

Abstract
Pulmonary fibrosis is a progressive lung disease hallmarked by increased fibroblast proliferation, amplified levels of extracellular matrix deposition and increased angiogenesis. Although dysregulation of angiogenic mediators has been implicated in pulmonary fibrosis, the specific rate-limiting angiogenic markers involved and their role in the progression of pulmonary fibrosis remains unclear. We demonstrate that bleomycin treatment induces angiogenesis, and inhibition of the central angiogenic mediator VEGF using anti-VEGF antibody CBO-P11 significantly attenuates bleomycin-induced pulmonary fibrosis in vivo. Bleomycin-induced nitric oxide (NO) was observed to be the key upstream regulator of VEGF via the PI3k/Akt pathway. VEGF regulated other important angiogenic proteins including PAI-1 and IL-8 in response to bleomycin exposure. Inhibition of NO and VEGF activity significantly mitigated bleomycin-induced angiogenic and fibrogenic responses. NO and VEGF are key mediators of bleomycin-induced pulmonary fibrosis, and could serve as important targets against this debilitating disease. Overall, our data suggests an important role for angiogenic mediators in the pathogenesis of bleomycin-induced pulmonary fibrosis.
Keywords: BLEOMYCIN, PULMONARY FIBROSIS, ANGIOGENESIS, VEGF, NITRIC OXIDE
Pulmonary fibrosis is a progressive lung disease with no proven therapeutic intervention and a limited array of biomarkers. Pulmonary fibrosis is characterized by prominent fibroblast proliferation and amplified deposition of extracellular matrix (ECM) [Lu et al., 2010]. The pathogenesis of pulmonary fibrosis is not clear, with evidence for involvement of abnormal repair and aberrant vascular remodeling associated with angiogenesis in both animal models and tissue specimens from patients [Nobel and Norman, 2003; Steurer et al., 2007]. The existence of angiogenesis in pulmonary fibrosis was first described by Turner-Warwick, who demonstrated that neovascularization led to anastomoses between the systemic and pulmonary microvasculature of patients with widespread pulmonary fibrosis [Turner-Warwick, 1963]. Other reports also have demonstrated vascular remodeling in pulmonary fibrosis, suggesting that neovascularization enhances fibrosis [Peao et al., 1994; Renzoni et al., 2003; Cosgrove et al., 2004]. Studies using both animal models and tissue specimens from patients with pulmonary fibrosis suggest an imbalance in the levels of angiogenic chemokines as compared with angiostatic chemokines, favoring net angiogenesis [Keane et al., 1997; Keane et al., 1999a; Keane et al., 1999b; Keane et al., 2001]. However, in spite of the several studies linking angiogenesis to pulmonary fibrosis, the specific mechanistic details underlying angiogenic modulation in pulmonary fibrosis remain unclear.
Vascular endothelial growth factor (VEGF) plays an important role in vascular permeability and angiogenesis by inducing migration, proliferation, elongation, network formation and branching of endothelial cells [Carmeliet and Collen, 2000; Helmlinger et al., 2000]. VEGF is expressed abundantly by various cells in adult human lungs including alveolar epithelial cells, clara cells, macrophages and fibroblasts [Fehrenbach et al., 1999]. VEGF is reported to be dysregulated in patients with fibrotic diseases such as idiopathic myelofibrosis and diffused lung fibrosis [Meyer et al., 2000; Steurer et al., 2007]. Recent reports also suggest that pathological changes in the lung are accompanied by elevated expression of other angiogenic mediators such as plasminogen activator inhibitor-1 (PAI-1), chemokine receptor (CXCR) and Akt, and cytokines such as interleukin-8 (IL-8), and IL-1β, all of which may play a critical role in pulmonary fibrosis [Fichtner et al., 2004; Russo et al., 2009; Russo et al., 2011; Bhandary et al., 2013]. While these factors have been shown to be important in pulmonary fibrosis, the mechanisms by which these mediators are regulated also remains poorly understood. Free radicals such as nitric oxide (NO) are also known to play an important role in the development of pulmonary fibrosis [Kinnula et al., 2005; Noguchi et al., 2014]. Nitric oxide (NO) is an important signaling molecule produced endogenously from L-arginine in a reaction catalyzed by NO synthases (NOSs) [Azad et al., 2006; Iyer et al., 2014]. NO is known to play an important role in maintaining respiratory homeostasis, and therefore even subtle changes in its rate of production may critically impact cellular homeostasis [Ricciardolo et al., 2004]. Several reports have implicated NO in various physiological processes, including angiogenesis in a manner consistent with a pro-angiogenic phenotype.
In this study, we investigated the role of angiogenesis in the pathogenesis of pulmonary fibrosis by analyzing the effect of angiogenic mediators such as NO and VEGF on bleomycin-induced pulmonary fibrosis. The overall objective of this study is to analyze (a) the effect of bleomycin on angiogenesis and important angiogenic mediators, (b) the key angiogenic signaling pathways and mediators that drive bleomycin-induced fibrosis, and (c) the role of NO and VEGF in the regulation of bleomycin-induced angiogenesis in the context of pulmonary fibrosis.
MATERIALS AND METHODS
CHEMICALS AND REAGENTS
Antibodies against phospho-Akt and total Akt were obtained from Cell Signaling Technology, Inc. (Beverly, MA). Antibodies for collagen type III were from Fitzgerald (Concord, MA). Antibodies for PAI-1, CXCR-1/2, β-actin, and horseradish peroxidase (HRP)-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). IL-8 and VEGF immunoassay kits, a neutralizing antibody against VEGF (CBO-P11), and its nonspecific control antibody were from R&D Systems (Minneapolis, MN). All other chemicals and reagents including bleomycin sulfate, 4,5-diaminofluorescein diacetate (DAF-DA), NO donor sodium nitroprusside (SNP), NO inhibitors aminoguanidine (AG) and 2-(4-carboxyphenyl)-4,4,5,5-tetramethyl-imidazoline-1-oxy-3-oxide (PTIO), and PI3K/Akt inhibitor LY-294002 were obtained from Sigma Chemical, Inc. (St. Louis, MO).
CELL CULTURE
Human lung CRL-1490 fibroblasts (ATCC; Manassas, VA) were maintained in Eagle’s Minimum Essential Medium (MEM) (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Hyclone), 100 U/ml penicillin (Hyclone), 100 mg/ml streptomycin (Hyclone) and Amphotericin B (Invitrogen). Human umbilical vein endothelial cells (HUVECs) were cultured in MCDB131 media (Gibco) supplemented with 25% FBS, 0.05% brain bovine extract (BD Biosciences), 0.25% endothelial cell growth supplement (BD Biosciences), 0.1% heparin (Sigma), 1% L-glutamine (Sigma) and 0.1% gentamicin sulphate (Sigma). HUVECs between passages two and seven were used. Both CRL-1490 and HUVEC cell lines were cultured at 37°C in 5% CO2 incubator, and were passaged at preconfluent densities using a solution containing 0.05% trypsin (Sigma) and 0.5 mM EDTA (Invitrogen).
ANIMAL MODEL
For the animal studies, 6–8 weeks old C57BL/6 mice were used (Jackson Laboratories, Bar Harbor, ME). Mice were housed in a barrier facility with specific pathogen-free conditions, and all experiments were performed using protocols approved by the Old Dominion University (ODU) animal facility. Briefly, mice were first anesthetized with isoflurane. In the first set of experiments, either bleomycin sulfate or equal volumes of saline as control was administered intranasally. In a separate set, bleomycin-treated mice were co-treated every other day by intraperitoneal injection of CBO-P11 starting at day 0 and were continued until the mice were euthanized. Mice were euthanized at various time points after bleomycin instillation. Bronchoalveolar lavage (BAL) fluid was collected after the trachea was exposed and cannulated with a 20-gauge catheter. After instillation of 1 ml of cold sterile PBS three times through the trachea into the lung, BAL fluid was recovered to 90% of the original volume. The BAL fluid was centrifuged for 10 min at 1500 rpm and the cell-free supernatant was stored at −80°C. The lungs were perfused with 5 ml of cold saline through the left ventricle and surgically removed. The left lungs were used to evaluate the fibrotic score by histological examination, and the right lungs were homogenized to analyze protein expression levels.
HISTOPATHOLOGY
Mice were euthanized and the left lung fixed with 10% formalin overnight and embedded in paraffin. Paraffin sections (3 μm thick) were stained with hematoxylin-eosin (H&E). The pathological grade of fibrosis was evaluated under 10X magnification. Histological assessment of the extent and severity of pulmonary fibrosis was independently determined by one pathologist and two researchers in a blind study using the Ashcroft method [Ashcroft et al., 1988] on sections of the left lung stained with H&E.
COLLAGEN DETECTION ASSAY
Soluble collagen levels were quantified by Sircol® Assay (Biocolor Ltd, Belfast, UK) as per manufacturer’s protocol. Briefly, mice lung homogenates or cell samples were mixed with 1 ml Sircol dye for 30 min then centrifuged to pellet collagen. The pellet was washed and resuspended in Alkyne solution and the absorption was measured at 550 nm (Synergy HI Hybrid Reader, BioTek).
ENZYME-LINKED IMMUNOSORBENT ASSAYS (ELISA)
The expression levels of VEGF and IL-8 proteins were quantified using ELISA. Briefly, supernatant from treated cells or lavage fluid from saline, bleomycin and CBO-P11 treated mice were collected and analyzed for VEGF or IL-8 protein levels using respective Quantikine ELISA kits (R&D Systems, Minneapolis, MN) per manufacturer’s protocol. Briefly, samples or reference standards (100 μl) were added to each well of a microplate pre-coated with monoclonal antibody specific to VEGF or IL-8 and incubated overnight at 4°C. After washing out unbounded proteins, an HRP-conjugated polyclonal secondary antibody was added to the wells and incubated for 2 h at room temperature. After washing and adding 100 μl of substrate solution, optical density was determined at 450 nm (Synergy HI Hybrid Reader, BioTek).
ANGIOGENESIS TUBE FORMATION ASSAY
Growth factor-reduced matrigel (Becton Dickinson) was diluted 1:1 with serum free media (SFM), placed in 48-well plates (175 μl/well), and allowed to set at 37°C for 60 min. HUVECs (5 × 104) suspended in 2.5% dialyzed FBS medium containing specific treatments were added to each well and incubated at 37°C in 5% CO2 for 24 h. The morphological changes were observed and photographed using phase contrast microscope under 5X magnification. The number of nodes formed was scored manually from at least five different fields for each treatment.
MESSENGER RNA EXPRESSION PROFILING AND QUANTATIVE RT-PCR
Expression of angiogenesis-related mRNAs in CRL-1490 cells was determined by RT-PCR. Cells were treated, lysed and mRNA isolated using the RNeasy Mini Kit (Qiagen). cDNA was prepared from extracted mRNA (1 μg) using the RT2 First Strand Kit (Qiagen). cDNA combined with RT2 SYBR Green Master Mix (Qiagen) was added to individual RT-PCR array wells (SABiosciences) for analysis. Data was collected using the iCycler RT-PCR system (BioRad). Gene arrays for angiogenesis was assayed for mRNA expression, and the data was normalized to blank housekeeping genes.
CELL PROLIFERATION ASSAY
Cellular proliferation in CRL-1490 cells was quantitatively determined by assessing incorporation of CyQUANT® (Invitrogen) DNA binding fluorescent dye. Cells were seeded in 96-well plates at a density of 5,000 cells/well in 10% MEM, and were treated after 24 h as indicated and incubated for various time points. Subsequently, cells were incubated in each well with 50 μl of 1X CyQUANT® for 60 min at 37°C. The fluorescence intensity of each sample was measured at excitation/emission wavelengths of 485/535 nm respectively.
NO DETECTION
Intracellular NO production was determined by spectrofluorometry using NO-specific probe DAF-DA. After specific traetments, cells (1 × 106/ml) were incubated with the probe (10 μM) for 30 min at 37°C, after which they were washed, resuspended in PBS, and analyzed for DAF fluorescence at excitation and emission wavelengths of 488 nm and 538 nm, respectively (Synergy HI Hybrid Reader, BioTek).
WESTERN BLOT ANALYSIS
Mice lung homogenates or cell lysates were resolved on a 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. The protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce Biotechnology, Rockford, IL), and equal amount of protein was loaded per sample. The membrane was blocked with TBS-T (0.1% Tween-20 in TBS) containing 5% dry milk, and incubated with primary antibody overnight at 4°C. After three washes with TBS-T, the membrane was incubated with HRP-conjugated secondary antibody for 1 h and then washed with TBS-T. Immunoreactive proteins were detected by chemiluminescence (Supersignal® West Pico, Pierce, Rockford, IL) and quantified by imaging densitometry, using myImageAnalysis Software (Thermo). Mean densitometry data from independent experiments was normalized to results obtained from untreated control cells.
STATISTICAL ANALYSIS
The data represent mean ± S.E.M from three or more independent experiments. Statistical analysis was performed using Student’s t-test to evaluate the measurements at a significance level of P< 0.05.
RESULTS
BLEOMYCIN INDUCED ANGIOGENESIS AND VEGF PRODUCTION
The capability of bleomycin to induce angiogenesis was analyzed by in vitro tube formation assay. HUVECs were treated with varying concentrations of bleomycin (0–25 mU/ml). Bleomycin induced angiogenesis in a dose-dependent manner, as assessed by the number of nodes (Fig. 1A). To correlate the angiogenic response to bleomycin-induced fibrogenesis, supernatant from fibroblasts treated with varying concentrations of bleomycin (0–25 mU/ml) were collected and tested for induction of angiogenesis in HUVECs (Fig. 1B and C). Supernatant from cells treated with bleomycin also showed a similar dose-dependent induction in angiogenesis.
Fig. 1.
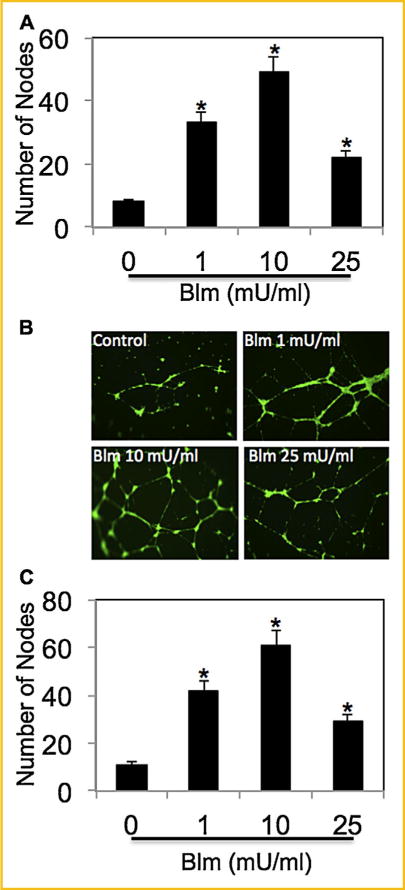
Bleomycin induces angiogenesis and VEGF production. (A) HUVECs were treated with varying concentrations of bleomycin (0, 1, 10, and 25 mU/ml) for 6 h, and analyzed for angiogenesis by in vitro tube formation assay. The number of nodes formed by the tubes were scored and plotted. (B) CRL-1490 cells were treated with varying concentrations of bleomycin (0–25 mU/ml) for 24 h. Supernatant of treated cells were used to induce angiogenesis in HUVECs for 6 h and analyzed for angiogenesis by in vitro tube formation assay. Representative micrographs after staining with calcein-AM are shown. (C) The number of nodes formed by the tubes were scored and plotted. Plots are mean ± S.E.M (n = 3). *P < 0.05 versus non-treated control.
We next assessed the effect of bleomycin exposure on VEGF. Bleomycin induced VEGF levels in a dose-dependent manner (Fig. 2A). The effect of bleomycin on fibroblast proliferation and collagen levels was assessed in order to confirm bleomycin-induced fibrogenic response. Fibroblast proliferation was induced by bleomycin in a dose-dependent manner (Fig. 2B). Analysis of total collagen content in the cell supernatants of bleomycin-treated samples by Sircol® assay showed a dose-dependent effect of bleomycin on induction of soluble collagen (Fig. 2C). Figure 2D shows that collagen type III, one of the most abundant proteins of the extracellular matrix (ECM), was induced by bleomycin treatment. Together, these results indicate that bleomycin was able to directly induce fibroblast proliferation and collagen production in CRL-1490 cells.
Fig. 2.
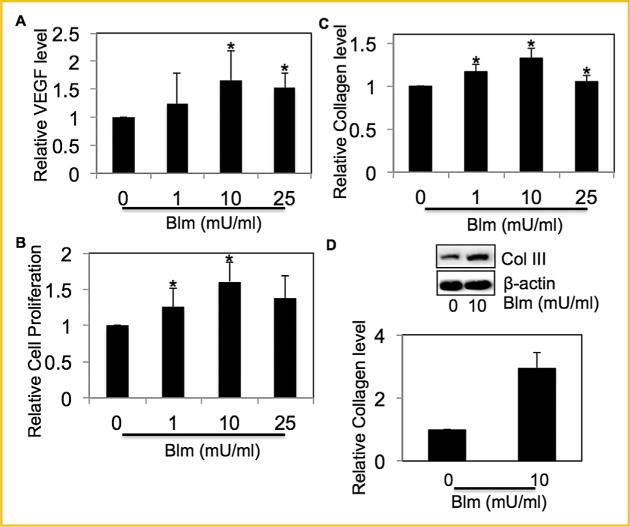
Effect of bleomycin on VEGF levels and fibrogenesis. (A) Supernatant from CRL-1490 cells treated with varying concentrations of bleomycin (0–25-mU/ml) for 24 h were collected and analyzed for VEGF by ELISA. (B) CRL-1490 cells were treated with varying concentrations of bleomycin (0–25 mU/ml) for 48 h, and cell proliferation was determined spectrofluorometrically using CyQUANT® dye. (C) Supernatant from CRL-1490 cells treated with varying concentrations of bleomycin (0–25 mU/ml) for 24 h was analyzed for soluble collagen levels by Sircol® Assay. (D) CRL-1490 cells were treated with bleomycin (10 mU/ml) for 24 h and analyzed for Collagen III levels by Western blotting. Western blots were reprobed with β-actin antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. Plots are mean ± S.E.M (n = 4). *P< 0.05 versus non-treated control.
BLEOMYCIN INDUCED VEGF LEVELS IN FIBROTIC MICE
We characterized\bleomycin-induced fibrotic response in mice. Lung histology data showed induction of interstitial fibrosis of the alveolar wall inmice receiving bleomycin (Fig. 3A). The extent of lung fibrosis was determined by quantitative histology according to Ashcroft’s method [Ashcroft et al., 1988] (Fig. 3B). Bleomycin-treated mice showed a dose-and time-dependent increase in collagen levels as compared to saline treated control mice (Fig. 3C). Levels of collagen III, one of the most abundant collagen proteins, were determined in bleomycin treated mice lung tissue homogenates by Western blotting (Fig. 3D). To validate the effect of bleomycin on VEGF, bleomycin-treated mice BAL fluid was analyzed for VEGF levels by ELISA. Bleomycin treatment induced significant levels of VEGF in mice BAL fluid in a dose- and time-dependent manner (Fig. 3E).
Fig. 3.
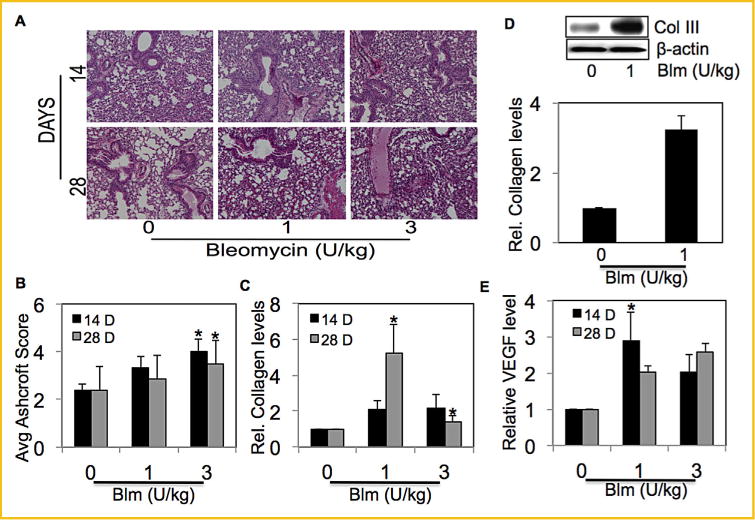
Bleomycin induced VEGF levels in fibrotic mice. (A) C57BL/6 mice were treated with bleomycin (1 and 3 U/kg body weight diluted in sterile saline) or equal volumes of saline as control, and were euthanized at various time points (14 days and 28 days). Representative immunohistochemical micrographs of mice lung tissue stained with H&E. (B) Calculated fibrosis score based on the histopathological assessment of H&E-stained slides. (C) Lung homogenate from treated mice was analyzed for soluble collagen levels by Sircol® Assay. (D) Mice treated with bleomycin (1 U/kg) or equal volumes of saline as control were euthanized at 28 days. Lung homogenate from treated mice was analyzed for collagen III levels by Western blotting. Western blots were reprobed with β-actin antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. (E) Mice were treated with bleomycin (1 and 3 U/kg body weight diluted in sterile saline) or equal volumes of saline as control, and were euthanized at various time points (14 days and 28 days). BAL fluid from treated mice was analyzed for VEGF by ELISA. Plots are mean ± S.E.M of five animals per group. *P< 0.05 versus non-treated control.
BLEOMYCIN-DEPENDENT INDUCTION OF VEGF IS REGULATED BY NO VIA THE PI3K/AKT PATHWAY
In response to bleomycin treatment, we observed an increase in NO levels and NOS-2 expression in fibroblast cells (Fig. 4A and B). Similar increase in NOS-2 levels was also observed in bleomycin-treated mice lung homogenates (Fig. 4C). Downregulation of Akt by PI3K inhibitor LY294002 confirmed our previous finding that treatment with bleomycin led to the phosphorylation of Akt through PI3K activation [Lu et al., 2010]. Interestingly, NO inhibitor AG significantly inhibited bleomycin-induced phospho-Akt levels indicating that NO is upstream of Akt (Fig. 4D). Furthermore, AG significantly inhibited bleomycin-induced VEGF levels and NO donor SNP showed an opposite effect. Pretreatment with PI3K/Akt inhibitor LY294002 significantly decreased bleo-mycin-induced VEGF levels confirming previously reported work by our group that VEGF is regulated by PI3K/Akt pathway in response to bleomycin treatment [Lu et al., 2010]. The VEGF inhibitor CBO-P11 was used as a negative control (Fig. 4E). Bleomycin-induced VEGF levels were also measured in BAL fluid of bleomycin-treated mice and compared with mice co-treated with VEGF inhibitor CBO-P11 (Fig. 4F). There was no significant effect of bleomycin on phospho-Akt and NOS-2 expression levels in mice tissue homogenates co-treated with VEGF inhibitor CBO-P11 (Supplemental Figures 1A and B). To delineate potential crosstalk between PI3K and NO, we used PI3K inhibitors LY294002 and Wortmannin, and assessed its effect on NO. PI3K inhibitors had no significant effect on bleomycin-induced NO or NOS-2 levels (Supplemental Figures 2A and B).
Fig. 4.
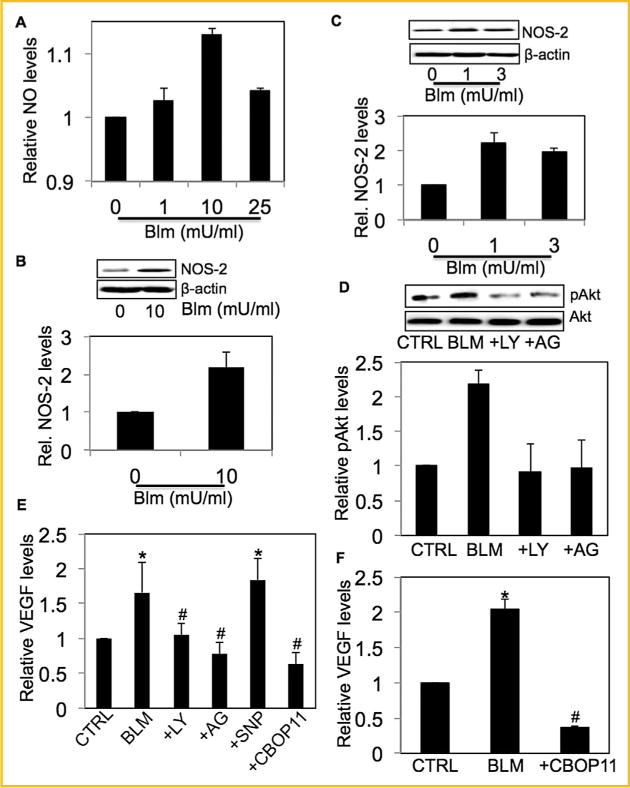
Bleomycin-dependent induction of VEGF is regulated by NO via the PI3K/Akt pathway (A) Cells were treated with bleomycin (0–25 NmU/ml)for 1 h and analyzed for DAF fluorescence. Plots show relative fluorescence intensity over non-treated control. (B) CRL-1490 cells were treated with bleomycin (10 mU/ml) for 3 h and analyzed for NOS-2 levels by Western blotting. (C) Mice treated with bleomycin (1 U/kg) or equal volumes of saline as control were euthanized at 28 days. Lung homogenate from treated mice was analyzed for NOS-2 levels by Western blotting. (D) CRL-1490 cells were either left untreated or pretreated with LY294002 (10 μM) or AG (300 μM) for 1 h, followed by bleomycin treatment (10mU/ml) for 6 h and analyzed for pAkt levels by Western blotting. The blots were reprobed with pan-Akt antibody and analyzed for total Akt levels. (E) Cells were left untreated or pretreated with either LY294002 (10μM), AG (300 μM), SNP (500μg/ml) or CBO-P11 (10 μM) for 1 h, followed by bleomycin treatment (10mU/ ml) for 24 h. Cell supernatant was analyzed for VEGF levels by ELISA. (F) Mice treated with bleomycin (1 U/kg) in the presence or absence of CBO-P11 (0.3 mg/kg) or equal volumes of saline as control were euthanized at 28 days. BAL fluid from treated mice was analyzed for VEGF by ELISA. All blots were reprobed with β-actin antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. Plots are mean ±S.E.M (n =4). *P< 0.05 versus non-treated control. #P<0.05 versus non-treated control and corresponding bleomycin treatment.
INVOLVEMENT OF OTHER ANGIOGENIC PROTEINS IN RESPONSE TO BLEOMYCIN
To analyze the involvement of additional angiogenic mediators in response to bleomycin exposure, mRNA microarray analysis for expression of well-established angiogenic mediators was performed. The mRNA extracted from untreated lung fibroblasts (control) or fibroblasts treated with bleomycin were analyzed by qRT-PCR. Genes showing at least a twofold change inexpression in bleomycin-treated cells as compared to untreated control cells are listed (Fig. 5A). Based on the mRNA analysis, the effect of bleomycin was analyzed on a few specific angiogenic proteins. Treatment with bleomycin significantly induced PAI-1 (SERPINE1) levels both in vitro and in vivo (Fig. 5B and C). Pretreatment with AG, LY294002 or CBO-P11 downregulated bleomycin-induced PAI-1 expression levels in cells (Fig. 5B). Lung tissue homogenates from mice pre-treated with VEGF inhibitor CBO-P11 showed significant down-regulation of bleomycin-induced PAI-1 expression levels (Fig. 5C). Next, the effect of bleomycin on one of the most abundant interleukins IL-8 was analyzed. Bleomycin treatment increased IL-8 levels and CBO-P11 inhibited this effect both in vitro and in vivo (Fig. 5D and E). NO inhibitors AG and PTIO also showed an inhibitory effect on bleomycin-induced IL-8 levels in cells (Fig. 5D). IL-8 is a substrate of CXC receptors one and two, and the various interleukins and chemokines that were induced in response to bleomycin bound CXCR 1/2; therefore, the abundance of protein receptors CXCR 1/2 was analyzed. CXCR 1/2 expression was positively regulated by bleomycin both in vitro and in vivo (Supplemental Figures 3A and B). Both AG and CBO-P11 pre-treatment showed minimal effect on CXCR 1/2 expression levels in fibroblasts; however CBO-P11 pre-treatment showed downregulation of bleomycin-induced CXCR 1/2 expression levels in lung homogenates of co-treated mice.
Fig. 5.
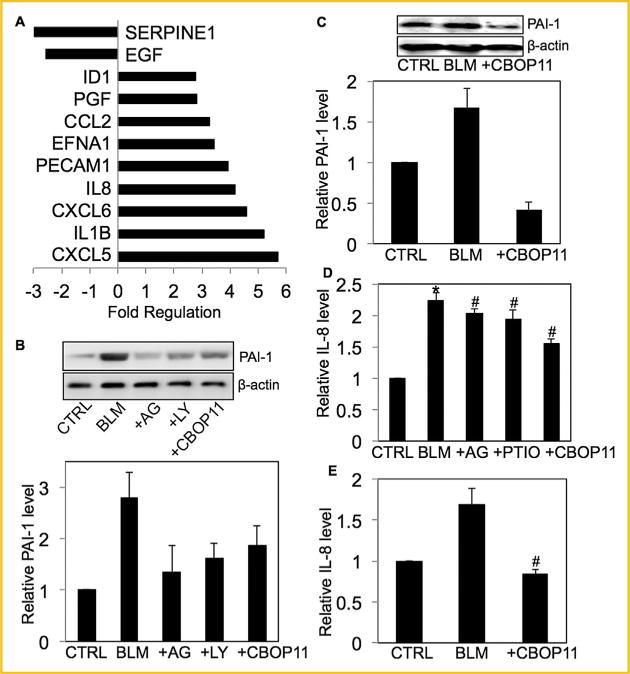
Involvement of other angiogenic proteins in response to bleomycin. (A) mRNA was extracted from untreated CRL-1490 cells or cells treated with bleomycin (10mU/ml) for 24 h and transcribed into cDNA. Abundance of specific genes was analyzed byqRT-PCR. Fold change between control and treated cells is shown. (B) Cells were left untreated or pretreated with AG (300 μM), LY294002 (10 μM)or CBO-P11 (10 μM) for 1 h, followed by bleomycin treatment (10 mU/ml) for 24 h and analyzed for PAI-1 levels by Western blotting. (B) Mice treated with bleomycin (1 U/kg) in the presence or absence of CBO-P11 (0.3 mg/kg) or equal volumes of saline as control were euthanized at 28 days. Lung homogenate from treated mice was analyzed for PAI-1 levels by Western blotting. (D) Cells were either left untreated or pretreated with either AG (300 μM), PTIO (300 μM) or CBO-P11 (10 μM)for 1 h, followed by bleomycin treatment (10 mU/ml) for 48 h. Cell supernatant were analyzed for IL-8 level by ELISA. (E) Mice treated with bleomycin (1 U/kg) in the presence or absence of CBO-P11 (0.3 mg/kg) or equal volumes of saline as control were euthanized at 28 days. BAL fluid from treated mice was analyzed for IL-8 levels by ELISA. Western blots were reprobed with β-actin antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. Plots are mean ± S.E.M of 5 animals per group. *P< 0.05 versus non-treated control. #P< 0.05 versus non-treated control and corresponding bleomycin treatment.
AG AND CBO-P11 INHIBITS BLEOMYCIN-INDUCED ANGIOGENESIS AND FIBROSIS
The effect of inhibitors of NO, Akt and VEGF was assessed on bleomycin-induced angiogenesis by in vitro tube formation assay. Supernatant from CRL-1490 cells treated with bleomycin in the presence or absence of AG, CBO-P11, and LY294002 were collected and used to induce angiogenesis in HUVECs. Pre-treatment with inhibitors significantly inhibited bleomycin-induced angiogenesis (Fig. 6A). To correlate bleomycin-induced angiogenic response to bleomycin-induced fibrogenesis, CRL-1490 cells were treated with bleomycin in the presence or absence of AG or CBO-P11, and cell growth monitored by CyQUANT® cell proliferation assay. Both AG and CBO-P11 significantly inhibited bleomycin-induced fibroblast proliferation (Fig. 6B). Similarly, pretreatment with AG and CBO-P11 significantly inhibited bleomycin-induced collagen levels in fibroblast cells as analyzed by Sircol® Assay and Western blotting (Fig. 6C and D). The effect of VEGF inhibitor CBO-P11 on bleomycin-induced fibrosis was confirmed in vivo. Bleomycin-induced collagen production was significantly inhibited in CBO-P11 pretreated mice (Fig. 6E and F). Lung histology data and quantitative histology showed a significant reduction in fibrosis in animals co-treated with CBO-P11, which brought the fibrotic response down to control levels (Fig. 6G and H).
Fig. 6.
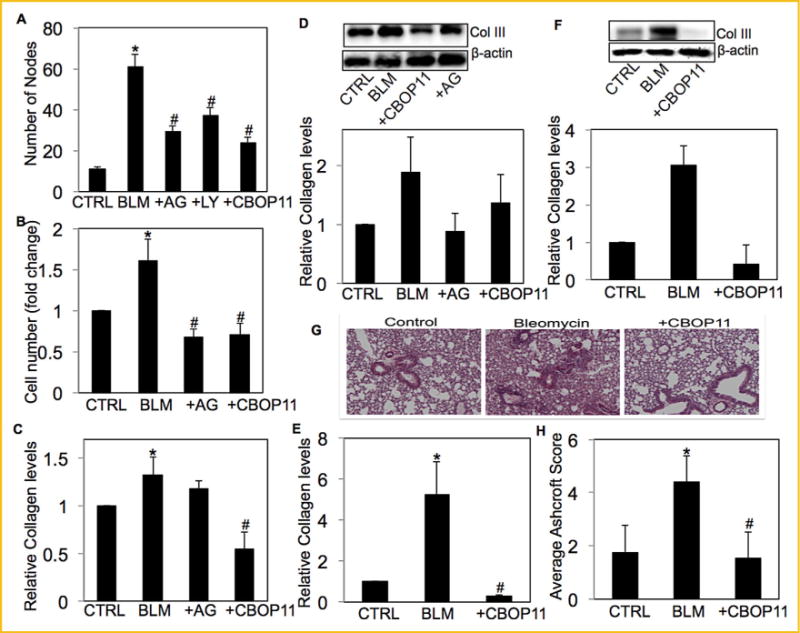
AG and CBO-P11 inhibits bleomycin-induced angiogenesisand fibrosis. (A) Cell supernatant from untreated CRL-1490 cells or cells treated with bleomycin (10mU/ml) in the presence or absence of AG (300 μM), LY294002 (10 μM) or CBO-P11 (10 μM) for 24h were used to induce angiogenesis in HUVECsfor 6 h and were analyzed by in vitro tube formation assay. (B) CRL-1490 cells were left untreated or were treated with bleomycin (10 mU/ml) in the presence or absence of either AG (300 μM) or CBO-P11 (10 μM) for 48 h and cell proliferation was determined spectrofluorometrically using CyQUANT® dye. (C) CRL-1490 cells left untreated or were treated with bleomycin (10mU/ml) in the presence or absence of either AG (300 μM) or CBO-P11 (10 μM) for 24 h and analyzed for soluble collagen levels by Sircol® Assay. (D) CRL-1490 cells left untreated or were treated with bleomycin (10mU/ml) in the presence or absence of either AG (300 μM) or CBO-P11 (10μM) for 24 h and analyzed for collagen III levels by Western blotting. (E) Mice treated with bleomycin (1 U/kg) in the presence or absence of CBO-P11 (0.3 mg/kg) or equal volumes of saline as control were euthanized at 28 days. Lung homogenatefrom treated mice was analyzed for soluble collagen levels by Sircol® Assay. (F) Lung homogenate from treated mice was analyzed for collagen III levels by Western blotting. (G) Representative immunohistochemical micrographs of treated mice lung tissue stained with H&E. (H) Calculated fibrosis score based on the histopathological assessment of H&E-stained slides. Western blots were reprobed with β-actin antibody to confirm equal loading of the samples. The immunoblot signals were quantified by densitometry. Plots are mean ±S.E.M (n = 3). *P< 0.05 versus non-treated control. #P<0.05 versus non-treated control and corresponding bleomycin treatment.
DISCUSSION
In pulmonary fibrosis, activation of fibroblast proliferation and accumulation of ECM during the repair process necessitates neovascularization, and increasing evidence supports the role of angiogenesis in pulmonary fibrosis [Selman et al., 1986; Brinckerhoff and Matrisian, 2002; Selman and Pardo, 2002]. VEGF is the central regulator of angiogenesis and has been widely reported to be overexpressed in fibrotic lungs [Carmeliet and Collen, 2000; Steurer et al., 2007). We demonstrate that bleomycin treatment not only potentiates an angiogenic response (Fig. 1) but also induces pro-angiogenic signaling molecules such as VEGF both in vitro and in vivo (Figs. 2A and 4F) that may promote vascularization. Furthermore, neutralization of VEGF by anti-VEGF antibody CBO-P11 significantly inhibited the fibrogenic effects of bleomycin (Fig. 6). Several strategies have been utilized to inhibit VEGF function, and CBO-P11 is considered to be one of the most potent inhibitors of VEGF [Zilberberg et al., 2003]. We have previously shown that PI3K/ Akt is the major cell survival pathway involved in bleomycin-induced pulmonary fibrosis that regulates VEGF protein [Lu et al., 2010]. Here we confirm that bleomycin-induced VEGF production was inhibited by treatment with PI3K/Akt inhibitor LY294002 (Fig. 4E). Furthermore, LY294002 significantly inhibited bleomycin-induced angiogenic response (Fig. 6A), indicating that PI3K/Akt may be one of the major regulatory pathways in bleomycin-induced VEGF production and angiogenesis.
Our data also demonstrates the involvement of the free radical NO in bleomycin-induced angiogenesis and fibrosis. NO has been shown to play a role in bleomycin–induced pulmonary fibrosis [Chen et al., 2001; Chen et al., 2003; Noguchi et al., 2014]. NO inhibitor AG showed significant inhibition of bleomycin-induced fibroproliferative and collagen-inducing effects (Fig. 6). Although the effect of NO on pulmonary fibrosis has been previously shown, its effect on bleomycin-induced angiogenesis has not been demonstrated. We report here for the first time that NO inhibitor AG significantly attenuated bleomycin-induced angiogenesis (Fig. 6A). Furthermore, AG significantly inhibited bleomycin-induced phospho-Akt and VEGF levels (Fig. 4), suggesting that modulation of angiogenesis by NO may be critical in bleomycin-induced fibrotic response. Furthermore, we observed no significant effect of PI3K inhibitors LY294002 and Wortmannin on bleomycin-induced NO (Supplemental Figure 2), indicating no that PI3K and NO work relatively independent of each other in exerting effects on VEGF.
The angiogenic process is known to involve an intricate cytokine network that activates and mediates interactions between multiple cell types. We investigated the involvement of other angiogenic mediators by mRNA analysis using qRT-PCR arrays for angiogenesis. The real-time mRNA data indicated regulation of various angiogenic mRNA (Fig. 5A). Notable examples include chemokines and cytokines such as CCL2, CXCL5, CXCL6, IL1B, and IL-8, many of which bind to CXCR 1 and 2, and VEGF homologues (Placental Growth Factor (PGF)) that promote angiogenesis. IL-8, one of the most abundant cytokines, is an angiogenesis promoting factor, and may stimulate blood vessel formation in fibrotic tissue [Fichtner et al., 2004; Keane et al., 1997]. Bleomycin increased IL-8 protein levels both in vitro and in vivo, which was the one of the most abundant mRNA expressed in fibroblast cells in response to bleomycin (Fig. 5). IL-8 binds CXC1/2 receptors, which represents a major class of receptors for proteins that promote angiogenesis. In addition, proteins transcribed from bleomycin-upregulated mRNAs such as CXCL6 binds CXCR1 while CXCL1 and CXCL5 bind CXCR2 (Fig. 5A). CXCR1/2 have been previously shown to be upregulated in response to bleomycin treatment [Kalayarasan et al., 2013]. While studies have focused on the regulation of various CXCR 1/2 substrates including IL-8 and IL-6, little has been reported in terms of bleomycin-mediated receptor modulation. We report that bleomycin treatment led to an increase in CXCR 1/2 levels in fibroblast cells as well as mouse lung tissue (Supplemental Figure 3). Bleomycin treatment downregulated mRNA levels of the pro-angiogenic mediator PAI-1 (Fig. 5A), whereas it increased PAI-1 protein levels, both in vitro and in vivo (Fig. 5). PAI-1 is reported to be upregulated in fibrotic tissue and its expression is mainly induced by TGFb and mediated by HIF1a [Ueno et al., 2011]. Another group reported that PAI-1 knockout mice did not develop pulmonary fibrosis when treated with bleomycin [Loskutoff and Quigley, 2000]. The contrasting effect on mRNA and protein levels indicates that PAI-1 protein expression may be regulated post-translationally in response to bleomycin exposure. PAI-1 has been shown to be regulated by PI3K/Akt pathway via HIF-1a [Kietzmann et al., 2003]. Pretreat-ment with LY294002 led to a decrease in bleomycin-induced PAI-1 levels (Fig. 5B), confirming that the PI3K/Akt pathway regulates this protein in response to bleomycin treatment.
We further explored the role of NO and VEGF in bleomycin-induced fibrogenesis and their interplay with other angiogenic mediators in response to bleomycin treatment by analyzing the effect of AG and CBO-P11 on various proteins. Pre-treatment with AG showed significant inhibition of bleomycin-induced PAI-1 levels but had no significant effect on bleomycin-induced CXCR1/2 levels (Supplemental Figure 3) in lung fibroblasts. In addition, NO inhibitors AG and PTIO significantly inhibited bleomycin-induced IL-8 levels (Fig. 5). Pre-treatment with CBO-P11 significantly inhibited bleomycin-induced PAI-1 and IL-8 protein levels in both in vitro and in vivo samples, suggesting that these proteins may be downstream of VEGF (Fig. 5). CBO-P11 showed minimal effect on bleomycin-induced CXCR 1/2 levels in vitro; however, pretreatment with CBO-P11 inhibited bleomycin-induced CXCR 1/2 levels in mice lung homogenates (Supplemental Figure 3). Overall, the regulation of CXCR1/2 by VEGF in response to bleomycin treatment was not conclusive. Furthermore, CBO-P11 significantly inhibited bleomycin-induced fibrogenic and angiogenic responses including cell proliferation, collagen production, and angiogenesis. Immunohistochemical analysis clearly showed that CBO-P11 has a protective effect in bleomycin-induced pulmonary fibrosis, thus establishing the significance of VEGF in pathogenesis of pulmonary fibrosis (Fig. 6).
In summary, our data provides evidence that in addition to fibrotic effects, bleomycin is capable of inducing an angiogenic response. We also demonstrate the significance of angiogenesis and angiogenic mediators in bleomycin-induced fibrosis. Importantly, NO and VEGF emerged as key players in response to bleomycin by regulating both fibrogenic and angiogenic responses. The involvement of NO in bleomycin-induced angio-genesis and its regulation of VEGF through PI3K/Akt pathway is a novel finding. The VEGF inhibitor CBO-P11 exerted protective effect in bleomycin-induced pulmonary fibrosis. Furthermore, we report novel effects of NO and VEGF in regulating PAI-1, IL-8 and CXCR 1/2 proteins, which are strong angiogenic inducers and partake in the progression of pulmonary fibrosis. Blocking angiogenesis and VEGF is considered an attractive strategy in inhibiting various forms of cancer and may be widely applied to other angiogenesis-related diseases. Since angiogenesis is a key characteristic of pulmonary fibrosis, understanding the mechanisms involved in cellular angiogenic responses and targeting key angiogenic mediators may aid in the development of more effective therapies against this fatal disease.
Supplementary Material
Acknowledgments
This work was supported by grants from National Institutes of Health (HL112630 and CA173069). We thank the Animal facility staff at ODU for maintaining the animals.
Grant sponsor: National Institutes of Health; Grant numbers: HL112630, CA173069.
Footnotes
Conflict of interest: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–470. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J Biolo Chem. 2006;281:34124–34134. doi: 10.1074/jbc.M602551200. [DOI] [PubMed] [Google Scholar]
- Bhandary YP, Shetty SK, Marudamuthu AS, Ji HL, Neuenschwander PF, Boggaram V, Morris GF, Fu J, Idell S, Shetty S. Regulation of lung injury and fibrosis by p53-mediated changes in urokinase and plasminogen activator inhibitor-1. Am J Pathol. 2013;183:131–143. doi: 10.1016/j.ajpath.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinckerhoff CE, Matrisian LM. Matrix metalloproteinases: A tail of a frog that became a prince. Nat Rev Mol Cell Biol. 2002;3:207–214. doi: 10.1038/nrm763. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Collen D. Molecular basis of angiogenesis. Role of VEGF and VE-cadherin. Annals of the New York Academy of Sciences. 2000;902:249–262. doi: 10.1111/j.1749-6632.2000.tb06320.x. discussion 262–244. [DOI] [PubMed] [Google Scholar]
- Chen XL, Huang SS, Li WB, Wang DH, Wang XL. Inhibitory effect of aminoguanidine on bleomycin-induced pulmonary toxicity in rat. Acta Pharmacol Sin. 2001;22:711–715. [PubMed] [Google Scholar]
- Chen XL, Li WB, Zhou AM, Ai J, Huang SS. Role of endogenous peroxynitrite in pulmonary injury and fibrosis induced by bleomycin A5 in rats. Acta Pharmacol Sin. 2003;24:697–702. [PubMed] [Google Scholar]
- Cosgrove GP, Brown KK, Schiemann WP, Serls AE, Parr JE, Geraci MW, Schwarz MI, Cool CD, Worthen GS. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: A role in aberrant angiogenesis. Am J Respir Crit Care Med. 2004;170:242–251. doi: 10.1164/rccm.200308-1151OC. [DOI] [PubMed] [Google Scholar]
- Fehrenbach H, Kasper M, Haase M, Schuh D, Muller M. Differential immunolocalization of VEGF in rat and human adult lung, and in experimental rat lung fibrosis: Light, fluorescence, and electron microscopy. Anatomical Rec. 1999;254:61–73. doi: 10.1002/(SICI)1097-0185(19990101)254:1<61::AID-AR8>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Fichtner F, Koslowski R, Augstein A, Hempel U, Rohlecke C, Kasper M. Bleomycin induces IL-8 and ICAM-1 expression in microvascular pulmonary endothelial cells. Exp Toxicol Pathol. 2004;55:497–503. doi: 10.1078/0940-2993-00345. [DOI] [PubMed] [Google Scholar]
- Helmlinger G, Endo M, Ferrara N, Hlatky L, Jain RK. Formation of endothelial cell networks. Nature. 2000;405:139–141. doi: 10.1038/35012132. [DOI] [PubMed] [Google Scholar]
- Iyer AK, Rojanasakul Y, Azad N. Nitrosothiol signaling and protein nitrosation in cell death. Nitric oxide. 2014;42:9–18. doi: 10.1016/j.niox.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalayarasan S, Sriram N, Soumyakrishnan S, Sudhandiran G. Diallylsulfide attenuates excessive collagen production and apoptosis in a rat model of bleomycin induced pulmonary fibrosis through the involvement of protease activated receptor-2. Toxicol Appl Pharmacol. 2013;271:184–195. doi: 10.1016/j.taap.2013.04.021. [DOI] [PubMed] [Google Scholar]
- Keane MP, Arenberg DA, Lynch JP, Whyte RI, Iannettoni MD, Burdick MD, Wilke CA, Morris SB, Glass MC, DiGiovine B, Kunkel SL, Strieter RM. The CXC chemokines, IL-8 and IP-10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol. 1997;159:1437–1443. [PubMed] [Google Scholar]
- Keane MP, Belperio JA, Arenberg DA, Burdick MD, Xu ZJ, Xue YY, Strieter RM. IFN-gamma-inducible protein-10 attenuates bleomycin-induced pulmonary fibrosis via inhibition of angiogenesis. J Immunol. 1999a;163:5686–5692. [PubMed] [Google Scholar]
- Keane MP, Belperio JA, Burdick MD, Lynch JP, Fishbein MC, Strieter RM. ENA-78 is an important angiogenic factor in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001;164:2239–2242. doi: 10.1164/ajrccm.164.12.2104106. [DOI] [PubMed] [Google Scholar]
- Keane MP, Belperio JA, Moore TA, Moore BB, Arenberg DA, Smith RE, Burdick MD, Kunkel SL, Strieter RM. Neutralization of the CXC chemokine, macrophage inflammatory protein-2, attenuates bleomycin-induced pulmonary fibrosis. J Immunol. 1999b;162:5511–5518. [PubMed] [Google Scholar]
- Kietzmann T, Samoylenko A, Roth U, Jungermann K. Hypoxia-inducible factor-1 and hypoxia response elements mediate the induction of plasminogen activator inhibitor-1 gene expression by insulin in primary rat hepatocytes. Blood. 2003;101:907–914. doi: 10.1182/blood-2002-06-1693. [DOI] [PubMed] [Google Scholar]
- Kinnula VL, Fattman CL, Tan RJ, Oury TD. Oxidative stress in pulmonary fibrosis: a possible role for redox modulatory therapy. Am J Respir Crit Care Med. 2005;172:417–422. doi: 10.1164/rccm.200501-017PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loskutoff DJ, Quigley JP. PAI-1, fibrosis, and the elusive provisional fibrin matrix. J Clin Invest. 2000;106:1441–1443. doi: 10.1172/JCI11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Azad N, Wang L, Iyer AK, Castranova V, Jiang BH, Rojanasakul Y. Phosphatidylinositol-3-kinase/akt regulates bleomycin-induced fibroblast proliferation and collagen production. Am J Respir Cell Mol Biol. 2010;42:432–441. doi: 10.1165/rcmb.2009-0002OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KC, Cardoni A, Xiang ZZ. Vascular endothelial growth factor in bronchoalveolar lavage from normal subjects and patients with diffuse parenchymal lung disease. J Lab Clin Med. 2000;135:332–338. doi: 10.1067/mlc.2000.105618. [DOI] [PubMed] [Google Scholar]
- Nobel JJ, Norman GK. Emerging information management technologies and the future of disease management. Dis Manage DM. 2003;6:219–231. doi: 10.1089/109350703322682531. [DOI] [PubMed] [Google Scholar]
- Noguchi S, Yatera K, Wang KY, Oda K, Akata K, Yamasaki K, Kawanami T, Ishimoto H, Toyohira Y, Shimokawa H, Yanagihara N, Tsutsui M, Mukae H. Nitric oxide exerts protective effects against bleomycin-induced pulmonary fibrosis in mice. Respir Res. 2014;15:92. doi: 10.1186/s12931-014-0092-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peao MN, Aguas AP, de Sa CM, Grande NR. Neoformation of blood vessels in association with rat lung fibrosis induced by bleomycin. Anatomical Rec. 1994;238:57–67. doi: 10.1002/ar.1092380108. [DOI] [PubMed] [Google Scholar]
- Renzoni EA, Walsh DA, Salmon M, Wells AU, Sestini P, Nicholson AG, Veeraraghavan S, Bishop AE, Romanska HM, Pantelidis P, Black CM, Du Bois RM. Interstitial vascularity in fibrosing alveolitis. Am J Respir Crit Care Med. 2003;167:438–443. doi: 10.1164/rccm.200202-135OC. [DOI] [PubMed] [Google Scholar]
- Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. Nitric oxidein health and disease of the respiratory system. Physiol Rev. 2004;84:731–765. doi: 10.1152/physrev.00034.2003. [DOI] [PubMed] [Google Scholar]
- Russo RC, Garcia CC, Barcelos LS, Rachid MA, Guabiraba R, Roffe E, Souza AL, Sousa LP, Mirolo M, Doni A, Cassali GD, Pinho V, Locati M, Teixeira MM. Phosphoinositide 3-kinase gamma plays a critical role in bleomycin-induced pulmonary inflammation and fibrosis in mice. J Leukocyte Biol. 2011;89:269–282. doi: 10.1189/jlb.0610346. [DOI] [PubMed] [Google Scholar]
- Russo RC, Guabiraba R, Garcia CC, Barcelos LS, Roffe E, Souza AL, Amaral FA, Cisalpino D, Cassali GD, Doni A, Bertini R, Teixeira MM. Role of the chemokine receptor CXCR2 in bleomycin-induced pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol. 2009;40:410–421. doi: 10.1165/rcmb.2007-0364OC. [DOI] [PubMed] [Google Scholar]
- Selman M, Montano M, Ramos C, Chapela R. Concentration, biosynthesis and degradation of collagen in idiopathic pulmonary fibrosis. Thorax. 1986;41:355–359. doi: 10.1136/thx.41.5.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman M, Pardo A. Idiopathic pulmonary fibrosis: An epithelial/ fibroblastic cross-talk disorder. Respir Res. 2002;3:3. doi: 10.1186/rr175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steurer M, Zoller H, Augustin F, Fong D, Heiss S, Strasser-Weippl K, Gastl G, Tzankov A. Increased angiogenesis in chronic idiopathic myelofibrosis: Vascular endothelial growth factor as a prominent angiogenic factor. Hum Pathol. 2007;38:1057–1064. doi: 10.1016/j.humpath.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Turner-Warwick M. Precapillary Systemic-Pulmonary Anastomoses. Thorax. 1963;18:225–237. doi: 10.1136/thx.18.3.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, Tanaka T, Iso T, Suga T, Kurabayashi M. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L740–L752. doi: 10.1152/ajplung.00146.2010. [DOI] [PubMed] [Google Scholar]
- Zilberberg L, Shinkaruk S, Lequin O, Rousseau B, Hagedorn M, Costa F, Caronzolo D, Balke M, Canron X, Convert O, Lain G, Gionnet K, Goncalves M, Bayle M, Bello L, Chassaing G, Deleris G, Bikfalvi A. Structure and inhibitory effects on angiogenesis and tumor development of a new vascular endothelial growth inhibitor. J Biolo Chem. 2003;278:35564–35573. doi: 10.1074/jbc.M304435200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.