

Abstract
Background
The lizards of the family Agamidae are one of the most prominent elements of the Australian reptile fauna. Here, we present a genomic resource built on the basis of a wild-caught male ZZ central bearded dragon Pogona vitticeps.
Findings
The genomic sequence for P. vitticeps, generated on the Illumina HiSeq 2000 platform, comprised 317 Gbp (179X raw read depth) from 13 insert libraries ranging from 250 bp to 40 kbp. After filtering for low-quality and duplicated reads, 146 Gbp of data (83X) was available for assembly. Exceptionally high levels of heterozygosity (0.85 % of single nucleotide polymorphisms plus sequence insertions or deletions) complicated assembly; nevertheless, 96.4 % of reads mapped back to the assembled scaffolds, indicating that the assembly included most of the sequenced genome. Length of the assembly was 1.8 Gbp in 545,310 scaffolds (69,852 longer than 300 bp), the longest being 14.68 Mbp. N50 was 2.29 Mbp. Genes were annotated on the basis of de novo prediction, similarity to the green anole Anolis carolinensis, Gallus gallus and Homo sapiens proteins, and P. vitticeps transcriptome sequence assemblies, to yield 19,406 protein-coding genes in the assembly, 63 % of which had intact open reading frames. Our assembly captured 99 % (246 of 248) of core CEGMA genes, with 93 % (231) being complete.
Conclusions
The quality of the P. vitticeps assembly is comparable or superior to that of other published squamate genomes, and the annotated P. vitticeps genome can be accessed through a genome browser available at https://genomics.canberra.edu.au.
Electronic supplementary material
The online version of this article (doi:10.1186/s13742-015-0085-2) contains supplementary material, which is available to authorized users.
Keywords: Pogona vitticeps; Dragon lizard; Central bearded dragon; Agamidae, Squamata, Next-generation sequencing
Data description
The central bearded dragon, Pogona vitticeps, is widespread through the arid and semi-arid regions of eastern central Australia. This lizard adapts readily to captivity, lays large clutches of eggs several times per season, and is kept as a favoured pet species in Europe, Asia and North America. The karyotype of P. vitticeps is typical of most Australian agamids, consisting of six pairs of macrochromosomes and ten pairs of microchromosomes (2n = 32) [1]. The sex determining mechanism is one of female heterogamety (ZZ/ZW) and the sex chromosomes are a pair of microchromosomes [2]. Sex determination, a primary driver for our interest in generating this genome sequence, is complex in this species, involving an interaction between the influences of incubation environment and the ZZ/ZW genotype [3, 4].
Samples and sequencing
DNA samples were obtained from a blood sample taken from a single male Pogona vitticeps (Fabian, UCID 001003387339) verified as a ZZ male using sex-linked polymerase chain reaction (PCR) markers [3] and cytological examination [2]. This work was undertaken in accordance with the Australian Capital Territory Animal Welfare Act 1992 and the approval of the University of Canberra Animal Ethics Committee. DNA was extracted and purified using standard protocols and transported to BGI-Shenzhen, China for sequencing. 13 insert libraries were constructed with insert sizes of 250 bp, 500 bp, 800 bp, 2 kbp (x2), 5 kbp, 6 kbp, 10 kbp (x2), 20 kbp (x2) and 40 kbp (x2) and subjected to paired-end sequencing on an Illumina HiSeq 2000 platform to generate 317 Gbp of raw sequence (Table 1). After filtering for low-quality reads and duplicated reads arising from PCR amplification during library construction, 146.38 Gbp of data were retained for genome assembly. This amount of data represents an average read depth of 82.7 (Table 1), assuming a genome size of 1.81 pg, as estimated for a female P. vitticeps by flow cytometry [5]. This mass converts to a genome size of 1.77 Gbp [6].
Table 1.
Summary of sequencing data derived from paired-end sequencing of 13 insert libraries using an Illumina HiSeq 2000 platform
| Raw data | Filtered data | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Insert size (bp) | Accession numbers | Nunber of libraries | Read length (bp) | Raw data (Gbp) | Average read depth (X) | Physical coverage (X) | Read length (bp) | Filtered data (Gbp) | Average read depth (X) | Physical coverage (X) |
| 250 | ERR409943 | 1 | 150 | 55.17 | 31.17 | 25.97 | 125 | 42.49 | 24.01 | 24.00 |
| ERR409944 | ||||||||||
| 500 | ERR409945 | 1 | 150 | 34.32 | 19.39 | 32.32 | 125 | 23.66 | 13.37 | 26.72 |
| ERR409946 | ||||||||||
| 800 | ERR409947 | 1 | 150 | 46.28 | 26.15 | 69.72 | 125 | 32.2 | 18.19 | 60.63 |
| 2,000 | ERR440173 | 2 | 49 | 38.39 | 21.69 | 442.64 | 49 | 18.19 | 10.28 | 209.73 |
| ERR409948 | ||||||||||
| 5,000 | ERR409949 | 1 | 49 | 17.48 | 9.88 | 503.95 | 49 | 6.56 | 3.71 | 188.99 |
| 6,000 | ERR409950 | 1 | 49 | 17.43 | 9.85 | 603.01 | 49 | 6.01 | 3.4 | 208.00 |
| 10,000 | ERR409951 | 2 | 49 | 34.94 | 19.74 | 2,014.60 | 49 | 7.89 | 4.46 | 455.00 |
| ERR409952 | ||||||||||
| 20,000 | ERR409953 | 2 | 49 | 38.53 | 21.77 | 4,443.48 | 49 | 6.63 | 3.75 | 764.38 |
| ERR409954 | ||||||||||
| 40,000 | ERR409955 | 2 | 49 | 34.4 | 19.44 | 7,932.30 | 49 | 2.75 | 1.55 | 633.29 |
| ERR409956 | ||||||||||
| 13 | 316.94 | 179.06 | 16,067.99 | 146.38 | 82.7 | 2,570.74 | ||||
Read depth was calculated on the basis of a genome size of 1.77 Gbp. Average read depth, number of times on average a particular base is included in a read. Physical coverage, the number of times on average a particular base is spanned by a paired read
Reads from the short-insert libraries (250, 500 and 800 bp) were decomposed into short sequences of length k (k-mers, with k = 17) using Jellyfish version 1.1 [7]. The histogram of k-mer copy number (Fig. 1) was strongly bi-modal, the first mode with a copy number that was half that of the second, which reflects the high level of heterozygosity in this wild-caught lizard (0.85 % of single nucleotide polymorphisms [SNPs] plus sequence insertions or deletions [indels]). The second mode in the k-mer graph was used to obtain an estimate of the genome size using the formula:
where L is the read length (125 bp), k is the k-mer length (17 bp), there are 98.35 Gbp of sequence data, and the mode is taken from the k-mer graph (48.5, Fig. 1). Our sequence-based estimate of 1.768 Gbp agrees well with the estimate of 1.77 Gbp for the ZW genome that was previously made using flow cytometry data [4].
Fig. 1.
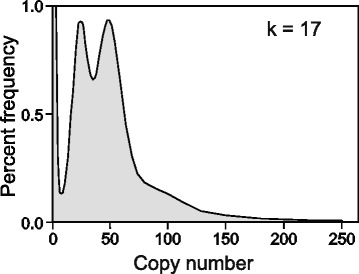
K-mer spectrum for the genome sequence of a male Pogona vitticeps (ZZ). Sequencing DNA derived from the short-insert libraries (250, 500, 800 bp) yielded 98.35 Gbases of clean data in the form of 125 bp reads, which generated 76.89x109 17-mer sequences. The solid line shows the k-mer spectrum (percentage frequency against k-mer copy number). The second mode (copy number 48.5) represents homozygous single copy sequence, whereas the first mode (24.5), half the copy number of the first, represents heterozygous single copy sequence. Heterozygosity is high, which complicated assembly
Assembly
Assembly was performed with SOAPdenovo (version 2.03) [8, 9]. Briefly, the sequences derived from the short-insert libraries were decomposed into k-mers to construct the de Bruijn graph, which was simplified to allow connection of the remaining k-mers into contiguous sequence (contigs). We then aligned the paired-end reads from small and large-insert library sets to the contigs, calculated the support for relationships between contigs, assessed the consistent and conflicting relationships, and constructed scaffolds. Finally, we retrieved paired reads that mapped to a unique contig but had the other member of the pair located in a gap region. Reads falling in the same gap region were then assembled locally. The final assembly (European Nucleotide Archive [ENA] accession number ERZ094017) yielded a contig N50 of 31.3 kbp and a scaffold N50 of 2.3 Mbp (N50 meaning that 50 % of the genome sequence is contained in contigs, or scaffolds, equal to or greater than this length), with unclosed gap regions representing only 3.78 % of the assembly (Table 2).
Table 2.
Statistics for the assembly contigs and scaffolds (after gap filling)
| Contig | Scaffold | |||
|---|---|---|---|---|
| Size (bp) | Number | Size (bp) | Number | |
| N90 | 4,850 | 63,958 | 200,992 | 1,095 |
| N80 | 12,159 | 42,491 | 670,865 | 644 |
| N70 | 18,332 | 30,884 | 1,149,567 | 441 |
| N60 | 24,540 | 22,654 | 1,671,674 | 311 |
| N50 | 31,298 | 16,344 | 2,290,546 | 219 |
| Longest | 295,776 | 14,681,335 | ||
| Total size | 1,747,524,961 | 1,816,115,349 | ||
| ≥100 bp | 636,524 | 545,300 | ||
| ≥2 kbp | 79,002 | 4,356 | ||
| Gap ratio | 0 % | 3.78 % | ||
Reads from small-insert libraries that satisfied our filtering criteria were aligned to the assembly using the Burrows-Wheeler Aligner (BWA, version 0.5.9-R16) [10], allowing for eight mismatches and one indel. Of the total number of reads (797.4 M), 96.4 % could be mapped back to the assembled genome and they covered 98.4 % of the assembly excluding gaps. Bases in the assembled scaffolds had, on average, reads mapped with 55X read depth. These data suggest that we have assembled most of the P. vitticeps genome. In addition, we used the CEGMA package (version 2.4) [11] to map 248 core eukaryotic genes to our P. vitticeps assembly. Our assembly captured 99 % (246 of 248) of the core CEGMA genes, with 93 % (231) being complete. This is a higher assembly rate than that estimated for the green anole Anolis carolinensis assembly (AnoCar2.0), which captured 93.6 % (232) of the core genes, with 85.9 % (213) being complete.
Transcriptomes
We generated transcriptome data from the brain, heart, lung, liver, kidney, skeletal muscle and gonads of male and female P. vitticeps (Table 3). None of the seven animals from which we collected RNA was used in generating the genome sequence. Two sets of sequencing runs on two different male and female individuals were performed by BGI-Shenzen, producing 309,436,077 90 bp paired-end reads (ENA accession numbers ERR753524-ERR753530 and ERR413064-ERR413076). A third set of samples was sequenced by The Ramaciotti Centre, University of New South Wales, Australia, including a sex-reversed ZZ female, producing 89,687,526 101 bp paired-end reads (ENA accession numbers ERR413077- ERR413082). We assembled these datasets (from all seven individuals) into 595,564 contigs using Trinity (release r2013_08_14) [12] with default parameters (ENA accession number ERZ097159). Only the first set of RNA-seq reads was available for genome annotation (ENA accession numbers ERR753524-ERR753530) but we make the entire dataset, including our de novo assembly, available with this article (see ‘Availability of supporting data’ section).
Table 3.
Number of predicted genes with RNA-seq signals
| Specimen ID (tissue ID) | Accession number | Tissue | Genotype | Phenotype | RPKM >0 | RPKM >1 | RPKM >5 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Number | Ratio (%) | Number | Ratio (%) | Number | Ratio (%) | |||||
| 1003347859 | ERR753524 | Brain | ZZ | Intersex | 17,049 | 87.85 | 14,403 | 74.22 | 11,244 | 57.94 |
| (AA45100) | ||||||||||
| 1003338787 | ERR753525 | Brain | ZZ | Male | 16,934 | 87.26 | 14,467 | 74.55 | 11,359 | 58.53 |
| (AA60463) | ||||||||||
| 1003348364 | ERR753526 | Brain | ZW | Female | 17,121 | 88.23 | 14,526 | 74.85 | 11,474 | 59.13 |
| (AA60435) | ||||||||||
| 1003347859 | ERR753527 | Testes | ZZ | Intersex | 16,874 | 86.95 | 13,874 | 71.49 | 10,784 | 55.57 |
| (AA45100) | ||||||||||
| 1003347859 | ERR753528 | Ovary | ZZ | Intersex | 16,827 | 86.71 | 12,952 | 66.74 | 10,421 | 53.7 |
| (AA45100) | ||||||||||
| 1003338787 | ERR753529 | Testes | ZZ | Male | 17,963 | 92.56 | 14,951 | 77.04 | 11,311 | 58.29 |
| (AA60463) | ||||||||||
| 1003348364 | ERR753530 | Ovary | ZW | Female | 17,188 | 88.57 | 13,634 | 70.26 | 10,946 | 56.41 |
| (AA60435) | ||||||||||
| Combined | 18,833 | 97.05 | 17,646 | 90.93 | 15,974 | 82.31 | ||||
Gene expression levels were measured as RPKM (reads per kilobase of gene per million mapped reads). Ratios are based on a total of 19,406 annotated protein-coding genes
Annotation
Transposable elements and other repetitive elements were identified using a combination of homology, at both the DNA and protein levels, and de novo prediction. In the homology-based approach, we searched Repbase [13] for known transposable elements, used RepeatMasker [14] for DNA homology search against the Repbase database, and used WuBlastX to search against the transposable element protein database provided within RepeatProteinMask (bundled in RepeatMasker). In the de novo approach, we used RepeatModeler [15] and LTR_FINDER [16] to predict repetitive elements. Tandem repeats were identified using Tandem Repeats Finder [17]. The relative success of the different approaches is shown in Table 4. Overall, we identified about 690 Mbp of repetitive sequences accounting for 39.47 % of the genome, of which the predominant elements were long interspersed nuclear elements (LINEs, which accounted for 33 % of repetitive sequences representing 12.2 % of the genome) (Table 5).
Table 4.
The statistics for repeats in the P. vitticeps genome annotated by different methods
| Program | Total repeat length (bp) | Percentage of genome |
|---|---|---|
| Tandem Repeats Finder | 59,773,950 | 3.42 |
| Repeatmasker | 174,011,206 | 9.96 |
| Proteinmask | 157,050,977 | 8.99 |
| RepeatModeler | 592,771,829 | 33.92 |
| LTR Finder | 65,464,996 | 3.75 |
| Total | 689,687,572 | 39.47 |
Table 5.
Breakdown of repeat content of the Pogona vitticeps genome derived from RepeatMasker analysis
| Category | Repbase TEs | TE proteins | de novo | Combined TEs | ||||
|---|---|---|---|---|---|---|---|---|
| Length (bp) | % of genome | Length (bp) | % of genome | Length (bp) | % of genome | Length (bp) | % of genome | |
| DNA | 25,035,683 | 1.43 | 6,450,126 | 0.37 | 56,943,252 | 3.26 | 70,663,766 | 4.04 |
| LINE | 124,676,466 | 7.13 | 132,747,210 | 7.60 | 191,015,014 | 10.93 | 213,508,152 | 12.22 |
| SINE | 20,281,741 | 1.16 | - | 0.00 | 54,941,907 | 3.14 | 57,180,364 | 3.27 |
| LTR | 7,613,766 | 0.44 | 17,931,338 | 1.03 | 16,104,019 | 0.92 | 28,021,391 | 1.60 |
| Other | 24,327 | 0.00 | - | 0.00 | - | 0.00 | 24,327 | 0.00 |
| Unknown | 761,119 | 0.04 | - | 0.00 | 283,563,847 | 16.23 | 284,276,315 | 16.27 |
| Total | 174,011,206 | 9.96 | 157,050,977 | 8.99 | 627,828,869 | 35.93 | 657,625,603 | 37.63 |
Abbreviations: LINE long interspersed nuclear element, LTR long terminal repeat, SINE short interspersed nuclear element, TE transposable element
We combined homology-based, de novo and transcriptome-based methods to predict gene content of the assembly. In the homology-based prediction, the assembly was annotated by generating reference sets of A. carolinensis, Gallus gallus and Homo sapiens proteins, and aligning the reference sets to the assembly using TBLASTN (version 2.2.23; E-value ≤ 1 × 10−5). The resultant homologous genome sequences were then aligned against matching proteins using Genewise (version wise2-2-0) [18] to define gene models. In the de novo prediction, we randomly selected 1,000 genes with intact open reading frames (ORFs) as predicted by the homology-based approach to train the Augustus gene prediction tool (version 2.5.5) [19] with the parameters appropriate to P. vitticeps. The de novo gene prediction was then performed with Augustus applied to the genome after repeat sequences were masked as described above. In the transcriptome-based approach, we mapped transcriptome reads to the assembly using TopHat (version 1.3.1) [20], which can align reads across splice junctions. These mapped reads were assembled into transcripts using Cufflinks (version 1.3.0) [21] and then merged across samples (n = 7, Table 3) into a single transcriptome annotation using the Cuffmerge option.
The results of the three approaches were combined into a non-redundant gene set of 19,406 protein-encoding genes, 63 % of which included intact ORFs (Table 6). Most of the predicted genes were supported by RNA-seq signals (Table 3).
Table 6.
Characteristics of predicted protein-coding genes in the Pogona vitticeps assembly and comparison with Anolis carolensis, Gallus gallus and Homo sapiens
| Gene set | Total | Intact ORF | Single exon gene | Gene length (bp) | mRNA length (bp) | Exons per gene | Exon length (bp) | Intron length (bp) | |
|---|---|---|---|---|---|---|---|---|---|
| Homolog | Anolis carolinensis | 16,009 | 2,583 | 1,668 | 23,021 | 1,524 | 8.57 | 178 | 2,839 |
| Gallus gallus | 12,727 | 2,068 | 1,509 | 27,608 | 1,558 | 9.06 | 172 | 3,232 | |
| Homo sapiens | 13,544 | 2,456 | 1,250 | 32,551 | 1,699 | 9.75 | 174 | 3,528 | |
| Combined | 18,033 | 3,263 | 2,180 | 26,631 | 1,577 | 8.93 | 177 | 3,160 | |
| De novo (Augustus) | 32,110 | 32,110 | 6,767 | 14,109 | 1,125 | 6.07 | 185 | 2,561 | |
| Transcriptome | 22,986 | 14,555 | 2,951 | 12,511 | 1,214 | 6.99 | 174 | 1,885 | |
| Merged | 19,406 | 12,172 | 1,999 | 26,215 | 1,642 | 9.24 | 178 | 2,984 | |
| Other species | Anolis carolinensis | 17,805 | 4,280 | 1,372 | 23,469 | 1,526 | 9.55 | 160 | 2,566 |
| Gallus gallus | 16,736 | 7,777 | 1,684 | 21,314 | 1,438 | 9.35 | 154 | 2,379 | |
| Homo sapiens | 21,849 | 20,905 | 2,602 | 46,301 | 1,635 | 9.44 | 173 | 5,293 | |
Except for the columns headed Total, Intact ORF and Single exon gene, the values presented are means.
Abbreviation: ORF open reading frame
To assign gene names to each predicted protein-coding locus, we mapped the 19,406 genes to an Ensembl library collated from A. carolinensis, chicken G. gallus, human H. sapiens, western clawed frog Xenopus tropicalis and zebrafish Danio rerio. The name associated with the best hit for each P. vitticeps gene was assigned to each of 19,083 genes. Most of these genes (16,510) mapped to a homolog even at high stringency (>80 % of protein length aligned).
Bacterial artificial chromosome library
A large-insert genomic DNA bacterial artificial chromosome (BAC) library was constructed from DNA from a wild-caught female dragon lizard (TC1542) confirmed to have the ZW genotype using sex-linked PCR markers [3, 4] and cytologically [3]. The library is estimated to represent 6.3× of genome coverage, and is comprised of 92,160 clones with an average insert size of 120 kbp. This resource is commercially available through Amplicon Express (Pullman, WA, USA; http://ampliconexpress.com).
Anchoring sequences to chromosomes
Our previously published cytogenetic map of P. vitticeps consisted of 87 BACs that were mapped to the macrochromosomes (64 BACs) and microchromosomes (23 BACs) [1]. We mapped an additional 80 BACs, extending the set to 125 markers on macrochromosomes and 42 on microchromosomes. Sequence scaffolds were anchored to chromosomes by 52 loci, contained in the BACs, that are conserved in homologous syntenic blocks across amniotes (A. carolinensis, G. gallus, H. sapiens). By using gene synteny information 37.9 % (670 Mbp) of the sequenced genome has been assigned to chromosomes (Deakin et al., unpublished data).
Sex chromosome sequences
The sex of P. vitticeps is determined by a combination of chromosomal constitution and influence of environmental temperature on the developing embryo. P. vitticeps has female heterogamety (with ZZ male and ZW female individuals), and the Z and W chromosomes are among the ten pairs of microchromosomes [2]. Sex chromosome heteromorphy is evident by C-banding, but the degree of differentiation of the Z and W chromosomes is slight [2]. The sex chromosomes of P. vitticeps are not homologous to the sex chromosomes of chicken (G. gallus) or other reptiles so far examined [22]. The ZZ genotype is reversed to a female phenotype at high incubation temperatures [3, 4].
Our laboratory has previously identified a sex-linked sequence using amplified fragment length polymorphism screening and genome walking [4, 23]. Five contiguous BAC clones containing sex-linked markers that map to the sex chromosome pair were sequenced to reveal 352 kbp of P. vitticeps sex chromosome sequence [24]. This region contained five protein-coding genes (oprd1, rcc1, znf91, znf131 and znf180) and several major families of repetitive sequences (long terminal repeat [LTR] and non-LTR retrotransposons, including chicken repeat 1 [CR1] and bovine B LINEs [Bov-B LINEs]) [1, 24].
More recently, we amplified micro-dissected W-chromosome fragments to yield many sex chromosome sequence tags that were reciprocally mapped to their Z homologs (Matsubara et al., unpublished data). All putative sex chromosome scaffolds were confirmed to co-localize with the known ZW-BAC Pv3-L07 when physically mapped (Deakin et al., unpublished data). In this way we identified 12.8 Mbp of the Z chromosome (on three scaffolds) and increased the number of confirmed sex chromosome genes to 240 (Deakin et al., unpublished data).
GC content and isochore structure
We investigated patterns of GC content variation in the P. vitticeps genome using two approaches. First, we examined the absolute GC content in non-overlapping 5 kbp windows for several genomes (P. vitticeps, A. carolinensis [25], Burmese python Python bivittatus [26], king cobra Ophiophagus hannah [27], western painted turtle Chrysemys picta [28],Chinese softshell turtle Pelodiscus sinensis [29], saltwater crocodile Crocodylus porosus [30] chicken G. gallus, mouse Mus musculus, domestic dog Canis familiaris [31] and western clawed frog X. tropicalis [32]; Table 7; Fig. 2). We then examined variation in GC composition for these same genomes at increasing spatial scales (5, 10, 20, 40, 80, 160 and 320 kbp windows; Fig. 3). We also looked at different subsets of the P. vitticeps genome, including macrochromosomes and microchromosomes, and the Z chromosome (Fig. 4a), by restricting the analysis to scaffolds that have been physically mapped (Deakin et al., unpublished data).
Table 7.
Comparison of mean GC content for available tetrapod genomes
| Organism | Genome version | Mean GC | SD |
|---|---|---|---|
| Pogona vitticeps | pvi1.1.Jan.2013 | 0.418 | 0.037 |
| P. vitticeps - microchromosomes | pvi1.1.Jan.2013 | 0.445 | 0.050 |
| P. vitticeps - macrochromosomes | pvi1.1.Jan.2013 | 0.409 | 0.029 |
| P. vitticeps - Z chromosome | pvi1.1.Jan.2013 | 0.469 | 0.037 |
| Xenopus tropicalis | JGI_4.2 | 0.398 | 0.038 |
| Anolis carolinensis | AnoCar2 | 0.403 | 0.032 |
| Canis familiaris | CanFam3.1 | 0.413 | 0.069 |
| Mus musculus | GRCm38 | 0.417 | 0.046 |
| Gallus gallus | Galgal4 | 0.416 | 0.059 |
| Crocodylus porosus | croc_sub2 | 0.442 | 0.050 |
| Pelodiscus sinensis | PelSin_1.0 | 0.441 | 0.053 |
| Chrysemys picta | ChrPicBel3.0.1 | 0.437 | 0.055 |
| Python bivittatus | python_5.0 | 0.396 | 0.042 |
| Ophiophagus hannah | GCA_000516915.1 (NCBI) | 0.386 | 0.040 |
Abbreviation: SD standard deviation
Fig. 2.
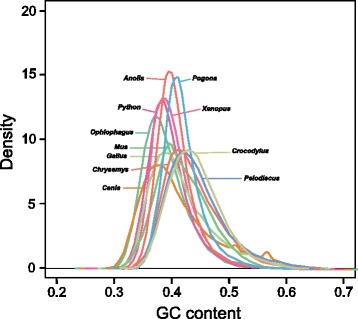
Distribution of GC content in 5 Kbp windows for a range of vertebrates including Pogona vitticeps
Fig. 3.
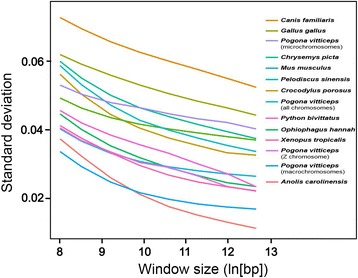
Variation in GC content among windows for various genome sequences with increasing window size (5, 10, 20, 40, 80, 160, and 320 Kb windows). The relationship for Pogona vitticeps is disaggregated to macrochromosomes, microchromosomes and the Z sex chromosome for comparison. Scale of X axis is natural logarithm. Pogona macrochromosomes share the lack of isochore structure reported for the Anolis genomeᅟ
Fig. 4.
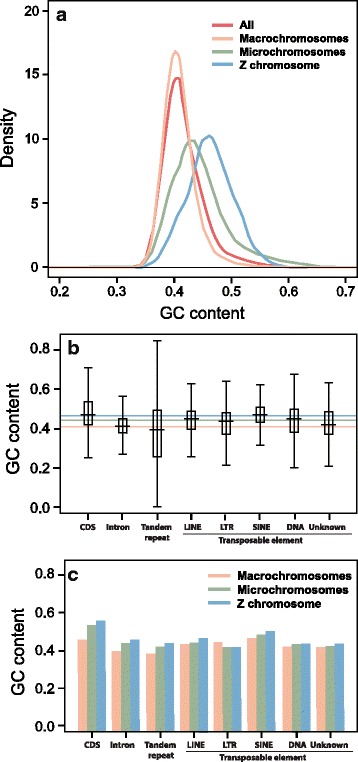
Analysis of GC content in Pogona vitticeps. a, Distribution of GC content in all chromosomes, macrochromosomes, microchromosomes and the Z chromosome, calculated with a non-overlap 5-kb sliding windows ; b, GC content of various components of the genome, in comparison with the average GC content for macrochromosomes (red line), microchromosomes (green line) and the Z chromosome (blue line) ; c, GC content of the macrochromosomes, microchromosomes and Z chromosomes broken down for various components of the genome
The macrochromosomes of P. vitticeps are largely devoid of variation in GC content at small (5 kbp) spatial scales. In fact, P. vitticeps macrochromosomes are more uniform in terms of GC distribution than is the A. carolinensis genome (standard deviation 0.029 versus 0.032 respectively; Table 7). With the exception of the Z microchromosome, P. vitticeps microchromosomes possess a heterogeneous distribution of GC-rich sequences over 5 kbp windows (Fig. 4a). In this regard, P. vitticeps microchromosomes resemble those of birds but differ markedly from those of A. carolinensis, whose GC content more closely resembles that of the macrochromosomes [33]. Intriguingly, the Z microchromosome of P. vitticeps has an average GC content comparable to that of coding sequences and short interspersed nuclear elements (SINEs) (Fig. 4b), which suggests that this chromosome may be enriched in these GC-rich components of the genome. However, with the exception of LTR transposable elements, all components (CDS, introns, tandem repeats and transposable elements) showed greater GC content if they resided on the Z chromosome than elsewhere (Z chromosome > microchromosomes > macrochromosomes; Fig. 4c), suggesting that there are other, as yet unidentified, reasons for the observed variation in GC content across different chromosome classes.
When variation in GC distribution is considered over larger spatial scales (tens to hundreds of kbp, Fig. 3), the P. vitticeps macrochromosomes are similar to the A. carolinensis genome, which lacks substantial variation in GC composition, a striking departure from isochore patterns seen in mammals and birds [33]. The Z chromosome, too, lacks substantial heterogeneity over larger spatial scales, which perhaps reflects the uniform distribution of repetitive elements along its length [24]. Only the autosomal microchromosomes of P. vitticeps bear any similarity in GC distribution to the other sauropsid genomes examined. The P. vitticeps genome, therefore, has compositional patterns distinct from that of A. carolinensis, which indicates that different processes have shaped the genomes of the two lizards since they shared a common ancestor 144 million years ago.
Comparison with other assemblies
P. vitticeps and A. carolinensis had similar scaffold N50 values (2.29 Mbp and 4.03 Mbp, respectively). These values for P. vitticeps are surprisingly good, as its genome was assembled from short read sequences, whereas that of A. carolinensis was generated using Sanger sequencing. Our assembly compares well to nine other sauropsid genomes, including those of two squamates, two turtles and three crocodilians (Table 8).
Table 8.
Comparison of sequencing platform, assembler, and assembly statistics for the reptiles for which a genome sequence is available
| Bearded dragon | Burmese python | King cobra | Saltwater crocodile | Chicken | Green anole | American alligator | Gharial | Chinese softshell turtle | Green sea turtle | Western painted turtle | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pogona vitticeps | Python bivittatus | Ophiophagus hannah | Crocodylus porosus | Gallus gallus | Anolis carolinensis | Alligator mississippiensis | Gavialis gangeticus | Pelodiscus sinensis | Chelonia mydas | Chrysemys picta | |
| Assembler | SOAP deNovo | SOAP deNovo | CLC NGS Cell (version 2011) | AllPaths (version R41313) | Celera Assembler (version 5.4) | Arachne (version 3.0.0) | Allpaths (version R41313)a | SOAP deNovo | SOAP deNovo | SOAP deNovo | Newbler |
| Sequence method | Illumina HiSeq 2000 | Illumina GAIIx & HiSeq 2000, Roche 454 | Illumina HiSeq | Illumina GAII & HiSeq 2000 | Sanger, Roche 454 | Sanger | Illumina GAII & HiSeq 2000 | Illumina GAII | Illumina HiSeq 2000 | Illumina HiSeq 2000 | Roche 454, Illumina, Sanger |
| Average read depth | 85.5X | 20X | 28X | 74X | 12X | 7.1X | 68X | 109X | 105.6X | 110X | 15X |
| Genome size (Gbp) | 1.77 | 1.44 | 1.36–1.59 | 2.12 | 1.20 | 2.17 | 2.88 | 2.21 | 2.24 | 2.6 | |
| Total bases in contigs (excluding unknown bases, Ns) | 1,747,541,145 | 1,384,532,810 | 1,380,486,984 | 2,088,185,434 | 1,032,841,023 | 1,701,336,547 | 2,129,643,287 | 2,198,585,703 | 2,106,622,020 | 2,110,365,500 | 2,173,204,098 |
| Total bases in scaffolds | 1,816,115,349 | 1,435,035,089 | 1.66 Gbp | 2,120,573,303 | 1,046,932,099 | 1,799,143,587 | 2,174,259,888 | 2,270,567,745 | 2,202,483,752 | 2,208,410,377 | 2,365,766,571 |
| No. of scaffolds (>100 bp) | 543,500 | 39,113 | - | 23,365 | 16,847 | 6,645 | 14,645 | 9,317 | 19,904 | 140,023 | 78,631 |
| N50 scaffold (kbp) | 2,291 | 214 | 226 | 204 | 12,877 | 4,033 | 509 | 2,188 | 3,351 | 3,864 | 6,606 |
| No. of contigs (>100 bp) | 636,524 | 274,244 | 816,633 | 112,407 | 27,041 | 41,986 | 114,159 | 177,282 | 205,380 | 274,367 | 262,326 |
| N50 contig (kbp) | 31.2 | 10.7 | 5.2 | 32.7 | 279 | 79.9 | 36 | 23.4 | 22.0 | 29.2 | 21.3 |
| Repeat content | 39.5 | 31.8 | 35.2 | 37.5 | 9.4 | 34.4 | 37.7 | 37.6 | 42.47 | 37.35 | 9.82 |
| No. protein-coding genes | 19,406 | 17262 | - | 13,321 | 15,508 | 17,472 | 23,323 | 14,043 | 19,327 | 19,633 | -- |
Information is taken from the NCBI database (http://www.ncbi.nlm.nih.gov/assembly), with additional data from the primary papers in which the findings were originally published. aManual scaffolding
The gene parameters listed in Table 6 compare well to those of other vertebrates (see also Fig. 5).
Fig. 5.
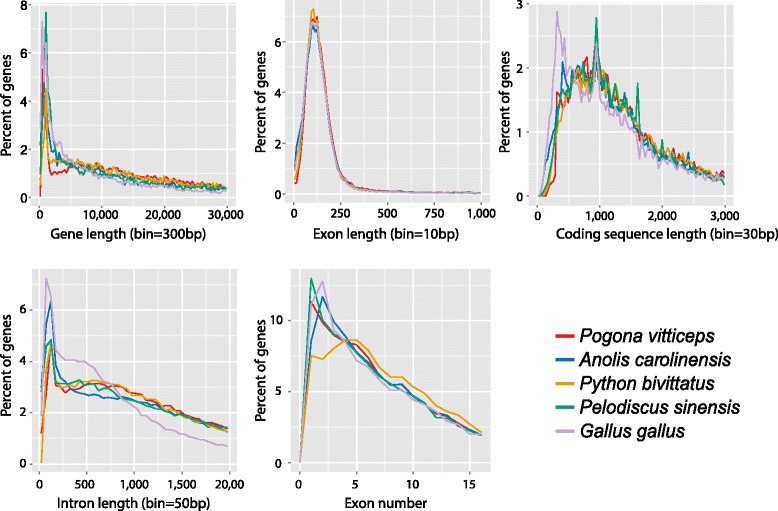
Comparisons of gene parameters among Pogona vitticeps, Gallus gallus, Python bivittatus, Anolis carolinensis, and Pelodiscus sinensis genomes
Concluding remarks
The quality of the P. vitticeps assembly is comparable to that of other published squamate genomes. This genome assembly, coupled with the availability of a BAC library and the development of a high-density physical map for each chromosome, provides an unparalleled resource for accelerating research on sex determination, major histocompatibility complex evolution, and the evolution of adaptive traits in squamates to complement the advances brought about by the sequencing of the A. carolinensis genome [25].
Availability of supporting data
The genomic and transcriptomic sequence reads and assemblies have been deposited in the ENA under the project accession number PRJEB5206 (see Additional file 1 for a complete list of accession numbers). The genome sequence has been submitted to GigaDB [34] along with other supporting resources, including:
SoapDeNovo2 pvi1.1.Jan2013 genome assembly (ENA accession number ERZ094017)
Trinity de novo transcriptome assembly (ENA accession number ERZ097159)
Peptide and coding sequences for the pvi1.1.Jan2013 assembly
Gene annotations and repeat annotations for the scaffolds
Sequence Read Archive accession numbers for all sequencing runs.
The annotated P. vitticeps genome sequence can be accessed through a publicly available genome browser [35].
Acknowledgements
We are indebted to the BGI-Shenzhen, China, for its contribution to the sequencing of the P. vitticeps genome and subsequent bioinformatics work. The Institute for Applied Ecology at the University of Canberra, Australia provided access to resources and facilities. The Institute, the Faculty of Applied Science and the Office of the PVC Research, University of Canberra, and China National GeneBank-Shenzhen contributed funding in support of this project. Tariq Ezaz provided advice and supervision of the cytogenetic work undertaken by Kazumi Matsubara.
Abbreviations
- BAC
Bacterial artificial chromosome
- bp
Base pair
- ENA
European Nucleotide Archive
- indel
Sequence insertion or deletion
- k-mer
Short sequence of length k
- LINE
Long interspersed nuclear element
- LTR
Long terminal repeat
- N50
50 % of the genome sequence is contained in contigs (or scaffolds) equal to or greater than this length
- ORF
Open reading frame
- SINE
Short interspersed nuclear element
- SNP
Single nucleotide polymorphism
Additional file
ENA accession numbers. (XLSX 18 kb)
Footnotes
Arthur Georges and Qiye Li contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AG and QL led the analyses and the project. AG, JAMG, JD and PW contributed funds to enable this genome to be sequenced and worked with GZ and QL on the approach to generating the data and the analyses. GZ and QL provided oversight of the sequencing and analysis through the BGI-Shenzhen, China. JL conducted the genome survey and genome assembly, assessment of assembly quality, synteny analysis, SNP identification and analysis of sex-linked scaffolds. ZW undertook the gene and repeat annotation. SS, JAMG and AG provided access to the P. vitticeps BAC library and associated resources. JD oversaw the cytological work of KM, who undertook the work necessary to identify the chromosomal sex of Fabian, complemented by work of CH and XZ using sex-linked markers. DO’M and HP handled the bioinformatics work required to deliver the genome sequence through the browser. PZ analysed the transcriptomes and construct transcript models that were used for gene annotation. MF and YZ undertook the CG and isochore analyses. All authors contributed to the preparation of the manuscript. All authors read and approved the final manuscript.
Authors’ information
GZ, AG and JAMG are members of the Genome 10K Consortium.
Contributor Information
Arthur Georges, Email: georges@aerg.canberra.edu.au.
Qiye Li, Email: liqiye@genomics.cn.
Jinmin Lian, Email: lianjinmin@genomics.org.cn.
Denis O’Meally, Email: omeally@gmail.com.
Janine Deakin, Email: janine.deakin@canberra.edu.au.
Zongji Wang, Email: wangzongji@genomics.cn.
Pei Zhang, Email: zhangpei@genomics.cn.
Matthew Fujita, Email: mkfujita@gmail.com.
Hardip R. Patel, Email: hardip.patel@anu.edu.au
Clare E. Holleley, Email: clare@holleley.net
Yang Zhou, Email: zhouyang@genomics.cn.
Xiuwen Zhang, Email: xiuwen.zhang@canberra.edu.au.
Kazumi Matsubara, Email: kazumi.matsubara@iae.canberra.edu.au.
Paul Waters, Email: p.waters@unsw.edu.au.
Jennifer A. Marshall Graves, Email: j.graves@latrobe.edu.au.
Stephen D. Sarre, Email: stephen.sarre@canberra.edu.au
Guojie Zhang, Email: zhanggj@genomics.cn.
References
- 1.Young MJ, O’Meally D, Sarre SD, Georges A, Ezaz T. Molecular cytogenetic map of the central bearded dragon Pogona vitticeps (Squamata: Agamidae) Chromosom Res. 2013;21:361–74. doi: 10.1007/s10577-013-9362-z. [DOI] [PubMed] [Google Scholar]
- 2.Ezaz T, Quinn AE, Miura I, Sarre SD, Georges A, Graves JAM. The dragon lizard Pogona vitticeps has ZZ/ZW micro-sex chromosomes. Chromosom Res. 2005;13:763–76. doi: 10.1007/s10577-005-1010-9. [DOI] [PubMed] [Google Scholar]
- 3.Holleley CE, O’Meally D, Sarre SD, Graves JAM, Ezaz T, Matsubara K, et al. Sex reversal triggers the rapid transition from genetic to temperature dependent sex. Nature. 2015;523:79–82. doi: 10.1038/nature14574. [DOI] [PubMed] [Google Scholar]
- 4.Quinn AE, Georges A, Sarre SD, Guarino F, Ezaz T, Graves JAM. Temperature sex reversal implies sex gene dosage in a reptile. Science. 2007;316:411. doi: 10.1126/science.1135925. [DOI] [PubMed] [Google Scholar]
- 5.MacCulloch RD, Upton DE, Murphy RW. Trends in nuclear DNA content among amphibians and reptiles. Comp Biochem Physiol. 1996;113B:601–5. doi: 10.1016/0305-0491(95)02033-0. [DOI] [Google Scholar]
- 6.Doležel J, Bartoš J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytometry. 2003;51A:127–8. doi: 10.1002/cyto.a.10013. [DOI] [PubMed] [Google Scholar]
- 7.Marcais G, Kingsford C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 2011;27:764–70. doi: 10.1093/bioinformatics/btr011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li R, Fan W, Tian G, Zhu H, He L, Cai J, et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463:311–7. doi: 10.1038/nature08696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20:265–72. doi: 10.1101/gr.097261.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parra G, Bradnam K, Korf I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics. 2007;23:1061–7. doi: 10.1093/bioinformatics/btm071. [DOI] [PubMed] [Google Scholar]
- 12.Grabherr M, Haas B, Yassour M, Levin J, Thompson D, Amit I, et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110:462–7. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 14.Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0 1996–2010. Institute for Systems Biology, Seattle, WA, USA. 1996. http://www.repeatmasker.org. Accessed 20-Dec-14 2014.
- 15.Smit AFA, Hubley R. RepeatModeler Open-1.0. 2008–2015. . Institute for Systems Biology, Seattle, WA, USA. 2008. http://www.repeatmasker.org. Accessed 20-Dec-2014 2014.
- 16.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35:W265–8. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–80. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birney E, Clamp M, Durbin R. GeneWise and Genomewise. Genome Res. 2004;14:988–95. doi: 10.1101/gr.1865504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl. 2):215–25. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 20.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ezaz T, Moritz B, Waters PD, Graves JAM, Georges A, Sarre SD. The ZW sex microchromosomes of an Australian dragon lizard share no homology with those of other reptiles or birds. Chromosom Res. 2009;17:965–73. doi: 10.1007/s10577-009-9102-6. [DOI] [PubMed] [Google Scholar]
- 23.Quinn AE, Ezaz T, Sarre SD, Graves JAM, Georges A. Extension, single-locus conversion and physical mapping of sex chromosome sequences identify the Z microchromosome and pseudo-autosomal region in a dragon lizard. Pogona vitticeps Heredity. 2010;104:410–7. doi: 10.1038/hdy.2009.133. [DOI] [PubMed] [Google Scholar]
- 24.Ezaz T, Azad B, O’Meally D, Young MJ, Matsubara K, Edwards MJ, et al. Sequence and gene content of a large fragment of a lizard sex chromosome and evaluation of candidate sex differentiating gene R-spondin1. BMC Genomics. 2013;14:899. doi: 10.1186/1471-2164-14-899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alfoldi J, di Palma F, Grabherr M, Williams C, Kong L, Mauceli E, et al. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature. 2011;477:587–91. doi: 10.1038/nature10390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castoe T, de Koning A, Hall K, Card D, Schield D, Fujita M, et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci U S A. 2013;110:20645–50. doi: 10.1073/pnas.1314475110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vonk F, Casewell N, Henkel C, Heimberg A, Jansen H, McCleary R, et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc Natl Acad Sci U S A. 2013;110:20651–6. doi: 10.1073/pnas.1314702110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shaffer H, Minx P, Warren D, Shedlock A, Thomson R, Valenzuela N, et al. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 2013;14:R28. doi: 10.1186/gb-2013-14-3-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Z, Pascual-Anaya J, Zadissa A, Li W, Niimura Y, Huang Z, et al. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet. 2013;45:701–6. doi: 10.1038/ng.2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.St John J, Braun E, Isberg S, Miles L, Chong A, Gongora J, et al. Sequencing three crocodilian genomes to illuminate the evolution of archosaurs and amniotes. Genome Biol. 2012;13:415. doi: 10.1186/gb-2012-13-1-415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirkness EF, Bafna V, Halpern AL, Levy S, Remington K, Rusch DB, et al. The dog genome: survey sequencing and comparative analysis. Science. 2003;301:1898–903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- 32.Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V, et al. The genome of the Western clawed frog Xenopus tropicalis. Science. 2010;328:633–6. doi: 10.1126/science.1183670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujita MK, Edwards SV, Ponting CP. The Anolis lizard genome: An amniote genome without isochores. Genome Biol Evol. 2010;3:974–84. doi: 10.1093/gbe/evr072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Georges A, Li Q, Lian J, O’Meally D, Deakin J, Wang Z et al.. Genome of the Australian dragon lizard Pogona vitticeps. 2015. GigaScience Database. http://gigadb.org/dataset/100166. [DOI] [PMC free article] [PubMed]
- 35.Georges A, O’Meally D, Genomics@UC . The Pogona vitticeps genome browser (pvi1.1 Jan 2013). Institute for Applied Ecology. Canberra: University of Canberra; 2015. [Google Scholar]