

Abstract
Tuberous sclerosis complex (TSC) is caused by inactivating mutations in either TSC1 or TSC2 and is characterized by uncontrolled mTORC1 activation. Drugs that reduce mTOR activity are only partially successful in the treatment of TSC, suggesting that mTOR-independent pathways play a role in disease development. Here, kinome profiles of wild-type and Tsc2−/− mouse embryonic fibroblasts (MEFs) were generated, revealing a prominent role for PAK2 in signal transduction downstream of TSC1/2. Further investigation showed that the effect of the TSC1/2 complex on PAK2 is mediated through RHEB, but is independent of mTOR and p21RAC. We also demonstrated that PAK2 over-activation is likely responsible for the migratory and cell cycle abnormalities observed in Tsc2−/− MEFs. Finally, we detected high levels of PAK2 activation in giant cells in the brains of TSC patients. These results show that PAK2 is a direct effector of TSC1-TSC2-RHEB signaling and a new target for rational drug therapy in TSC.
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder characterized by the presence of benign tumours in the brain and in other vital organs such as the kidneys, heart, eyes, lungs, and skin, and is associated with severe neurological and behavioral symptoms. Up to 90% of patients suffer from epilepsy, and almost 50% have intellectual disabilities1. TSC is caused by a heterozygous mutation in either TSC12,3 or TSC24,5 which encode for hamartin (TSC1) and tuberin (TSC2), respectively6. These two proteins interact with each other7 to form a functional heterodimeric complex that controls different aspects of cellular metabolism by its GTPase activating function towards the Ras homolog enriched in brain protein (RHEB)8. In a normal situation, the TSC1-TSC2 complex inactivates RHEB by stimulating the conversion of RHEB-GTP to RHEB-GDP, which subsequently downregulates the mammallian target of rapamycin (mTOR) signaling pathway9. In the presence of a mutation in either TSC1 or TSC2, the TSC1-TSC2 complex is unable to form and RHEB is kept in a permanently active, GTP-bound state, leading to constitutive activation of the mTOR pathway and, as a consequence, to uncontrolled cell growth. The importance of mTOR signaling downstream of the TSC genes is further illustrated by the observation that upregulation of TSC1 following ischaemia induces protective autophagia via inhibition of the mTOR pathway and can protect neurons from ischaemia-induced cell death10.
mTOR is an important regulator of cellular metabolism and has multiple downstream targets including p70S6 kinase and the eukaryotic initiation factor 4E (eIF4E)-binding proteins 1 and 2 (4E-BP1 and 4E-BP2)11. Various molecules are potent inhibitors of mTOR and are used clinically to combat disease characterised by excessive mTOR activity, including TSC. Rapamycin and its analogues such as sirolimus and everolimus, play a prominent role in this respect. Rapamycin is a macrolide antibiotic that blocks mTOR signaling by forming an inhibitory complex with the cytosolic FK-binding protein-12 (FKBP-12)12. The Rapamycin-FKBP12 complex directly binds and inhibits mTOR when present in a protein complex consisting of mTOR, Raptor, LST8, PRAS and DEPTOR, together known as mTORC1, but not when present in the protein complex mTORC2 (consisting of mTOR, Rictor, LST8, mSIN1, DEPTOR and Protor). mTORC1 inhibition by rapamycin is now firmly established as a therapeutic option in various diseases, especially transplantation medicine and various forms of cancer. Indeed, in two prospective studies sirolimus proved to be safe and effective for the treatment of TSC-associated renal angiomyolipoma13,14. Furthermore, everolimus substantially and significantly reduced the volume of subependymal giant cell astrocytomas in an open-label phase 1/2 study15, while in a larger double-blind placebo-controlled phase III study, it was shown to reduce tumour size and seizure frequency in a significant fraction of patients with TSC and associated subependymal giant cell astrocytoma16. Unfortunately, some clinical manifestations of TSC, especially lung function14 and neurological symptoms, do not or only partially react to prolonged treatment with rapamycin analogues. Although none of the everolimus-treated patients progressed, only 27 (35%) of the 78 patients in the everolimus-treated group of the phase III study (versus none of the placebo group) was considered to have a response in terms of a reduction in the total volume of all subependymal giant cell astrocytomas16. Thus mTOR inhibition was only partially effective in these patients. Furthermore, TSC associated tumours are highly autophagia dependent, and as rapamycin analogues are well-known to stimulate autophagia, their administration will provide a pro-survival stimulus that will limit their own efficacy, contributing to the partial clinical response seen for human angiomyolipomas and subependymal giant cell astrocytomas to mTORC1 inhibition17. Thus, alternative treatments are urgently called for.
The potential of prolonged rapamycin treatment to cause full mTOR inhibition is undisputed, and hence the failure of mTOR inhibition to counteract all manifestations of TSC indicates that mTOR may well not be the sole molecular mediator of the effects of TSC1/2 deficiency on the cellular phenotype14. Obviously, knowledge of TSC1-TSC2-regulated pathways acting in parallel with mTOR activation should prove exceedingly useful for designing superior clinical treatments. These considerations prompted us to contrast signal transduction in Tsc2−/− and wild-type mouse embryonic fibroblasts (MEFs), employing a peptide array approach that provides a comprehensive description of cellular kinase activity by comparing the phosphorylation of 1024 different kinase substrates18,19. We have previously shown that kinome profiling is a powerful tool to define therapeutically interesting kinases without a priori knowledge of the pathways affected in different pathogeneses, since it allows the generation of complete descriptions of cellular kinase activity in a single experiment20,21. Our results reveal the role of TSC2 in cellular signal transduction and uncover a previously unrecognised action of this protein in the down regulation of PAK2-dependent signaling, which is independent of mTOR. Thus PAK2 constitutes a new downstream target of the TSC1-TSC2 complex and provides a potential novel avenue for treatment of TSC.
Results
The influence of genetic deletion of Tsc2 on the cellular kinome in MEFs
To gain insight into the influence of genetic deletion of Tsc2 on cellular signal transduction, we compared the kinome of Tsc2−/− MEFs to that of wild-type MEFs. To this end, cells were serum-starved for two hours before lysates were prepared, as this would be expected to increase signaling of the AMPK signal transduction cassette22, and thus accentuate differences in signal transduction between the two different genetic backgrounds. Kinome profiles were then generated by incubating each cell lysate with a peptide array containing different kinase substrates in the presence of 33P-γ-ATP. Three technical replicates of two biological replicates of each sample were performed. The arrays incorporated substantial amounts of radioactivity (scans not shown). The technical quality of the profiles was good as the average Pearson product moment was well in excess of 0.85 between both the technical and the biological replicas for all cell types analysed for each condition. From a qualitative comparison, it appeared that the profile of the Tsc2−/− MEFs was substantially similar to the wild-type control (Supplementary figure S1). However, specific differences were also observed between the profiles of the two conditions (Fig. 1). As expected, the canonical effect of Tsc2 deletion, an upregulation of mTOR-dependent signaling, is clearly manifest: mTOR substrate peptides as well as substrates for p70S6 kinase, a canonical target for mTOR-dependent signal transduction12,23,24, showed increased phosphorylation when incubated with lysates obtained from Tsc2−/− MEFs (Fig. 1). Strikingly, AMPK activity, which negatively regulates mTOR through the TSC1-TSC2 complex25, is much stronger in the Tsc2−/− MEFs as well (Fig. 1), possibly reflecting a compensatory response to mTOR activation or a reflection of ATP depletion by over-activation of mTOR dependent signaling by this ADP/AMP-stimulated kinase. There is also a substantial activation of the Chk1/Chk2 cell cycle checkpoint signaling network in the Tsc2−/− MEFs (Fig. 1), which may represent a p53-dependent endogenous feed-back mechanism activated by loss of Tsc2 to limit cell cycle progression, and may explain why in experimental animals genetic loss of Tsc2 enhances tumorigenesis in a p53-deficient context26. (Of note, while some Tsc2−/− mouse models described are p53 deficient27, the Tsc−/− MEFS used in the current study retain p53 expression—see supplementary Figure S2). Furthermore, this observation corresponds well with the fact that the AMPK activator AICAR, which acts upstream of the TSC1-TSC2 complex11, activates the p53 pathway in a rapamycin-sensitive fashion28. Most interestingly, an up-regulation of the Ras-related C3 botulinum toxin substrate 1/p21 activated kinase 2 (RAC/PAK2) pathway was observed in Tsc2−/− MEFs. This was evident from increased phosphorylation of substrates directly measuring the activity of this kinase (Fig. 1), but also from increased activity of possible indirect effectors of this kinase, like p125FAK, MKK3/6 and c-Src29, in the lysates of Tsc2−/− MEFs (Supplementary table 2).
Figure 1. Effect of genetic deletion of Tsc2 on signal transduction in mouse embryonic fibroblasts (MEFs) as determined by the phosphorylation of peptide arrays containing 1024 kinase substrates.

Graphical impression of the results obtained, representing three technical replicas performed on two biological replicates. Individual results for each technical replicate were first subjected to Markov state analysis. (i.e. is the peptide significantly more phosphorylated than the background?) and the number of reactions in which this is the case (an integer between zero [in none of the 6 reactions was significant radioactivity incorporated into the substrate peptide] and six [in all 6 reactions significant radioactivity was incorporated into the substrate peptide]) were determined per peptide for each of the experimental conditions (listed as Markov score, Supplementary Table 1). The Markov scores obtained were collapsed on signal categories that provide insight into the signaling differences between the Tsc2 proficient and deficient cells and are expressed in grey scale (representing the number of peptides phosphorylated as a fraction of all peptides in that category). Note the difference in mTOR signaling, PAK signaling and Chk1/2-related signaling.
Taking into account that PAKs represent a pleiotropic family of kinases implicated in a variety of cell physiological effects, especially in neuronal migration30; that TSC is often considered to be a disease of abnormal neuronal migration31; and that PAKs are highly druggable32, these results suggest the tantalising possibility that PAKs may be involved in the development of TSC. Further investigation was initiated to test this notion.
Increased activity of Pak2 upon genetic inactivation of the TSC1-TSC2 complex is independent of mTor in MEFs
The kinome profiles revealed higher Pak2 enzymatic activity in MEFs genetically deficient in Tsc2. Western blot analysis of wild-type and Tsc2−/− MEFS confirmed the Tsc2-negative status of the Tsc2−/− MEFs, as well as the increased phosphorylation of the canonical mTor targets p70S6K and S6. In addition, the observed increase in Pak2 enzymatic activity in Tsc2−/− MEFs corresponded to an increased autophosphorylation of Pak2, detected by using an antibody against phospho-serine 20, an autophosphorylation site associated with the activation of this kinase (Fig. 2A). To investigate the mTor-dependency of Pak2 activation in this genetic model, Tsc2−/− and wild-type MEFs were incubated with rapamycin (Fig. 2B). As expected, rapamycin caused a strong down-regulation of p70S6K and S6 phosphorylation and almost completely abrogated the effects of Tsc2 deficiency on this phosphorylation. Strikingly, however, Pak2 autophosphorylation measured in parallel was unaffected. Similarly, in Tsc1−/− MEFs, we observed a clear, rapamycin-sensitive up-regulation of p70S6K and S6 phosphorylation, whereas a strong, rapamycin-insensitive Pak2 auto-phosphorylation was seen in these cells, when compared to wild-type MEFs (Fig. 2C). Thus, our results show that the TSC1-TSC2 complex acts as a negative upstream regulator of Pak2 and that activation of this kinase following Tsc1 or Tsc2 deletion occurs in a mTor-independent fashion.
Figure 2. Pak2 phosphorylation is resistant to rapamycin treatment and is dependent of Rheb.

(A) Western blot analysis of wild-type and Tsc2−/− MEFs confirms the Tsc2-negative status of the Tsc2−/− MEFS and shows that the increased activity of Pak2 observed in the kinome profile of Tsc2−/− MEFs corresponds to increase autophosphorylation of Pak2 and increased phosphorylation of p70S6K and S6. (B) Treatment of MEFs for 1 hour with 20 nM rapamycin efficiently inhibits increased phosphorylation of p70S6K and S6 following deletion of Tsc2, but does not affect the increase in Pak2 autophosphorylation. (C) Western blot analysis of wild-type and Tsc1−/− MEFs shows that loss of Tsc1 provokes autophosphorylation of Pak2 and phosphorylation of p70S6K and S6. The latter two events are sensitive to a 1 hour treatment of the cells with 20 nM rapamycin, but Pak2 autophosphorylation is unaffected by this treatment. (D) Western blot analysis of wild-type and Rheb1−/− MEFs confirms the Rheb-negative status of the Rheb1−/− MEFS and shows decreased phosphorylation of Pak2, p70S6K and S6. (E) Over-expression of a Myc-tagged variant of Rheb in wild-type MEFs induces Pak2 activation. (F) Rheb knock down using a short hairpin RNA targeting Rheb, abrogates activation of Pak2 autophosphorylation in Tsc2−/− MEFs. A control hairpin shows no effect. All blots were run under the same conditions and analysed in a similar way.
Rheb regulates Pak2 activation in MEFs
In canonical signal transduction, loss of the TSC1-TSC2 complex influences cellular biochemistry through the subsequent constitutive activation of RHEB and mTOR. The observation that PAK2 activation as a consequence of the genetic loss of one of the components of the TSC1-TSC2 complex is independent of mTOR raises questions as to the role of RHEB in the TSC1-TSC2 dependent control of PAK2 activity. To answer this question we examined Pak2 autophosphorylation levels in Rheb1−/− MEFs (Fig. 2D). These cells displayed very low levels of autophosphorylated Pak2, demonstrating that constitutive Pak2 activity requires functional Rheb. Furthermore, exogenous expression of a Myc-tagged version of Rheb in wild-type MEFs was sufficient to cause increased Pak2 autophosphorylation (Fig. 2E). Finally, when Tsc2−/− MEFs were transfected with a short hairpin RNA targeting Rheb1, the stimulatory effect of genetic deletion of Tsc2 on Pak2 autophosphorylation was lost. A control short hairpin RNA did not effect either Rheb levels or Pak2 autophosphorylation (Fig. 2F). Taken together, these results indicate that Rheb is a major regulator of the activation status of Pak2.
Regulation of Pak2 by the TSC1/2-Rheb signaling module is independent of Rac
As the the canonical regulator of activity of the family of p21-activated kinases is the RAC GTPase33, we next investigated the activity of this protein in Tsc2−/− MEFs. Interestingly, the genetic loss of Tsc2 in MEFs was not accompanied by increased Rac-GTP loading (Fig. 3A). If anything Rac-GTP levels were lower in Tsc2−/− MEFs, hinting at the existence of Pak2 negative feedback mechanisms on Rac activation. Thus, we decided to investigate whether loss of Tsc2 stimulated Pak2 autophosphorylation in the presence of NSC23766, an inhibitor of RAC GDP→GTP exchange34. Indeed treatment of wild-type MEFs for 30 min with 20 μM of NSC23766 efficiently impaired GTP loading of Rac (Fig. 3B). Importantly, NSC23766 treatment did not markedly affect the upregulation of Pak2 autophosphorylation in Tsc2−/− MEFs (Fig. 3C). Furthermore, treating cells with a combination of rapamycin and NSC23766 did not affect Pak2 autophosphorylation (although phosphorylation of S6 was highly sensitive to this treatment). Hence, the TSC1/2-Rheb signaling module controls Pak2 activity in a Rac and mTOR-independent fashion and represent a specific output of this signaling cascade.
Figure 3. Pak2 autophosphorylation in Tsc2−/− MEFs is independent of Rac activity.
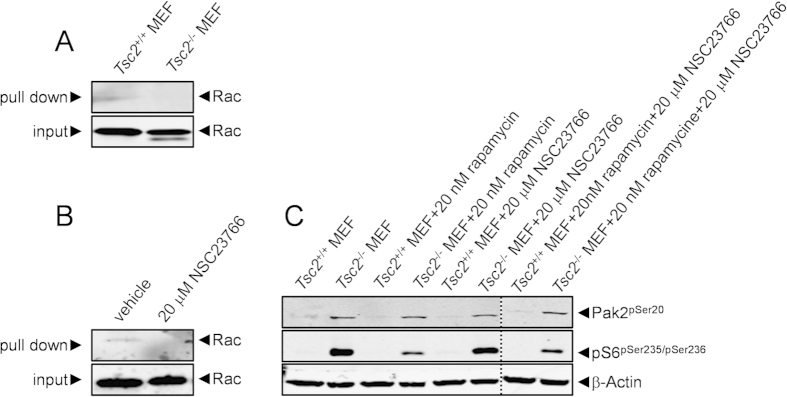
(A) The activation status of Rac was compared in wild-type and Tsc2−/− MEFs by pull down of Rac-GTP using recombinantly-produced PAK-Crib proteins. Tsc2−/− MEFs are not characterised by excessive Rac activity. (B) Rac-GTP pull down with recombinantly-produced PAK-Crib proteins was used to investigate the effect of a 30 min treatment of 20 μM NSC23766 on Rac activity. The treatment efficiently blocks GTP loading of Rac. (C) The effects of a 1 hour treatment with 20 nM rapamycin and a 30 min treatment with 20 μM NSC23766 on Pak2 autophosphorylation and S6 phosphorylation were investigated in wild-type and Tsc2−/− MEFs. Rapamycin efficiently inhibits S6 phosphorylation but has no effect on Pak2 autophosphorylation. NSC23766 affects neither Pak2 autophosphorylation nor S6 phosphorylation. The combination of rapamycin and NSC23766 (lanes cropped from the same blot) does also not influence Pak2 phosphorylation, nor is it more effective on S6 phosphorylation as compared to rapamycin alone. All blots were run under the same conditions and analysed in a similar way.
Pak2 affects the migration of Tsc2 −/− MEFs
Predicted consequences of exagerated PAK2 activation are cytoskeletal abnormalities and consequently impaired migratory responses. We visualised filamentous actin by performing rhodamin-phalloidin stainings. Wild-type MEFs were characterised by relatively modest amounts of filamentous actin and highly organised stess fibers (Fig. 4A). In contrast Tsc2−/− MEFs exhibited robust cytoskeletal reangerangements, including the formation of numerous lamellipodium-like networks, increased disorganisation of cytoskeletal actin, the appearance of prominent cortical actin structures, and the formation of many small microspikes (Fig. 4A). Furthermore, in scratch assays, wild-type MEFs polarised at the wound edge and migrated into the wound space, whereas the Tsc2−/− MEFS were much less proficient in this respect (Fig. 4B,C). In order to prove that the migratory defect obseved on the Tsc2−/− MEFs is induced by Pak2, we reduced Pak2 expression in these MEFs using a specific siRNA. We also used a non-targeting siRNA as control. We observed that in the Pak2 siRNA transfected MEFs cell migration was partly restored, while the control siRNA transfected MEFs showed no improvement (Fig. 4B,C). Efficient downmodulation of Pak2 upon siRNA treatment of Tsc−/− MEFs was shown by qPCR (Fig. 4D). This experiment furthermore demonstrates that the increased phosphorylation of Pak2 observed (Fig. 2A–C and Fig. 3C) in Tsc2−/− MEFs as compared to Tsc2+/+ MEFs is independent on Pak2 mRNA expression levels (lanes 1 and 2 in Fig. 4D) and thus relies on posttranslational modification of the Pak2 protein. In toto, these experiments show that Tsc2−/− MEFs demonstrate loss of migratory response which is partially dependent on Pak2.
Figure 4. Loss of Tsc2 results in Pak2 dependent wound healing defects.

(A) Filamentous actin was visualised by performing rhodamin-phalloidin stainings. Wild-type MEFs are characterised by a relatively modest amount of filamentous actin and highly organised stess fibers (osf; an example is indicated in the figure). Loss of Tsc2 coincides with robust cytoskeletal rearrangements, including the formation of numerous lamellipodium-like networks (lln), increased and disorganised cytoskeletal actin, the appearance of prominent cortical actin structures (ca) and the formation of many small microspikes (μs). Scale bars 20 μm. (B) Scratch wound assays performed in Tsc2−/− and wild-type MEFs shows that Tsc2−/− cells migrate slower than wild-type MEFs. Reducing Pak2 by siRNA results in partial restoration of migration in Tsc2−/− MEFs, whereas control siRNA does not. (C) Quantification of the difference in lebesgue measure between the initial (0 hours) and final (24 hours) of the apparent scratch area confirms impaired migration in Tsc2−/− MEFs (p < 0.001) and restoration thereof by Pak2 siRNA (p = 0.0137). (D) Quantitative PCR results demonstrating that levels of Pak2 mRNA expression are not affected in Tsc2−/− MEFs, and are reduced upon siPAK2 treatment, but not control siRNA (*<0.0005).
Lesions from TSC patients show increased PAK2 autophosphorylation
Independent confirmation of PAK2 activation as a downstream effect of loss of the TSC1-TSC2 complex came from experiments in which PAK2 autophosphorylation was investigated in the brains of TSC patients. Pathologically, TSC is characterized by the occurrence of multiple cortical tubers in the cerebral cortex which are thought to be responsible for many neuropsychiatric symptoms of TSC. Histologically, the most conspicuous feature of cortical tubers is the presence of giant cells, which show abnormal size and differentiation markers35. Strikingly, strong immunoreactivity for autophosphorylated PAK2 was observed in brain lesions from 5 patients carrying a TSC1 or TSC2 mutation (1 TSC1: 4 TSC2). The staining was restricted to the giant cell compartment and was cytoplasmic, as expected based on the normal localization of this kinase (Fig. 5A). Double-staining for autophosporylated PAK2 and phosphorylated S6 was also performed in three lesions (all patients carrying TSC2 mutations). The majority of giant cells showed high levels of phosphorylated S6, the golden standard for positive identification of giant cells with loss of heterozygosity for TSC1 or TSC236, and were strongly positive for PAK2 autophosphorylation (Fig. 5B). Thus PAK2 activation accompanies TSC loss in human disease.
Figure 5. PAK2 phosphorylation levels are increased in brain lesions from TSC patients.

(A) Immunohistochemistry data showing high levels of PAK2 autophosphorylation in giant cells of TSC patients (representative example of 5 patients). Scale Bars 1 cm; A, B: 80 μM. (B) Double-staining in lesions from TSC patients shows co-localization of p-PAK2 and p-S6 in a significant fraction of the giant cells (representative immunohistochemistry analysis of 3 patients; same magnification used as in panel A).
Discusion
The search for new and better therapeutical targets is a constant pursuit in the treatment of different disorders. This is especially the case for Tuberous Sclerosis Complex (TSC), as TSC symptoms are only partially resolved by current therapy. In particular, the treatment of the neuropsychiatric manifestations is as yet inadequate. So far, most attention in the search for therapeutics has been given to the mTORC1 complex as it constitutes the canonical target of the TSC1-TSC2 complex. Here, we provide evidence that PAK2 signaling constitutes a second signal transduction module downstream of the TSC1-TSC2-RHEB signal transduction complex, whose inhibition may be potentially useful, as strong PAK2 activation appears to be a characteristic of human disease, at least in the brain. A proposed possible diagram as to how PAK2 activation might be implicated in TSC is provided in Fig. 6.
Figure 6. Schematic representation of the TSC1-TSC2-RHEB network showing PAK2 as a new downstream target of RHEB.
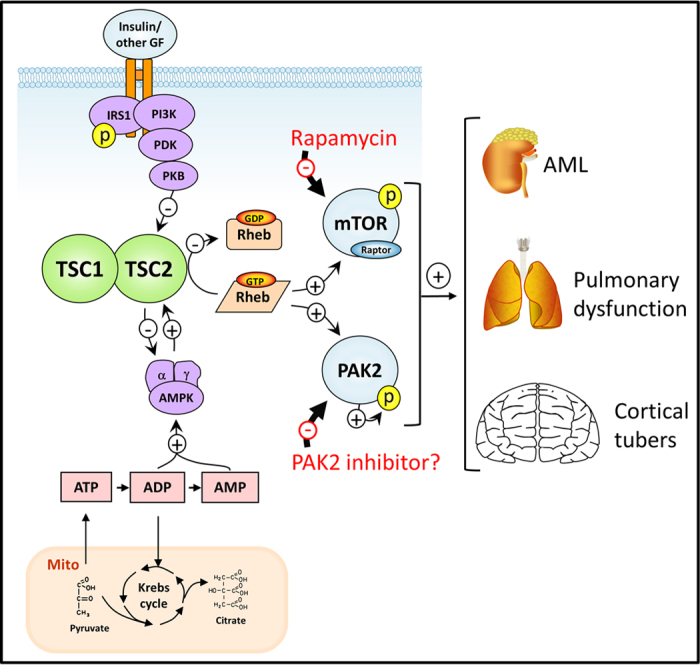
AML; angiomyolipoma, Mito; mitochondrion, GF; growth factor. Figure was designed by the authors, with no copyright owed elsewhere.
PAK2 is a serine/threonine kinase involved in cytoskeletal reorganization originally identified by its role in chemotactic signal transduction37, and is now well-established to be a mediator of directed cell migration in a variety of settings in the body38. In the context of TSC, to our knowledge no information has been reported hitherto, but it is well recognised that many of the psychoneurological manifestations of the disease involve aberrant migration of neuronal precursors39. In addition, loss of Tsc1 in mouse subventricular zone neural stem/progenitor cells leads to migration deficits that result in the development of nodular protusions and small tumours that present many features of the brain lesions found in TSC patients40. Thus, it is tempting to propose that at least certain aspects of TSC, especially those related to aberrant migration, may involve superactivation of PAK2, rather than that of mTOR. Intriguingly, LKB1, a canonical upstream element in TSC1-TSC2-RHEB-mTOR signaling41, is well-established to regulate cell motility and axon growth42, and thus it is possible that TSC1-TSC2 complex-regulated PAK2 activation may be involved in the pathophysiological effects of this important tumour suppressor as well. Indeed, a correlation between increased PAK phosphorylation and biallelic LKB1 inactivation has been reported both in non-small cell lung cancer43 and breast cancer44. Thus, the observation that PAK2 acts as a mTOR-independent effector of TSC1-TSC2-RHEB signaling is not only highly novel, but it also fits well with the body of available biomedical literature.
Interestingly, the PAKs constitute a family of highly druggable kinases and many inhibitors, including those that target PAK2, have been developed45. These compounds have yet to be tested in humans, but the relation between loss of functional TSC1/TSC2 and PAK2 hyperactivation makes it tempting to suggest that TSC could constitute a good candidate disease for such clinical trials. In any case, the current study has shown that the PAK2 kinase constitutes a bona fide mTOR-independent effector of the TSC1-TSC2 pathway and thus provides an important starting point for novel thinking on the development of new therapeutic avenues for dealing with TSC.
Materials and Methods
Kinome array analysis
The protocol of the kinome array has been provided before in extensive detail46. Cell lysates were prepared with m-PER lysis buffer (Thermo-Scientific) containing protease (Roche) and phosphatase inhibitors (Thermo-Scientific). Protein quantification was done using Bradford analysis (Bio-Rad) and 100 μg of protein was used for kinome analysis. After drying, the glass slides were exposed to a phosphor-imager plate for 72 hours and assessed with ScanAlyze software (http://rana.lbl.gov/EisenSoftware.htm). Spot density and individual background intensities were analysed using grid tools and data from two individual experiments were exported to an excel sheet for further analysis. Individual results for each technical replicate were first subjected to Markov state analysis (i.e. is the peptide significantly more phosphorylated than background) and the number of reactions in which this is the case (an integer number between zero [in none of the 6 reactions significant radioactivity in incorporated into the substrate peptide] and six [in all 6 reactions significant radioactivity was incorporated into the substrate peptide]) is listed for each peptide for each of the experimental conditions (listed as Markov score). These Markov scores were collapsed on signal categories that provide insight into the difference in signaling between the Tsc2 proficient and deficient cells.
Expression vectors
pcDNA-Myc-Rheb was kindly provided by Liz Henske (Division of Pulmonary and Critical Care Medicine, Department of Medicine, Brigham and Women’s Hospital, Boston, U.S.A). pLKO.1-shRheb was generated in the Biomics Center of the Erasmus Medical Center (Rotterdam, The Netherlands) using the MISSIONtm shRNA library (Sigma-Aldrich). The shRheb targeting sequence is CCTCAGACATACTCCATAGAT. pGEX4T-PAK-Crib vector contains the RAC-binding domain of PAK2 and was cloned using BamHI and EcoRI restriction sites.
Cell culture and transfection
Tsc1−/−, Tsc2−/− and wild-type MEFs were kindly provided by David Kwiatkowski (Division of Pulmonary and Critical Care Medicine, Department of Medicine, Brigham and Women’s Hospital, Boston, U.S.A). Rheb1−/− and wild-type MEFs were prepared as described before47. All MEFs were cultured in DMEM with high glucose content (Lonza), supplemented with 10% fetal calf serum (Sigma-Aldrich) and 1% penicillin/streptomycin (Gibco). Cells were cultured at 37oC and 5% CO2. For transient transfection, 300. 000 cells were seeded in 6 well plates. Transfections were performed 24 hours after seeding using Lipofectamine 2000 (Invitrogen). For the siRNA experiments, a SMARTpool: Accell siRNA specifically targeting Pak2 and a SMARTpool: Accell siRNA control were used (GE Healthcare, Dharmacon). Transfection was performed according to the manufacturers’ instructions 24 hours after seeding.
Treatments, cell lysates, and Western blot analysis
Before lysis, cells were incubated for 2 hours in serum free medium, after which they were treated with rapamycin (1 h, 20 nM) and/or NSC23766 (30 min, 20 μM, Tocris Bioscience). Stimulations were terminated by an ice-cold phosphate buffered saline wash and cells were incubated with lysis buffer (m-PER containing protease and phosphatase inhibitors) for 30 minutes on ice. Cell lysates were collected by scraping and cleared by centrifugation at 14 000 rpm for 10 minutes in a pre-cooled (4 °C) centrifuge. Cell lysates were stored at −80 °C before they were processed further for SDS-PAGE and immunoblotting. The following antibodies were used: TSC2 and actin (Santa Cruz Biotechnology); TSC1, MYC, p-PAK2, p-p70S6K and p-S6 (Cell Signaling); Rheb antibody was kindly provided by Richard Lamb (Department of Oncology, University of Alberta, Cross Cancer Institute, Edmonton, Alberta, Canada). Secondary antibodies used were IRDye 800CW Goat anti-Rabbit and IRDye 680RD Goat anti-Mouse (Li-Cor).
Pull-down assays
RAC activation48 was assessed using pull-down assays. Cells were lysed with 1X GST buffer (10% glycerol, 50 mM Tris-HCl pH7.4, 200 mM NaCl, 1% NP-40, 2 mM MgCl2, 1X protease inhibitors and 1X phosphatase inhibitors) and kept on ice. GST-PAK-Crib coupled beads were prepared by incubating GST-PAK-Crib with GST-beads (GE Healthcare) and 1X GST lysis buffer for 45 minutes at 4 °C. Beads were then washed 5 times with 1X GST buffer and incubated with 750 μg of total protein lysate for 45 minutes at 4 °C on a rotating wheel. Samples were washed 3 times with 1X GST buffer and the beads were resuspended in 20 μl Laemmli buffer followed by denaturation at 95 °C for 5 min. Samples were stored at −80 °C before being processed by SDS-PAGE followed by western blot analysis.
Microscopy and image analysis
For immunofluorescence analysis49, cells were cultured on cover slips. Two percent paraformaldehyde (PFA) was used as a fixative and the cells were permeabilised with 1% BSA and 0.1% Triton X-100 in PBS. Phalloidin-rhodamin (Santa Cruz Biotechnology) was added at a concentration of 1:200. Fluorescent images were made using a Leica (AOBS) microscope.
Migration Assays
Cells were plated in six-well plates and maintained in complete medium. Once the cells reached 80–90% confluence, a scratch was made in the center of the cell monolayer using a sterile plastic pipette tip. Cells were washed immediately with PBS to remove floating cellular debris and incubated for an additional 24 hours. The ability of the cells to migrate into the scratch area was assessed in standard growth conditions after 24 hours by comparing the 0- and 24-hour phase-contrast micrographs of six marked points along the scratched area on each plate. The total remaining scratch area was determined using ImageJ software (Rasband, W.S., ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA). Results are presented as the difference percentage of each time point. For the siRNA experiments, the scratch was made 72 hours after transfection, and cells were analysed as described above.
RNA extraction and RT-PCR
RNA extraction was performed using a RNA extraction kit (Qiagen) according to the manufacturers’ instructions. 500 ng of RNA was used as template for cDNA preparation with the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was performed in a IQ5 cycler PCR machine (Bio-Rad), using the Kapa SYBR Fast qPCR kit (Kapa Biosystems). Primers used are described in Supplementary table 1. Expression levels were corrected for expression of two house-keeping genes, Actb and Gapdh, averaged and presented as fold changes.
Tissue preparation and immunohistochemistry
Immunohistochemistry was performed on tissue samples from five TSC patients (M/F: 3/2; TSC1/TSC2: 1/4). All patients included in this study fulfilled the diagnostic criteria for TSC50 and were obtained from the archives of the Departments of Neuropathology of the Academic Medical Center (University of Amsterdam) and the University Medical Center Utrecht (UMCU). Tissue was obtained in accordance with the Declaration of Helsinki and the Academic Medical Center (AMC) Research Code.
Tissue was fixed in 10% buffered formalin and embedded in paraffin. Paraffin-embedded tissue was sectioned at 6 μm, mounted on organosilane-coated slides (Sigma, St. Louis, MO) and used for immunohistochemical staining. Paraffin-embedded sections were deparaffinized, re-hydrated, and incubated for 20 min in 0.3% H2O2 diluted in methanol to quench endogenous peroxidase activity. Antigen retrieval was performed by incubation for 10 min at 121 °C in citrate buffer (0.01 M, pH 6.0). Sections were washed with PBS and incubated for 30 min in 10% normal goat serum (Harlan Sera-Lab). Incubation with anti p-PAK2 antibody (Cell Signalling, 1:50) was done overnight at 4 °C. Thereafter, sections were washed in PBS and stained with ready-to-use Powervision peroxidase system (Immunologic) and 3,3′-diaminobenzidine (DAB; Sigma) as chromogen. Sections were counterstained with haematoxylin, dehydrated and coverslipped.
For double-labeling, after incubation with p-PAK2 antibody, sections were incubated with Brightvision poly-alkaline phosphatase (AP)-anti-Rabbit (Immunologic) for 30 minutes at room temperature. Sections were washed with PBS and the pH was adjusted with Tris-HCl buffer (0,1 M; pH 8,2). AP activity was visualized with the AP substrate kit I Vector Red (SK-5100, Vector laboratories Inc.). Sections were then incubated at 121 °C in citrate buffer (10 mM NaCi, pH 6.0) for 10 minutes, in order to remove the first primary antibody. Incubation with the second primary antibody (p-S6, Cell Signaling Technology, 1:50) was performed overnight at 4 °C. AP activity was visualized with the AP substrate kit III Vector Blue (SK-5300, Vector laboratories). Controls performed with depletion of the primary antibody showed no staining in any of the material tested (data not shown).
Additional Information
How to cite this article: Alves, M. M. et al. PAK2 is an effector of TSC1/2 signaling independent of mTOR and a potential therapeutic target for Tuberous Sclerosis Complex. Sci. Rep. 5, 14534; doi: 10.1038/srep14534 (2015).
Supplementary Material
Acknowledgments
The authors would like to thank Kaushal Parikh for technical assistance on the scratching assays and Gert van Cappellen for microscopy assistance. Financial Support: MMA is financially supported by the Foundation Michelle and the Fondation Jérôme Lejeune. GMF is financially supported by the Dutch Cancer Society (EMCR 2010-4737). EA is supported by the European Union Seventh Framework Programme FP7/2007-2013 under the project EPISTOP (grant agreement n: 602391). MPP is supported by NWO-ALW (840.12.001) and a personal ECCO grants.
Footnotes
Author Contributions M.M.A., G.M.F. and M.P.P. designed and planned the experiments; M.M.A., G.M.F., K.C.S.Q., S.G., J.A. and M.H.W. prepared and executed the experiments, J.S. analysed the kinome profiling data; M.M.A., G.M., K.C.S.Q., A.S., M.J.B., M.N., Y.E., E.A. and M.P.P. interpreted the data; M.M.A., G.M.F. and M.P.P. wrote the manuscript.
References
- Roach E. S. et al. Diagnostic criteria: tuberous sclerosis complex. Report of the Diagnostic Criteria Committee of the National Tuberous Sclerosis Association. J. Child Neurol. 7, 221–224 (1992). [DOI] [PubMed] [Google Scholar]
- Kandt R. S. et al. Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet. 2, 37–41 (1992). [DOI] [PubMed] [Google Scholar]
- van Slegtenhorst M. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277, 805–808 (1997). [DOI] [PubMed] [Google Scholar]
- Smith M., Yoshiyama K., Wagner C., Flodman P. & Smith B. Genetic heterogeneity in tuberous sclerosis. Map position of the TSC2 locus on chromosome 11q and future prospects. Ann. N. Y. Acad. Sci. 615, 274–283 (1991). [DOI] [PubMed] [Google Scholar]
- European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315 (1993). [DOI] [PubMed] [Google Scholar]
- Huang J. & Manning B. D. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 412, 179–190 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Slegtenhorst M. et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 7, 1053–1057 (1998). [DOI] [PubMed] [Google Scholar]
- Stocker H. et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol 5, 559–565 (2003). [DOI] [PubMed] [Google Scholar]
- Kwiatkowski D. J. Rhebbing up mTOR: new insights on TSC1 and TSC2, and the pathogenesis of tuberous sclerosis. Cancer Biol Ther 2, 471–476 (2003). [DOI] [PubMed] [Google Scholar]
- Papadakis M. et al. Tsc1 (hamartin) confers neuroprotection against ischemia by inducing autophagy. Nat Med 19, 351–357 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veelen W., Korsse S. E., van de Laar L. & Peppelenbosch M. P. The long and winding road to rational treatment of cancer associated with LKB1/AMPK/TSC/mTORC1 signaling. Oncogene 30, 2289–2303 (2011). [DOI] [PubMed] [Google Scholar]
- Chung J., Kuo C. J., Crabtree G. R. & Blenis J. Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69, 1227–1236 (1992). [DOI] [PubMed] [Google Scholar]
- Bissler J. J. et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358, 140–151 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies D. M. et al. Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 358, 200–203 (2008). [DOI] [PubMed] [Google Scholar]
- Krueger D. A. et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 363, 1801–1811 (2010). [DOI] [PubMed] [Google Scholar]
- Franz D. N. et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 381, 125–132 (2013). [DOI] [PubMed] [Google Scholar]
- Parkhitko A. et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci USA 108, 12455–12460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queiroz K. C. et al. Violacein induces death of resistant leukaemia cells via kinome reprogramming, endoplasmic reticulum stress and Golgi apparatus collapse. PLoS One 7, e45362 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baal J. W. et al. Comparison of kinome profiles of Barrett’s esophagus with normal squamous esophagus and normal gastric cardia. Cancer Res 66, 11605–11612 (2006). [DOI] [PubMed] [Google Scholar]
- Parikh K. & Peppelenbosch M. P. Kinome profiling of clinical cancer specimens. Cancer Res 70, 2575–2578 (2010). [DOI] [PubMed] [Google Scholar]
- Fuhler G. M., Diks S. H., Peppelenbosch M. P. & Kerr W. G. Widespread deregulation of phosphorylation-based signaling pathways in multiple myeloma cells: opportunities for therapeutic intervention. Mol Med 17, 790–798 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim C. T., Kola B. & Korbonits M. AMPK as a mediator of hormonal signalling. J Mol Endocrinol 44, 87–97 (2010). [DOI] [PubMed] [Google Scholar]
- Kuo C. J. et al. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358, 70–73 (1992). [DOI] [PubMed] [Google Scholar]
- Price D. J., Grove J. R., Calvo V., Avruch J. & Bierer B. E. Rapamycin-induced inhibition of the 70-kilodalton S6 protein kinase. Science 257, 973–977 (1992). [DOI] [PubMed] [Google Scholar]
- Kola B., Boscaro M., Rutter G. A., Grossman A. B. & Korbonits M. Expanding role of AMPK in endocrinology. Trends Endocrinol Metab 17, 205–215 (2006). [DOI] [PubMed] [Google Scholar]
- Kim S. H., Kowalski M. L., Carson R. P., Bridges L. R. & Ess K. C. Heterozygous inactivation of tsc2 enhances tumorigenesis in p53 mutant zebrafish. Dis Model Mech 6, 925–933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. Loss of Tsc1/Tsc2 activated mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J. Clin Invest 112, 1223–1233 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajkowicz A. & Rusin M. The activation of the p53 pathway by the AMP mimetic AICAR is reduced by inhibitors of the ATM or mTOR kinases. Mech Ageing Dev 132, 543–551 (2011). [DOI] [PubMed] [Google Scholar]
- Ridley A. J. Rho family proteins: coordinating cell responses. Trends Cell Biol 11, 471–477 (2011). [DOI] [PubMed] [Google Scholar]
- Kelly M. L. & Chernoff J. Getting smart about p21-activated kinases. Mol Cell Biol 31, 386–387 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte L. & Crino P. B. The neurobiology of the tuberous sclerosis complex. Neuromolecular Med 8, 531–546 (2006). [DOI] [PubMed] [Google Scholar]
- Eswaran J., Li D. Q., Shah A. & Kumar R. Molecular pathways: targeting p21-activated kinase 1 signaling in cancer—opportunities, challenges, and limitations. Clin Cancer Res 18, 3743–3749 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whale A., Hashim F. N., Fram S., Jones G. E. & Wells C. M. Signalling to cancer cell invasion through PAK family kinases. Front Biosci 16, 849–864 (2011). [DOI] [PubMed] [Google Scholar]
- Gao Y., Dickerson J. B., Guo F., Zheng J. & Zheng Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA 101, 7618–7623 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte L., Aronica E., Baybis M. & Crino P. B. Cytoarchitectural alterations are widespread in cerebral cortex in tuberous sclerosis complex. Acta Neuropathol 123, 685–693 (2012). [DOI] [PubMed] [Google Scholar]
- Crino P. B., Aronica E., Baltuch G. & Nathanson K. L. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 74, 1716–1723 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaus U. G., Morris S., Dong H. J., Chernoff J. & Bokoch G. M. Regulation of human leukocyte p21-activated kinases through G protein—coupled receptors. Science 269, 221–223 (1995). [DOI] [PubMed] [Google Scholar]
- Ye D. Z. & Field J. PAK signaling in cancer. Cell Logist 2, 105–116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova K. A. & Crino P. B. The tuberous sclerosis complex. Ann N Y Acad Sci 1184, 87–105 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J. et al. Tsc1 mutant neural stem/progenitor cells exhibit migration deficits and give rise to subependymal lesions in the lateral ventricle. Genes Dev 25, 1595–1600 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velden Y. U. & Haramis A. P. Insights from model organisms on the functions of the tumor suppressor protein LKB1: zebrafish chips in. Aging 3, 363–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes A. P. et al. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell 129, 549–563 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang S. et al. The tumor suppressor LKB1 regulates lung cancer cell polarity by mediating cdc42 recruitment and activity. Cancer Res 68, 740–748 (2008). [DOI] [PubMed] [Google Scholar]
- Jain P. et al. Integrin-binding Protein Nischarin Interacts with Tumor Suppressor Liver Kinase B1 (LKB1) to Regulate Cell Migration of Breast Epithelial Cells. J Biol Chem 288, 15495–15509 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman N. & Kissil J. Recent advances in the development of p21-activated kinase inhibitors. Cell Logist 2, 132–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh K., Peppelenbosch M. P. & Ritsema T. Kinome profiling using peptide arrays in eukaryotic cells. Methods Mol Biol 527, 269–280 (2009). [DOI] [PubMed] [Google Scholar]
- Goorden S. M. et al. Rheb is essential for murine development. Mol Cell Biol 31, 1672–1678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhler G. M. et al. Disturbed granulocyte macrophage-coloby stimulating factor priming of phosphatidylinositol 3,4,5-trisphosphate accumulation and Rac activation in fMLP-stimulated neutrophils from patients with myelodysplasia. J Leuk Biol 76, 254–262 (2004). [DOI] [PubMed] [Google Scholar]
- Peppelenbosch M. et al. Multiple signal transduction pathways regulate TNF-induced actin reorganization in macrophages: inhibition of Cdc42-mediated filopodium formation by TNF. J Immunol 162, 837–845 (1999). [PubMed] [Google Scholar]
- Gomez M., Sampson J. & Whittemore V. Tuberous Sclerosis Complex. Oxford University Press, Oxford, UK (1999). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.