally, in order to determine the Km and kcat values, GDO activity was assayed at various gentisate concentrations (0–500 mM). The Km value for gentisate was determined to be 26.64 μM and the kcat value was 161.29 s−1.
Figure 4. Enzyme kinetics of the purified GDO.
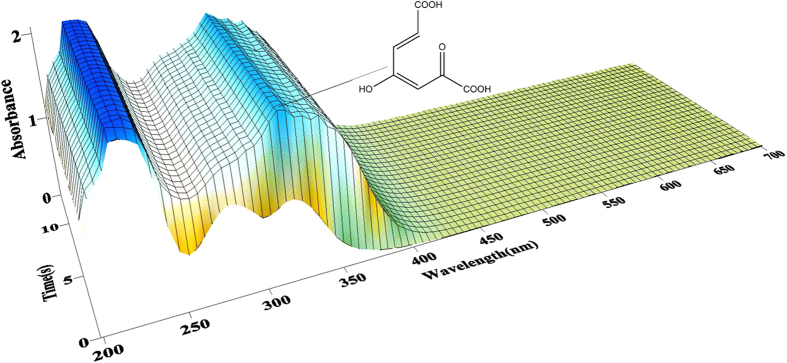
A mixture containing 0.46 mM gentisate in 0.1 M phosphate buffer, pH 7.4, was mixed with an equal volume of (2.4 mg/ml) GDO. Absorbance was measured at times ranging from 0 s to 10 s at 4 °C at the UV absorbance spectra, 330 nm.
Conserved His residue mutations
Multiple amino acid sequence alignment of GDO sequences from various strains showed the presence of four highly conserved His residues at positions 155, 157, 167, and 169. We designed primers to generate mutants with His to Ala amino acids changes. The resultant mutant plasmids (pET28a-H155A, pET28a-H157A, pET28a-H167A, and pET28a-H169A) were expressed in E. coli. The mutant proteins were then purified and the enzyme activity was assayed under the same conditions as for wild type GDO. No enzymatic activity was observed for the four mutant proteins compared to the wild type (Fig. S1).
18O isotope experiments
Two oxygen atoms were added to the degradation product of gentisate, maleylpyruvate. To determine the source of the two oxygen atoms on maleylpyruvate, radio labeled H2O or O2 were supplied to replace the unlabeled H2O or O2 in the reaction. The results (Fig. 5) confirmed that the two oxygen atoms added to the maleylpyruvate were from H2O and O2, one 18O labeled maleylpyruvate m/z 187.0126 was observed in the H218O (Fig. 5B) or 18O2 (Fig. 5C) experiments and two 18O labeled maleylpyruvate m/z 189.0177 were observed in the H218O and 18O2 (Fig. 5D) experiment. As a control, the product of the unlabeled H2O and O2 (Fig. 5A) experiment was analyzed. The labeling experiment results suggest that the two oxygen atoms added to the maleylpyruvate were from H2O and O2, respectively.
Figure 5. 18O isotope experiments.

(A) Control experiment, unlabeled H2O was used as a solvent and unlabeled O2 was used for the reaction. (B) Labeled H218O was used as a solvent and unlabeled O2 was used for reaction. (C) Unlabeled H2O was used as a solvent and labeled 18O2 was used for the reaction. (D) Labeled H218O was used as a solvent and labeled 18O2 was used for the reaction.
RT-qPCR analysis
The mRNA levels of the gene gdo in strain AD-3 grown in the presence or absence of phenanthrene and gentisic acid were compared using RT-qPCR. The transcript level of gene gdo has increased about 6.5 times in the process of degrading phenanthrene and 80.4 times in response to gentisic acid16. (Fig. S2).
Discussion
In recent years, the issue of PAHs pollution has initiated concern among the general public. Specifically, PAHs pollution at high concentrations, in high salinity conditions, and combined pollution17 has been of significant concern. Finding microorganisms with the ability to degrade PAHs in high salinity conditions is very important, however, in general, it is difficult for microorganisms to survive under these conditions. A PAH-degrading bacterial strain was previously isolated from highly saline petroleum-contaminated soil13 and named as halophilic Martelella AD-3. Previous studies indicated that strain AD-3 has a wide spectrum for PAHs substrate degradation though gentisate pathway and a broad range of salinity from 0.1–15%14. Therefore, dissecting the specific features of GDO from AD-3 may be helpful to understand the enzymatic mechanisms of GDO.
In this study, gentisate 1,2-dioxygenase from strain AD-3 was purified and characterized. The maximum enzyme activity was observed at 12% salinity. Optimal activity of GDO from Haloferax sp. D1227 was reported at 2 M KCl or NaCl (10.4% salinity)9. Halophilic proteins function well in high-salt conditions, possessing additional acidic residues (glutamic acid and aspartic acid)18. Compared to the amino acid sequence of GDO from Haloferax sp. D1227, the sequence from AD-3 contains 16.7% additional acidic residues. This may explain why the higher salinity of 12% was the best condition for its enzymatic activity.
The molecular weight of gentisate 1,2-dioxygenase from strain AD-3 (38 kDa) is similar to GDO from Ralstonia solanacearum GMI 1000 (38 kDa)19, Klebsiella pneumoniae (38 kDa)1, Sphingomonas sp. RW5 (38.5 kDa)4, Moraxella sp. VG45 (38 kDa)1, Polaromonas naphthalenivorans CJ2 (NagI3, 38.7 kDa)20, and E. coli O157:H7 (38.9 kDa)21. A prominent band of ~120 kDa was obtained by Native-PAGE analysis, therefore, we proposed the GDO protein might exist as a trimer protein. The result was the same as the previous reported proteins such as from Sphingomonas sp. RW54, E. coli O157:H721, Haloferax sp. D12279, Pseudomonas alcaligenes NCIB 98676. Gentisate 1,2-dioxygenase from strain AD-3 displayed a Km value (26.64 μM) for gentisate, higher than that from Sphingomonas sp. RW5 (15 μM)4, Xanthobacter polyaromaticivorans 127 W (18.6 ± 1.6 μM)5 and Polaromonas naphthalenivorans CJ2 (NagI3, 10 μM)20. Additionally, GDO from strain AD-3 has an optimal pH of 7.0, similar to that of Sphingomonas sp. RW54, and the same optimal temperature 30 °C as Ralstonia solanacearum GMI 100019. Above all, the enzyme characteristics of GDO from strain AD-3 are quite similar to those of Sphingomonas sp. RW54, with the protein sharing 34.4% amino acid sequence identity4.
A previous study reported that Fe2+ increased GDO activity by approximately 160% at Fe2+ concentrations between 0.05–0.10 mM, for Pseudomonas alcaligenes NCIMB 986710. At similar concentrations of Fe2+, the activity of GDO from E. coli O157:H7 was increased by 115%21. For the GDO from strain AD-3, the addition of Fe2+ to a final concentration of 0.25 mM only increased enzymatic activity by 34%. The addition of Fe2+ was also able to restore part activity of the comparely lost activity GDO from strain AD-3. Cu2+, Mn2+ can activate GDO from strain AD-3, however the addition of 1 mM Cu2+, or 10 mM Mn2+ can completely inactivate the GDO from K. pneumoniae21. The enzyme from Pseudomonas alcaligenes NCIB 9867 was inactivated by 5 mM Cu2+7. The crystal structure of E. coli-encoded GDO demonstrates the occupation of three of the possible six iron coordination sites by protein residues, His104, His106, and His14521. In this study, in order to assess whether the effect of Fe2+ on GDO from strain AD-3 is similar to that of others, we performed site-directed mutagenesis, changing each of the four highly conserved His residues to Ala residues. The results suggest that all four highly conserved His residues are crucial for GDO activity. However, in Pseudomonas alcaligenes NCIMB 986722, Silicibacter (Ruegeria) pomeroyi DSS-3 (AAV97252.1)11, Klebsiella pneumoniae M5a1, and Ralstonia sp. strain U21, site-directed mutagenesis was performed, changing His residues to Asp residues, based on the results of error-prone PCR mutagenesis of gdo from Pseudomonas alcaligenes NCIB 98676,23. It is possible that GDO could lose its activity when any one of the four His residues was replaced by Asp residues. His and Asp can carry different charges, so the results of the mutagenesis study are not completely convincing. The results in this present study provide further convincing evidence to demonstrate the importance of the four His residues by changing the four His residues to Ala residues.
In this study, we confirmed the source of the two oxygen atoms introduced into maleylpyruvate by 18O isotope experiments and subsequent LC-MS analysis. Results suggested that the two oxygen atoms are derived from H2O and O2. According to the result, we speculated acquiring the oxygen from the O2 is ‘easier’ than acquiring it from H2O. Because of the traces of 16O2, the one 18O labeled maleylpyruvate m/z 187.0126 was observed (Fig. 5D). No isotopic 18O form H218O was observed in Pseudomonas24. To E. coli O157, H721, Haloferax sp. D12278, K. pneumoniae M5a1, and Ralstonia sp. strain U225, the two oxygen atoms added to maleylpyruvate were reportedly derived only from O2. Here, we identified the source of the oxygen atoms by 18O isotope labeling experiments, providing a new understanding of the mechanism of gentisate degradation.
In summary, we cloned, sequenced, and characterized a novel gene gdo in the degradation of PAHs from Martelella strain AD-3. These findings will expand our knowledge of the mechanisms of gentisate 1,2-dioxygenase enzymatic activity.
Materials and Methods
Chemicals and media
Gentisic acid (98% purity) was purchased from Sangon Biotech (Shanghai, China). All other reagents and solvents used were analytical grade and the highest purity available. Luria-Bertani (LB) broth (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl) was used for both culturing and cloning. Solid agar plates were prepared with the addition of 1.5% (w/v) agar to the liquid medium. 18O2 and H218O were purchased from Shanghai Research Institute of Chemical Industry.
Bacterial strains, plasmids, and growth conditions
Escherichia coli DH5α was used as host for recombinant plasmids, and E. coli BL21(DE3) (Invitrogen, Carlsbad, CA, USA) was used for expression. The gene gdo was amplified from the halophilic Martelella strain AD-3. Gene expression plasmids pET28a were obtained from Invitrogen (Invitrogen, Carlsbad, CA, USA). All recombinant cells were grown in LB broth or LB agar plates (15% [w/v]) containing 50 mg/L kanamycin8.
Expression of His6-tagged GDO
The gene gdo was PCR amplified from the genomic DNA of strain AD-3 with Prime STAR HS DNA polymerase (Takara co. Ltd., Dalia, China)16. Primers were designed such that the forward primer contains an NdeI site and the reverse primer contains a HindIII site. The primer sequences were as follows, forward, 5′-GCCGCATATGAACATGATGATGCCTGAAGA-3′ and reverse, 5′-ATTAAGCTTTCATGCGTCTGCGTCTTCGACC-3′ (NdeI and HindIII recognition sites are underlined). PCR amplification was performed with 100-μl reaction mixtures containing 50 pmol of each primer, 10 μl of a deoxynucleoside triphosphate mixture, 250 ng of template DNA, and 50 μl of 2 × Prime Star buffer. PCR was carried out by using the following program, 5 min at 94 °C and then 30 cycles of 30 s at 94 °C, 30 s at 58 °C, and 2 min 30 s at 72 °C. The PCR products were purified, digested with NdeI and HindIII, and then ligated into pET28a (Novagen Corp., Germany), which had been double digested with the same restriction enzymes. The pET28a-gdo plasmid was transformed into E. coli BL21(DE3). DNA restriction enzymes and T4 DNA ligase were purchased from New England Biolabs26. E. coli BL21(DE3) cells containing pET28a-gdo, were cultured at 37 °C in LB medium containing 50 μg/ml kanamycin to an OD600 of 0.6–0.8. Then, after adding IPTG to a final concentration of 1 mM, the cultures incubated for 8 hours at 16 °C or 20 °C or 30 °C to express GDO. The cells were harvested and resuspended in binding buffer (25 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, pH 8.0) at an OD600 of 30. Cells were then lysed by sonication on ice, and centrifuged at 10,000 × g for 20 min to separate the soluble cell lysate from the insoluble membrane and protein aggregates5.
Purification of GDO
The supernatant was filtered through a 0.22 μm filter and the resultant filtrate was applied to a 5-ml column of Ni-NTA agarose (GE, Healthcare, Little Chalfont, UK), which had been equilibrated with the binding buffer. After a wash with 30 ml of washing buffer (25 mM Tris-HCl, 300 mM NaCl, 70 mM imidazole, pH 8.0), His6-tagged GDO was eluted from the column with elution buffer (25 mM Tris-HCl, 300 mM NaCl, 200 mM imidazole, pH 8.0). All purification steps were carried out at 4 °C.
Enzyme assays and protein determination
Gentisate 1,2-dioxygenase activity was spectrophotometrically assayed at 330 nm by measuring maleylpyruvate formation5. Activity was assayed in 1 ml of reaction mixture containing 0.46 mM gentisate in 0.1 M phosphate buffer, pH 7.4, at 23 °C with a UV-2550 spectrophotometer (Shimadzu, Kyoto, Japan). A molar extinction coefficient of 10.8 × 103 M/cm was used to calculate specific activity20. One enzyme unit was defined as the amount of enzyme that produces 1 mmol of maleylpyruvate per min at 23 °C7. Enzyme activity was assayed 5 min after adding the enzyme to the reaction mixture.
Rapid reaction kinetics
The enzyme kinetics was measured with an SFM 4000 stopped flow apparatus (BioLogic, France). Constant temperature was maintained in a JULABO model F21 temperature-controlled bath (Julabo, Seelbach, Germany). Spectral scans were recorded with a TIDAS S 300 K diode array detector (J&M Analytik AG, Germany)27. Experimental parameters were set as follows, enzyme/substrate solutions, 1/1; UV absorbance spectra, 330 nm; injection rate, 20 μl/s; detection times, 10 ms to 10 s; temperature, 4 °C. Additionally, the enzyme concentration was no lower than 5 mg/ml and the gentisic acid were the same as in the enzyme assays.
Site-directed mutagenesis
Site-directed mutagenesis was performed using a recombinant PCR method. Primers are listed in Table 1. Mutant genes were subcloned into a pET28a vector between the NdeI and HindIII restriction sites, separately. All mutant strains were analyzed by sequencing to confirm disruption of the target gene28.
Table 1. Primers for site-directed mutagenesis.
| Points | Sequence(5′–3′) |
|---|---|
| H155–F | AGCCCGTCATTCCCAGAATGCCTTCCGTTT |
| H155–R | CATTCTGGGAATGACGGGCTTCGGGCGCCGTTTC |
| H157–F | ACACCGTGCCTCCCAGAATGCCTTCCGTTT |
| H157–R | CATTCTGGGAGGCACGGTGTTCGGGCGCCG |
| H167–F | CTTCGCCGGCCACCACAACGAAACCGACCA |
| H167–R | CGTTGTGGTGGCCGGCGAAGTTCCAGCCGGG |
| H169–F | GCCCACAACGAAACCGACCAGCCCATGGCC |
| H169–R | GGTTTCGTTGTGGGCGCCGTGGAAGTTCCA |
DNA and amino acid sequence analysis
Amino acid sequences of GDO from other strains were obtained from GenBank. All homology searches were carried out on the NCBI BLAST server (http,//www.ncbi.nlm.nih.gov/BLAST) with the nucleotide BLAST and protein BLAST. These obtained GDO sequences were then compared with the sequence from AD-3. Conserved binding domain searches were performed using Vector NTI DNA analytical software (version 11.0).
18O isotope experiments
To ascertain the origin of the two oxygen atoms added onto maleylpyruvate, H218O was added to the solvent for the gentisate reaction or 18O2 was added to the anaerobic environment. Then, the assay mixtures of dried GDO powder and gentisic acid dissolved in H218O were incubated at 30 °C for 20 min. Another assay mixture dissolved in H216O was incubated at the same temperature for 20 min, injected with 18O2 at the beginning. As a control, another mixture dissolved in H216O was prepared for reacting in the 16O2 atmosphere. After termination of the reaction by adding 2 volumes of ethanol, samples were analyzed using liquid chromatography-electrospray ionization-mass spectrometry (LC-ESI-MS) (samples were prepared by filtration)28, the negative mode of ESI-MS analysis was used to monitor the products.
Liquid chromatography-mass spectrometry (LC-MS) analysis
Identification of GDO degradation products was carried out by LC-MS (Agilent, Pale Alto, CA, USA) using ESI in the negative ion mode. Substrate solutions were prepared by adding two volumes of ethanol to precipitate the enzyme and filtered through a 0.22-μm Millipore filter 80 min after adding 0 μl or 1 μl GDO enzyme. Then, the samples were automatically injected (5 μl) into the high performance liquid chromatography system. Separation was achieved by ion-pair chromatography on a Luna C18 5 μm column (4.6 × 150 mm, Keystone Scientific, Bellefonte, PA), at 30 °C. Eluent A was deionized H2O (18 MΩ/cm, 0.05% formic acid [v/v]), and eluent B was acetonitrile (99.9%). The working conditions were 90% eluent A and eluent B 10% [v/v] at an injection rate of 0.4 ml/min. The UV absorbance spectra were obtained at 330 nm, on-line16.
RT-qPCR analysis
Total RNA was isolated from strain AD-3, which was cultured in glycerol medium or induced medium with the presence of phenanthrene or gentisic acid, using an RNeasy mini kit (TIANGEN, China)16. Reverse transcription PCR (RT-PCR) was performed using a Prime Script one-step RT-PCR kit (Takara, Japan). Quantitative PCR reactions were carried out using 0.2 ml qPCR tubes (Bio-Rad) and a Chromo 4 real-time PCR thermocycler (Bio-Rad). Reaction was performed by using 20-μl reaction mixtures containing 9 μl PerfeCTa SYBR Green Fast Mix (TIANGEN, China), 0.4 μl of each primer, 10 ng DNA sample and DNase free water to a final volume of 20 μl. The standard curve for each pair of primer was constructed with a tenfold dilution of genomic DNA from strain AD-326. The 16S rRNA gene was used as control. For gene gdo, the thermocycler program used for qPCR was as follow: 95 °C for 3 min, 34 cycles of 20 s at 95 °C, 10 s at 63.2 °C/50.9 °C, and 15 s at 68 °C, then 95.0 °C for 1 min, 55.0 °C 1 min and 5 s, stored at 95 min. The primers for RT-qPCR were 5′-AAGAGGTAAGTGGAATTG-3′ and 5′-CAGTAATGGACCAGTAAG-3′ for 16S rRNA, 5′-ATGATGATGCCTGAAGACA-3′ and 5′-GAGCGGATTGAGGTGATT-3′ for the gene gdo. The threshold cycle (Ct) values for gene gdo from three different conditions, were normalized to the reference gene, 16S rRNA gene. The relative expression level was calculated by using the 2 –ΔΔCt method, where ΔΔCt = (Ct target—Ct 16S rRNA) INDUCTION—(Ct target—Ct 16S rRNA) CONTROL16.
Additional Information
How to cite this article: Huang, L. et al. Identification and Characterization of a Novel Gentisate 1,2-Dioxygenase Gene from a Halophilic Martelella Strain. Sci. Rep. 5, 14307; doi: 10.1038/srep14307 (2015).
Supplementary Material
Acknowledgments
This research was supported by the National Natural Science Foundation of China (41301329 and 51378208), the Chinese National Science Foundation for Excellent Young Scholars (31422004), the Shanghai Municipal Natural Science Foundation (13ZR1410900) and the Open Funding Project of the State Key Laboratory of Bioreactor Engineering.
Footnotes
Author Contributions C.C., L.H. and H.T. conceived and designed the project and experiments. L.H., H.H. and J.S. performed the experiments. L.H., C.C. and H.T. wrote the paper. P.X., Y.L., K.L. and Q.L. reviewed the manuscript. All authors reviewed the paper.
References
- Luo S., Liu D. Q., Liu H. & Zhou N. Y. Site-directed mutagenesis of gentisate 1,2-dioxygenases from Klebsiella pneumoniae M5a1 and Ralstonia sp. strain U2. Microbiol Res 161, 138–144 (2006). [DOI] [PubMed] [Google Scholar]
- Xu Y., Yan D. Z. & Zhou N. Y. Heterologous expression and localization of gentisate transporter Ncg12922 from Corynebacterium glutamicum ATCC 13032. Biochem Biophys Res Co 346, 555–561 (2006). [DOI] [PubMed] [Google Scholar]
- Gennaro P. D. et al. Identification and characterization of genes involved in naphthalene degradation in Rhodococcus opacus R7. Appl Microbiol Biotechnol 87, 297–308 (2010). [DOI] [PubMed] [Google Scholar]
- Werwath J., Arfmann H. A., Pieper D. H., Timmis K. N. & Wittich R. M. Biochemical and genetic characterization of a gentisate 1,2-dioxygenase from Sphingomonas sp. strain RW5. J Bacteriol 180, 4171–4176 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S. I., Morikawa M., Takano K., Imanaka T. & Kanaya S. Gentisate 1,2-dioxygenase from Xanthobacter polyaromaticivorans 127W. Biosci Biotechnol Biochem 71, 192–199 (2007). [DOI] [PubMed] [Google Scholar]
- Chua C. H., Feng Y. M., Yeo C. C., Khoo H. E. & Poh C. L. Identification of amino acid residues essential for catalytic activity of gentisate 1,2-dioxygenase from Pseudomonas alcaligenes NCIB 9867. FEMS Microbiol Lett 204, 141–146 (2001). [DOI] [PubMed] [Google Scholar]
- Feng Y. M., Khoo H. E. & Poh C. L. Purification and characterization of gentisate 1,2-dioxygenases from Pseudomonas alcaligenes NCIB 9867 and Pseudomonas putida NCIB 9869. Appl Environ Microbiol 3, 946–950 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairley D. J., Wang G., Rensing C., Pepper I. L. & Larkin M. J. Expression of gentisate 1,2-dioxygenase (gdoA) genes involved in aromatic degradation in two Haloarchaeal genera. Appl Microbiol Biotechnol 73, 691–695 (2006). [DOI] [PubMed] [Google Scholar]
- Fu W. & Oriel P. Gentisate 1,2-dioxygenase from Haloferax sp. D1227. Extremophiles 2, 439–446 (1998). [DOI] [PubMed] [Google Scholar]
- Yeo C. C., Tan C. L., Gao X. L., Zhou B. & Pho C. L. Characterization of hbzE-encoded gentisate 1,2-dioxygenase from Pseudomonas alcaligenes NCIMB 9867. Res Microbiol 158, 608–616 (2007). [DOI] [PubMed] [Google Scholar]
- Chen J. et al. J. Crystal structure and mutagenic analysis of GDOsp, a gentisate 1,2-dioxygenase from Silicibacter pomeroyi. Protein Sci 17, 1362–1373 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C. Z. et al. Metabolic pathway for degradation of anthracene by halophilic Martelella sp. AD-3. Int Biodeter Biodegr 89, 67–73 (2014). [Google Scholar]
- Feng T. C. et al. Phenanthrene biodegradation by halophilic Martelella sp. AD-3. J Appl Microbiol 113, 779–789 (2012). [DOI] [PubMed] [Google Scholar]
- Cui C. Z. et al. Genome sequence of Martelella sp. strain AD-3, a moderately halophilic polycyclic aromatic hydrocarbon-degrading bacterium. Genome announc 2, 01189–01113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montersino S. & Berkel W. Functional annotation and characterization of 3-hydroxybenzoate-6-hydroxylase from Rhodococcus Jostii RHA1. Biochim Biophys Acta 1824, 433–442 (2012). [DOI] [PubMed] [Google Scholar]
- Yu H., Tang H. Z., Zhu X. Y., Li Y. Y. & Xu P. Molecular mechanism of nicotine degradation by a newly isolated strain, Ochrobactrum sp. strain SJY1. Appl Environ Microbiol 81, 272–281 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J. et al. Polycyclic aromatic hydrocarbons and ecotoxicological characterization of sediments from the huaihe river, China. J Environ Monit 13, 597–604 (2011). [DOI] [PubMed] [Google Scholar]
- Dennis P. P. & Shimmin L. C. Evolutionary divergence and salinity-mediated selection in Halophilic archaea. Microbiol Mol Biol Rev 61, 90–104 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D., Zhu S. & Ni J. Purification and characterisation of a gentisate 1,2-dioxygenase from Ralstonia solanacearum GMI 1000. Ann Microbiol 57, 307–312 (2007). [Google Scholar]
- Lee H. J. et al. Gentisate 1,2-dioxygenase, in the third naphthalene catabolic gene cluster of polaromonas naphthalenivorans CJ2, has a role in naphthalene degradation. Microbiology 157, 2891–2903 (2011). [DOI] [PubMed] [Google Scholar]
- Adams M. A., Singh V. K., Keller B. O. & Jia Z. H. Structural and biochemical characterization of gentisate 1,2-dioxygenase from Escherichia coli O157:H7. Mol Microbiol 61, 1469–1484 (2006). [DOI] [PubMed] [Google Scholar]
- Tan C. L., Yeo C. C., Khoo H. E. & Poh C. L. Replacement of tyrosine 181 by phenylalanine in gentisate 1,2-dioxygenase I from Pseudomonas alcaligenes NCIMB 9867 enhances catalytic activities. J Bacteriol 187, 7543–7545 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraroni M. et al. The salicylate 1, 2-dioxygenase as a Model for a conventional gentisate 1,2-dioxygenase, crystal structures of the G106a mutant and its adducts with gentisate and salicylate. FEBS J 280, 1643–1652 (2013). [DOI] [PubMed] [Google Scholar]
- Harpel M. R. & Lipscomb J. D. Gentisate 1,2-dioxygenase from pseudomonas. Purification, characterization, and comparison of the enzymes from Pseudomonas testosteroni and Pseudomonas acidovorans. J Biol Chem 265, 22187–22196 (1990). [PubMed] [Google Scholar]
- Zhou N. Y., Fuenmayor S. L. & Williams P. A. Nag genes of Ralstonia (formerly Pseudomonas) sp. strain U2 encoding enzymes for gentisate catabolism. J Bacteriol 183, 700–708 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. Z. et al. Novel nicotine oxidoreductase-encoding gene involved in nicotine degradation by Pseudomonas putida strain S16. Appl Environ Microbiol 75, 772–778 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Hausinger R. P., Tang H. Z. & Xu P. Mechanism of the 6-hydroxy-3-succinoyl-pyridine 3-monooxygenase flavoprotein from Pseudomonas putida S16. J Biol Chem 289, 29158–29170 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. Z. et al. A novel NADH-dependent and FAD-containing hydroxylase is crucial for nicotine degradation by Pseudomonas putida. J Biol Chem 286, 39179–39187 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.