

Abstract
Our previous amino-acid substitutions at the postulated lipid-exposed transmembrane segment M4 of the Torpedo californica acetylcholine receptor (AChR) focused on the alpha subunit. In this study we have extended the mutagenesis analysis using single tryptophan replacements in seven positions (I288, M291, F292, S294, L296, M299 and N300) near the center of the third transmembrane domain of the gamma subunit (γM3). All the tryptophan substitution mutants were expressed in Xenopus laevis oocytes following mRNA injections at levels close to wild type. The functional response of these mutants was evaluated using macroscopic current analysis in voltage-clamped oocytes. For all the substitutions the concentration for half-maximal activation, EC50, is similar to wild type using acetylcholine. For F292W, L296W and M299W the normalized macroscopic responses are 2- to 3-fold higher than for wild type. Previous photolabeling studies demonstrated that these three positions were in contact with membrane lipids. Each of these M3 mutations was co-injected with the previously characterized αC418W mutant to examine possible synergistic effects of single lipid-exposed mutations on two different subunits. For the γM3/αM4 double mutants, the EC50s were similar to those measured for the αC418W mutant alone. Tryptophan substitutions at positions that presumably face the interior of the protein (S294 and M291) or neighboring helices (I288) did not cause significant inhibition of channel function or surface expression of AChRs.
Keywords: Acetylcholine receptor, M3 Transmembrane segment, Macroscopic response, Voltage-clamp
Introduction
The nicotinic acetylcholine receptor (AChR) from muscle and electric ray organ (e.g., Torpedo californica) is an integral membrane protein comprised of four homologous polypeptide subunits in the stoichiometry of α2βγδ (for review see Karlin & Akabas, 1995; Arias, 1998; Changeux & Edelstein, 1998). A consensus model for the AChR topology, obtained from hydrophobicity profiles for protein sequences deduced from cDNA sequences, indicates that each subunit contains at least four membrane-spanning regions (MSRs) denoted M1 through M4 with both N- and C-terminals located on the extracellular side (DiPaola, Czajkowski & Karlin, 1989). The primary and secondary structure of the MSRs may be crucial for the gating and permeation properties of the channel. Most of the studies that combine pharmacological, biochemical, site-directed mutagenesis and electrophysiological data on MSRs have focused on M2 (Giraudat et al., 1986, 1987, 1989; Hucho, Oberthur & Lottspeich, 1986; Imoto et al., 1986, 1988, 1991; Charnet et al., 1990; Villaroel, Herlitze & Sakmann, 1991; Pedersen et al., 1992; White & Cohen, 1992). The M2 and M1 segments from each subunit associate around a central axis to form part of the aqueous ion channel. In contrast, the M4 and M3 segments have been shown to have the largest contact with lipid (Blanton & Cohen, 1992, 1994).
Previous findings demonstrated that a single mutation at the postulated protein-lipid surface of the AChR could produce dramatic effects on the closing transition of the channel (Lee et al., 1994; Lasalde et al., 1996; Ortiz-Miranda et al., 1997; Bouzat et al., 1998; Tamamizu et al., 1999, 2000). These results suggest a possible role of the lipid-exposed regions of the AChR on the gating mechanism. The data gathered from M4 mutations suggest that Van der Waals and perhaps dipole interactions at the periphery of the AChR with the lipid interface could play a significant role in the overall mechanism of the AChR channel gating.
The overall spatial organization of the AChR has been deduced from cryoelectron microscopy of Torpedo electric organ. A 9-Å resolution structure obtained by micrograph image reconstruction of frozen Torpedo postsynaptic membranes reported by Unwin (1993) suggested that the M1, M3 and M4 domains are β structures. The most recent image reconstruction of frozen Torpedo postsynaptic membranes reported at 4.6-Å resolution does not add any more information on the secondary structure of these M3 and M4 transmembrane domains (Miyazawa et al., 1999). On the other hand, the pattern of 3-trifluoromethyl-3-m-[125I]-iodophenyldiazirine (125I-TID)-labeling on the M3 and M4 domains is compatible with an α helical structure (Blanton and Cohen, 1994). An NMR study of a synthetic peptide corresponding to the M3 transmembrane segment of the Torpedo alpha subunit also suggests an α-helical structure (Lugovskoy et al., 1998). Another FTIR study using hydrogen/deuterium exchange also suggested an α-helical structure for all four transmembrane segments of Torpedo AChR (Baenzinger & Méthot, 1995). Recently, a functional analysis of tryptophan substitutions between positions α412 and α425 in the alpha-subunit M4 transmembrane segment suggested a potential α-helical structure (Tamamizu et al., 2000).
In this work, we extend the mutational analysis of the putative M3 transmembrane domain of Torpedo nAChR further by mutating seven positions to tryptophan at single sites. Four of these amino acids have been shown to be in contact with the membrane lipids (Blanton & Cohen, 1994). We also examined the combined effect of multiple lipid-exposed mutations in α and γ subunits on the functional response of the AChR.
The rationale of these experiments is to examine the functional contribution of lipid-exposed positions on different subunits to the conformational transitions of the AChR. These experiments provide new information on how hydrophobic contacts at different subunits can modulate the allosteric properties of the AChR. The AChR M3 mutants were expressed in Xenopus laevis oocytes and analyzed by the whole-cell voltage-clamp method. The present results illustrate the functional contribution of lipid-protein interactions to the dynamics of the AChR channel function.
Materials and Methods
Mutations on the γ Subunit of Torpedo nAChR
Site-directed mutagenesis on the γ subunit of Torpedo californica nAChR was carried out by mismatch amplification using two sequential PCRs (Horton & Pease, 1991). Mutagenic primers containing the desired codon replacement (TGG (Trp) instead of wild-type codon) were extended 11–13 bases on each side of the mismatched region. Each PCR mix contained 100 μl of 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 1.5 mM MgCl2, 0.01% gelatin, 200 nM of each deoxynucleotide, 100 ng DNA template, 1.0 μg of each primer, and 2.0 units of Taq DNA polymerase. Amplification reactions were performed in a DNA thermal cycler (Perkin Elmer-Cetus) programmed for 30 cycles of a three-step protocol; 1 min at 94°C, 2 min at 43°C and 3 min at 72°C. These mutagenic fragments were fused to generate a single fragment containing the mutation and then amplified a second time by PCR reaction. The desired PCR products were purified from excised gel slices with Gene-Clean (Bio 101, La Jolla, CA). These DNA fragments were used in a second fusion PCR.
The 500 bp fusion product was purified from the gel with Gene-Clean and digested with SalI and BspEI. After digestion, the 400 bp mutagenized fragment was inserted into the γ subunit gene using T4 DNA ligase (Promega, Madison, WI). After the plasmid and the mutagenic fragment were ligated with T4 ligase, competent αDH5 E. coli cells were transformed. Tryptophan replacements at positions I288, M291 and S294 were performed using the QuickChange Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). Mutations were confirmed by sequencing at the DNA Sequencing Facility c/o Section of Evolution and Ecology, University of California, Davis, CA.
Expression in Xenopus laevis Oocytes
RNA transcripts were synthesized in vitro as described by Lee et al. (1994). The RNA transcripts (10 ng/oocyte at the concentration of 0.2 μg/μl) of α-, β-, γ-, and δ-subunit at a 2:1:1:1 ratio were injected into Xenopus oocytes. The concentrations of RNA mixes were increased up to 0.8 μg/μl to give 40 ng/oocyte depending upon the level of expression for single-channel recording because some of the mutant AChRs were not expressed as much as wild-type.
Voltage-clamping
ACh-induced currents were recorded with a two-electrode voltage-clamp 3–5 days after mRNA injection with the Gene Clamp 500 amplifier (Axon Instruments, Foster City, CA). Electrodes were filled with 3 M KCl and had a resistance of less than 2 MΩ. Impaled oocytes in the recording chamber were perfused at a rate of 0.5 ml/sec with MOR2 buffer in mM: 82 NaCl, 2.5 KCl, 5 MgCl2, 1 Na2HPO4, 5 N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 0.2 CaCl2, pH 7.4. ACh solutions were made from calcium-free MOR2 to avoid activation of an endogenous Ca2+-dependent Cl− current (Barish, 1983; Mishina et al., 1984). For dose-response curves each oocyte was held at a holding potential of −70 mV. Membrane currents were digitized at 0.5–2 kHz and filtered at 0.1 kHz by an Axon TL-1 interface (Axon Instruments) and recorded by Whole Cell Program 2.3 software (kindly provided by Dr. John Demspter) running on a Pentium III-based computer. Prism version 3.0 (Graphpad Software, San Diego, CA) software was utilized for data analysis and fitting. Dose-response data were collected from peak currents at seven ACh concentrations (0.1 μM–300 μM). The data were fitted by the equation Y =100/(1 + (EC50/A)n). The EC50 and Hill coefficient values for individual oocytes were averaged to generate final estimates. The normalized whole-oocyte currents were analyzed by the unpaired t-test from the program Instat (GraphPAD Software, San Diego, CA) to determine if the differences were statistically significant.
125I-α-Bungarotoxin Binding Assay
The expression of nAChR in the oocyte membrane was assayed by measuring the binding of 125I-α-bungarotoxin (Amersham Life Sciences, Arlington Heights, IL) to intact oocytes according to Lee et al. (1994). From 6–35 oocytes per mutation were incubated in 10 nM 125I-α-bungarotoxin with 5 mg/ml bovine serum albumin in MOR2 at room temperature for 2 hours and the excess toxin was removed by washing 5 times with 1 ml of MOR2. A standard curve was obtained by counting 0–20 μl of 1 nM 125I-α-bungarotoxin solution (equivalent to 0–20 fmol). Noninjected oocytes were used as a background for nonspecific binding.
Normalized Macroscopic AChR Response
To determine the normalized functional response to ACh, the 125I-α-bungarotoxin binding assay was performed immediately following voltage-clamping. The protocol to normalize the ACh-induced response is to apply a single ACh concentration of 300 μM per oocyte and then normalize the amount of current measured to the toxin binding sites in the oocyte surface (nA/fmol). This ACh concentration was selected based on the dose-response curve of the wild-type Torpedo AChR (Ortiz-Miranda et al., 1997). This is defined as the peak of the ACh-induced current (nA) per fmol of surface α-bungarotoxin binding sites.
RESULTS
Expression of Mutant AChRs
Figure 1 shows a hypothetical helical model illustrating the seven positions of the third transmembrane domain of the γ-subunit of Torpedo californica nAChR that were replaced by tryptophan. The RNA transcribed from each mutated γ-subunit DNA along with wild type α-, β- and δ-subunit RNA was injected into Xenopus laevis oocytes. The level of cell-surface expression of mutant AChRs was assessed by the 125I-α-bungarotoxin (125I-BgTx) binding assay. As summarized in the Table, the expression levels of all seven mutations were normal compared to the wild type (2.04 ± 0.48 fmols). M291W, a mutant that presumably faces the interior of the protein, has the highest expression levels (2.53 ± 0.47, n =3) of all mutants. Two other mutations facing the interior of the protein (I288W and S294W) also did not appear to affect the functional assembly and oligomerization of the AChR.
Fig. 1.

Hypothetical helical model of the M3 transmembrane segment of the Torpedo californica nAChR γ-subunit. Residues on the right-hand side, F292, L296, M299 and N300 have been shown to be facing the membrane lipid interface (Blanton & Cohen, 1994). The same photo-labeling study detected no labeling at positions I288, M291, and S294, suggesting that these three positions may be facing neighboring transmembrane segments towards the interior of the protein.
Table.
Mutations in the third transmembrane domain of the gamma subunit
| AChR Type | Expression (fmols) | EC50 (μM) | Hill coefficient | Normalized Response (nA/fmol) |
|---|---|---|---|---|
| Wild Type | 2.04 ± 0.48 (9) | 23.62 ± 1.65 (5) | 1.17 ± 0.63 (5) | 454 ± 86 (5) |
| γI288W | 1.78 ± 0.32 (7) | 43.67 ± 2.23 (7) | 1.08 ± 0.86 (7) | 182 ± 43 (7) |
| γM291W | 2.53 ± 0.47 (3) | 31.37 ± 1.86 (3) | 1.11 ± 0.77 (3) | 532 ± 99 (3) |
| γF292W | 2.09 ± 0.70 (9) | 32.59 ± 1.15 (9) | 1.17 ± 0.17 (9) | 945 ± 56 (7) |
| γS294W | 1.69 ± 0.44 (4) | 41.75 ± 1.91 (4) | 1.05 ± 0.68 (4) | 510 ± 97 (4) |
| γL296W | 0.94 ± 0.31 (4) | 36.84 ± 1.37 (4) | 0.99 ± 0.25 (4) | 1241 ± 162 (3) |
| γM299W | 1.06 ± 0.40 (7) | 57.46 ± 1.35 (7) | 1.08 ± 0.22 (7) | 1227 ± 141 (3) |
| γN300W | 1.72 ± 0.42 (4) | 42.97 ± 1.52 (4) | 1.02 ± 0.42 (4) | 362 ± 25 (4) |
| αC418W | 1.27 ± 0.50 (12) | 3.2 ± 1.65 (9) | 1.14 ± 0.23 (9) | 979 ± 61 (9) |
| αC418W/γF292W | 1.44 ± 0.35 (2) | 7.15 ± 2.56 (3) | 0.68 ± 0.40 (3) | 981 ± 145 (3) |
| αC418W/γL296W | 1.01 ± 0.32 (6) | 2.13 ± 1.38 (6) | 0.94 ± 0.29 (3) | 1226 ± 61 (3) |
| αC418W/γM299W | 1.40 ± 0.36 (3) | 5.32 ± 1.86 (5) | 0.85 ± 0.46 (3) | 580 ± 146 (3) |
Data are expressed as mean ± SEM with the number of experiments in parentheses.
These results contrast with previous data where tryptophan replacements in the alpha M4 domain facing the interior of the protein or neighboring subunits resulted in reduced expression (Tamamizu et al., 2000). The L296W and M299W mutations, which are presumably in contact with the membrane lipids (Blanton & Cohen, 1994), displayed low but still substantial expression levels (0.94 ± 0.31, n =4 and 1.06 ± 0.40, n =7 respectively) of all seven single M3 mutations tested. These results suggest that these seven positions of the M3 transmembrane domain can accommodate a bulky hydrophobic side chain without significant disruption of receptor assembly. We also evaluated the coexpression of the novel M4 αC418W mutation with the three gain-in-function M3 mutations. The expression levels of the γF292W (2.09 ± 0.70, n =9) was reduced about 31% when coexpressed with αC418W to 1.44 ± 0.35, n =3. In contrast, the coexpression of the γM299W with the αC418W produced a significant increase in the surface expression of about 32%. The coexpression of the γL296W with the αC418W did not produce any significant change in the expression of the nAChR.
Ion Channel Properties of Single M3 Mutant AChRs
To examine the ion-channel function of the mutants, the ACh-induced currents were measured by two-electrode voltage-clamp. Currents were normalized to the fmoles of surface α-bungarotoxin binding sites as described in Methods. Figure 2 shows families of current traces of ACh-induced currents for wild type and seven γM3 single tryptophan mutants AChRs. ACh concentrations used for the generation of the family of currents were 0.1, 1.0, 3.0, 10, 30, 100 and 300 μM ACh at −70 mV. After recording macroscopic currents, the amount of surface AChRs was estimated for individual oocytes. We next evaluated the consequences of these γM3 mutants using normalized responses and macroscopic current analysis.
Fig. 2.

Macroscopic-current responses of wild type and γM3 mutants. Family of ACh-induced macroscopic currents derived from individual oocytes for wild type and each γM3 mutants recorded with the two-electrode voltage-clamp. Oocytes in the recording chamber were perfused at a rate of 0.5 ml/sec with MOR2 buffer containing the agonist. ACh induced currents were detected at a membrane potential of −70 mV digitized at 2 kHz. The corresponding mutation is indicated at the top of each family of currents. After recording macroscopic currents, the amount of surface AChRs was normalized to the surface α-BTX binding for each individual oocyte. Their corresponding expression levels in fmols of nAChRs for each individual oocyte are included along with each family of currents. The ACh concentrations used to generate the current traces are: 0.1, 1, 3, 10, 30, 100 and 300 μM. Some mutations did not show any current responses at 0.1 μM ACh.
αC418W Coexpression Experiments
To evaluate the contribution of lipid-exposed mutations on multiple subunits to AChR function we co-injected the αC418W mutation (Lee et al., 1994; Ortiz-Miranda, 1997) with γM3 mutations that increase the macroscopic current. Figure 3 shows families of ACh-induced currents for the three M3 mutants (F292W, L296W and M299W) coexpressed with the αC418W. As for the single γM3 mutations, ACh concentrations used for the generation of the family of currents were 0.1, 1.0, 3.0, 10, 30, 100 and 300 μM ACh at −70 mV. Coexpression of the F292W and L296W produced a gain in macroscopic response while coexpression with the M299W produced a loss in macroscopic response.
Fig. 3.

ACh-induced macroscopic currents for the coexpression of the αC418W and γM3 double mutants. (A) Family of currents derived from individual oocytes for the αM4 mutation C418W. (B) C418W/F292W, (C) C418W/L296W and (D) C418W/M299W mutants. The corresponding mutation is indicated at the top of each family of currents. After recording macroscopic currents, the amount of surface AChRs was normalized to the surface α-BTX binding for each individual oocyte. Their corresponding expression levels in fmols of nAChRs for each individual oocyte were included along with each family of currents. The ACh concentrations used to generate the current traces are: 0.1, 1, 3, 10, 30, 100 and 300 μM.
Normalized Macroscopic Responses
The normalized macroscopic responses (nA/fmols) for the seven single γM3 mutants are shown in the Table. We found three mutations that display normalized current peak values at 300 μM ACh higher than wild type (454 nA/fmol): F292W, L296W and M299W (946 nA/fmol, 1,242 nA/fmol and 1,227 nA/fmol respectively). These three amino-acid positions have been shown to be in contact with the lipid interface (Blanton & Cohen, 1994). The normalized response of these three mutations is higher than the previous C418W mutant (979 nA/fmol) in the alpha M4 transmembrane segment (Tamamizu et al., 2000). I288W exhibited the lowest normalized response (183 ± 43 nA/fmol) among these seven mutations. According to the hypothetical helical structure the position I288 might be facing an adjacent transmembrane segment. The N300W also showed a reduction in the normalized response (362 ± 25 nA/fmol) compared to wild type. M291W and S294W, which are presumably facing the interior of the protein, produced a normalized response slightly higher than wild type (532 ± 99 nA/fmol, n =3 and 510 ± 97 nA/fmol, n =4 respectively). Coexpression of the αC418W with the γF292W and γL296W did not produce any significant change in the normalized response of the single γM3 mutants, however, coexpression with the M299W produced a 46% reduction in the normalized response.
Effects of γM3 Mutations on the Dose-Response Curves for ACh
Dose-response curves for single γM3 mutant nAChRs are shown in Fig. 4A. The EC50s and Hill coefficient of the wild type and M3 mutants are summarized in the Table. Mutations γF292, γL296, and γM299 showed a slight increase in the EC50 for ACh, although the Hill coefficients of those mutants were not significantly different from wild type. However, coexpression of αC418W with the γM3 mutations that produced increases in macroscopic response (Fig. 4B) displayed significant reductions in the EC50s of the multiple mutants (see Table).
Fig. 4.

Dose-response curves for X. laevis oocytes expressing wild type and single M3 (A) and αC418W coexpressed with three γM3 mutant AChRs (B) were expressed as ACh-induced currents (nA) per fmol of surface α-bungarotoxin binding sites. Dose-response data were collected from peak currents at seven ACh concentrations (0.1–300 μM). The data were fitted using a curve of the form Y =100/(1 + (EC50/A)n) and nonlinear regression curves. Data points for each ACh concentration are mean ± SEM of individually measured oocytes. Each data point is an average of 4 to 25 oocytes.
Discussion
In the present paper, we analyzed the contribution of seven positions of the M3 transmembrane segment of the Torpedo californica γ-subunit to AChR function. The rationale for replacing each of these positions with a tryptophan side chain is based on previous results from our laboratory, which demonstrated that this bulky hydrophobic residue produced significant effects on the AChR channel gating kinetics when located at lipid-exposed regions (Lee et al., 1994; Lasalde et al., 1996; Ortiz-Miranda et al., 1997; Tamamizu et al., 1999, 2000). These seven positions were selected based on their spatial location at the center of the M3 domain of the gamma subunit.
Photoaffinity-labeling studies have previously shown that four of these positions (F292, L296, M299 and N300 in Fig. 1) are in contact with the membrane lipid (Blanton & Cohen, 1994). The same photolabeling studies detected no labeling at positions I288, M291 and S294, suggesting that these three positions may be facing neighboring transmembrane segments towards the interior of the protein. We analyzed the pattern of functional consequences of γM3 mutations on the AChR function and the functional assembly in the oocyte surface membrane. 125I-α-Bungarotoxin binding assays on the Xenopus laevis oocytes expressing wild-type and mutant AChRs demonstrated that tryptophan residues could be accommodated at these seven M3 positions of the γ subunit without significant loss of functional expression. Previously, we found that tryptophan substitutions in the alpha M4 domain can dramatically affect the functional assembly and the oligomerization of the AChR (Tamamizu et al., 1999, 2000). The present results suggest that there might be structural differences involving helix-helix packing between the gamma M3 domain and the alpha M4.
We normalized the responses of all mutations to compare their relative contribution to the macroscopic current. The present data show that tryptophan replacement at positions F292, L296 and M299 produced a substantial gain in macroscopic response to ACh. Furthermore, these single M3 mutations display higher macroscopic responses than the double αM4 mutation C418W (Lee et al., 1994), since there are two alpha-subunits per receptor. The L296W has the most prominent macroscopic response (1,241 nA/fmol). If we assume a hypothetical alpha-helical structure for the gamma M3 domain, this position will be located at the same membrane depth of the C418W in the α-subunit. The M299W has the second most prominent macroscopic response (1,068 nA/fmol). Thus, in the γ-subunit, a single tryptophan substitution in the M3 domain produced a higher normalized macroscopic response than a lipid-exposed tryptophan replacement in the αM4 domain. This result suggests that the stoichiometry of the mutation does not necessarily determine the macroscopic response of a lipid-exposed mutation. The F292W has the lowest volume increase from the three gamma mutations and it produced a 2-fold increase in the ACh-induced macroscopic response (946 nA/fmol) at 300 μM ACh. These three mutations demonstrate that lipid-exposed positions play a significant role in the channel gating mechanism.
A periodic tryptophan analysis of the alpha M4 transmembrane segment strongly suggested an alpha-helical structure for this domain (Tamamizu et al., 2000). Tryptophan substitutions at positions C418 and V425 that were previously shown to face the lipid interface (Blanton & Cohen, 1994) produced dramatic gains in functional responses. In contrast to previous gain-in-function lipid-exposed mutations, the three γM3 mutants displayed only a slight increase in the EC50s for ACh. The shape of the normalized dose-response curves of the present M3 mutations differs from the previously characterized alpha M4 mutation C418W (Fig. 4B) and V425W mutation (Tamamizu et al., 2000). The gain-in-function response of the αC418W and V425W mutants is clearly indicated from the reduction in the EC50 for ACh and the hyperbolic shape of the dose-response curve. However, this is not the pattern that we observe for the present M3 mutations. The F292W, L296W and M299W mutations displayed a slight increase in their EC50s compared to wild type. The shape of the normalized dose-response curves for these three mutants appear to be similar to the wild type with a higher level of response at higher ACh concentrations, and the Hill coefficients of these three mutants showed no significant differences from wild type (see Table). The present mutations have no effect on the single-channel conductance (Cruz-Martin et al., 2000), suggesting that the open channel probability of these mutations is higher than wild type at agonist concentrations above 30 μM. All the positions that were found to increase the macroscopic response after replacement with a tryptophan side chain are lipid-exposed except the N300. Previously we found that the increase in macroscopic response produced by tryptophan replacements at lipid-exposed positions in the alpha M4 domain diminished below the center of the bilayer (Lasalde et al., 1996). In that study a tryptophan replacement at position C412 in the alpha M4 domain did not show any increase in macroscopic response. Based on the evidence suggesting that the alpha M4 segment is an alpha helix together with the primary structure of the gamma M3 domain, we speculate that the γN300 position may have a similar location and membrane depth penetration as the αC412 in the M4 domain. If we hypothesize an α-helical structure for the gamma M3 domain, the γN300 may be located at the same depth penetration in the membrane bilayer as the αC412 in the M4 transmembrane segment, and this could provide a possible explanation on the lack of response of this mutant. Further mutagenesis analysis of this transmembrane domain is ongoing in our laboratory to test this hypothesis.
Mutations facing the interior of the protein (M291W and S294W) were not as inhibitory as those in the alpha M4 segment (Tamamizu et al., 2000). However, the I288W mutation, which is facing a neighboring transmembrane segment (based on the model used, see Fig. 5), resulted in substantial inhibition. A very interesting observation is that backside mutations like M291W and S294W in the γM3 did not show a marked inhibition of expression as observed in the alpha and beta subunits (Guzman et al., 2000). One hypothesis is that packing of this domain against other neighboring transmembrane segments is less strained compared to the alpha and beta M4 domains. Another hypothesis is that the interaction of the gamma M3 transmembrane domain with the interior of the protein is more flexible than in other subunits like the alpha M3. We speculate that this flexibility may arise from differences in the secondary structure between the gamma-M3 and the alpha-M4 transmembrane segments. These results indicate that the relative position of the mutated tryptophan residues relative to other transmembrane domains and possibly to the membrane bilayer might be a critical factor in the channel-gating mechanism.
Fig. 5.
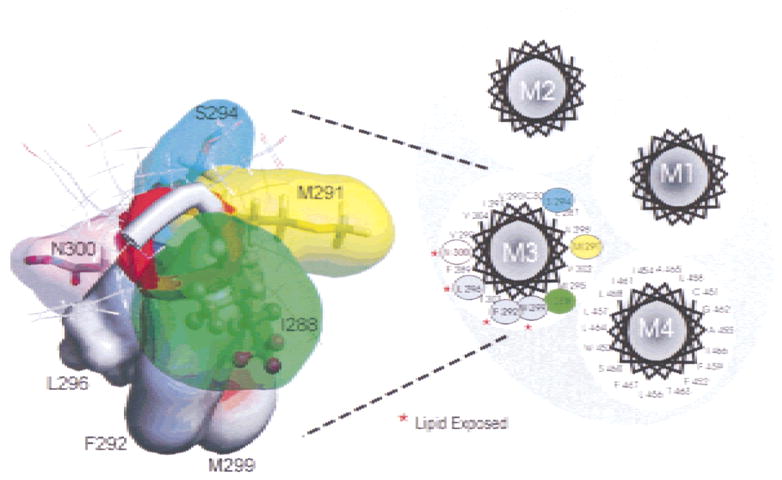
Topological model of the four transmembrane domains (M1–M4) of the Torpedo californica gamma subunit using an α-helical segment. On the left a theoretical α-helical model of the γM3 shows the positions examined in this study. Tryptophan substitutions at positions F292, L296 and M299, which are presumably exposed to the lipid, produced significant increase in the macroscopic response of the AChR.
Another aspect that we examined is the combined effect of multiple lipid-exposed mutations in αM4 and γM3 on the functional response of the AChR. As shown in Fig. 5 the coexpression of the αC418W with γF292W and γL296W mutations did not produce any significant increase in the macroscopic response of combined mutations. According to the dose-response curves in Fig. 4B, the αC418W/γF292W and αC418W/γL296W produced gain in function responses; the EC50s of the single γ mutants (i.e., F292W and L296W) were reduced from 32.6 ± 1.2 to 7.15 ± 2.56 and from 36.8 ± 1.4 to 2.13 ± 1.4, respectively. In contrast, coexpression of the αC418W with the γM299W produced a 52% decrease on the macroscopic response of this double mutant. Also, the EC50 of the γ M299W mutant was significantly reduced from 57.5 ± 1.4 to 5.32 ± 1.86 μM ACh when coexpressed with the αC418W. This reduction in the normalized response of the αC418W/γM299W mutant at 300 μM ACh is possible due to a fast desensitization that can lead to an apparent reduction of the number of functional receptors. The main observation of the multiple mutations experiments is that the allosteric transitions of the AChR can be altered by lipid-exposed mutations, clearly indicated by the significant reduction of the EC50s when the αC418W mutant was coexpressed with the γF292W and γL296W and γM299W mutants. The Hill coefficients of αC418W/γL296W (0.94 ± 0.29) and αC418W/γM299W (0.85 ± 0.46) are not significantly different from wild type (1.17 ± 0.63). However, the αC418W/γF292W shows a significant reduction in the EC50 and Hill coefficient, which suggest that this multiple mutant altered the allosteric transitions of the AChR.
The present results provide additional evidence of the functional role of lipid-exposed positions on the M3 transmembrane domain of the AChR function. There seems to be an exclusive group of lipid-exposed positions on the AChR that are very sensitive to hydrophobic substitutions. An increase in the volume or hydrophobicity of this type of positions will increase the AChR function via a mechanism that remains to be established. Recently, elegant work by Sine’s group demonstrated that the cause of a congenital myasthenic syndrome was the result of the mutation V285I in the M3 of the α subunit (Wang et al., 1999) previously denoted to face the interior of the protein (Blanton & Cohen, 1994). The increase in the side chain volume at this position resulted in an inhibition of channel function. Thus, the M3 domain has been implicated in the gating machinery of the AChR.
The AChR has proven to be an excellent model of an allosteric protein (for review see Changeux & Edelstein, 1998). One of the predictions of the occurrence of allosteric interactions in the AChR is that a mutation occurring at a pivotal position between the ligand binding site and the ion channel pore may affect the channel gating properties or dose-response relationship. The present results illustrate the functional contribution of lipid-exposed residues of the gamma M3 transmembrane domain to the conformational transitions of the AChR. Specific contacts of amino-acid side chains with the lipid interface may represent another allosteric mode of interaction that remains to be defined. Finally, defining the structure-function relationship of lipid-exposed positions may lead to an understanding of the dynamic role of lipid protein interactions on the conformational transitions of the AChR.
Acknowledgments
This research was supported by National Institutes of Health grants GM56371, GM08102-27 and MARC MBRS program.
References
- Arias HB. Binding sites for exogenous and endogenous non-competitive inhibitors of the nicotinic acetylcholine receptor. Bio-chim Biophys Acta. 1998;1376:173–220. doi: 10.1016/s0304-4157(98)00004-5. [DOI] [PubMed] [Google Scholar]
- Baenzinger JE, Méthot N. Fourier transform infrared and hydrogen/deuterium exchange reveal an exchange-resistant core of alpha-helical peptide hydrogens in the nicotinic acetylcholine receptor. J Biol Chem. 1995;270:29129–29137. doi: 10.1074/jbc.270.49.29129. [DOI] [PubMed] [Google Scholar]
- Barish ME. A transient calcium-dependent chloride current in the immature Xenopus oocyte. J Physiol. 1983;342:309–325. doi: 10.1113/jphysiol.1983.sp014852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton MP, Cohen JB. Mapping the lipid-exposed regions in the Torpedo californica nicotinic acetylcholine receptor. Biochemistry. 1992;31:3738–3750. doi: 10.1021/bi00130a003. [DOI] [PubMed] [Google Scholar]
- Blanton MP, Cohen JB. Identifying the lipid-protein interface of the Torpedo nicotinic acetylcholine receptor: secondary structure implications. Biochemistry. 1994;33:2859–2872. doi: 10.1021/bi00176a016. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Roccamo AM, Garbus I, Barrantes FJ. Mutations at lipid-exposed residues of the acetylcholine receptor affect its gating kinetics. Mol Pharmcol. 1998;54:146–153. doi: 10.1124/mol.54.1.146. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- Charnet P, Labarca C, Leonard RJ, Vogelaar NJ, Czyzyk L, Gouin A, Davison N, Lester HA. An open-channel blocker interacts with adjacent turns of alpha helices in the nicotinic acetylcholine receptor. Neuron. 1990;4:87–95. doi: 10.1016/0896-6273(90)90445-l. [DOI] [PubMed] [Google Scholar]
- Cruz-Martín A, Rojas LV, Mercado JL, McNamee MG, Lasalde JA. Tryptophan substitutions in lipid exposed positions of the M3 transmembrane domain of the Torpedo californica nicotinic acetylcholine receptor alter ion channel kinetics. Biophys Congr. 2000;78:358A. [Google Scholar]
- Dipaola M, Czajkowski C, Karlin A. The sidedness of the COOH terminus of the acetylcholine receptor delta subunit. J Biol Chem. 1989;264:15457–15463. [PubMed] [Google Scholar]
- Giraudat J, Dennis M, Heidmann T, Chang JY, Changeux JP. Structure of the high affinity binding site for noncompetitive blockers of the acetylcholine receptor: serine-262 of the δ subunit is labeled by [3H]chlorpromazine. Proc Natl Acad Sci USA. 1986;83:2719–2723. doi: 10.1073/pnas.83.8.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudat J, Dennis M, Heidmann T, Hamont PY, Lederer F, Changeux JP. Structure of the high-affinity site for non-competitive blockers of the acetylcholine receptor: [3H]chlor-promazine labels homologous residues in the β and δ chains. Biochemistry. 1987;26:2410–2418. doi: 10.1021/bi00383a003. [DOI] [PubMed] [Google Scholar]
- Giraudat J, Gali J, Revah F, Changeux JP, Haumont P, Lederer F. The noncompetitive blocker [3H]chlorpromazine labels segment M2 but not segment M1 of the nicotinic acetylcholine receptor alpha-subunit. FEBS Lett. 1989;253:190–198. doi: 10.1016/0014-5793(89)80957-3. [DOI] [PubMed] [Google Scholar]
- Guzmán G, Santiago J, Marti R, Rojas L, García-Arraras JE, Lasalde-Dominicci JA. Effect of tryptophan substitutions in the alpha M3 transmembrane domain of the Torpedo californica nicotinic acetylcholine receptor. Soc Neurosci Abstr. 2000;26:372A, 137.13, 372. doi: 10.1021/bi992835w. [DOI] [PubMed] [Google Scholar]
- Horton RM, Pease LR. Recombination and mutations of DNA sequencing using PCR. In: McPherson MJ, editor. Directed mutagenesis. IRL Press; NY: 1991. pp. 217–246. [Google Scholar]
- Hucho F, Oberthur W, Lottspeich F. The ion channel of the nicotinic acetylcholine receptor is formed by the homologous helices MII of the receptor subunits. FEBS Lett. 1986;205:137–142. doi: 10.1016/0014-5793(86)80881-x. [DOI] [PubMed] [Google Scholar]
- Imoto K, Methfessel C, Sakmann B, Mishina M, Mori Y, Konno T, Fukuda M, Kurosaki M, Bujo H, Fujita Y, Numa S. Location of a δ subunit region determining ion transport through the acetylcholine receptor. Nature. 1986;324:670–674. doi: 10.1038/324670a0. [DOI] [PubMed] [Google Scholar]
- Imoto K, Busch C, Sakmann B, Mishina M, Konno T, Nakai JM, Bujo H, Mori Y, Fukuda M, Numa S. Rings of negatively charged amino acids determine the acetylcholine receptor channel conductance. Nature. 1988;335:645–648. doi: 10.1038/335645a0. [DOI] [PubMed] [Google Scholar]
- Imoto K, Konno T, Nakai J, Wang F, Mishina M, Numa S. A ring of uncharged polar amino acids as a component of channel constriction in the nicotinic acetylcholine receptor. FEBS Lett. 1991;289:193–200. doi: 10.1016/0014-5793(91)81068-j. [DOI] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- Lasalde JA, Tamamizu S, Butler DH, Vibat CRT, Hung B, McNamee MG. Tryptophan substitutions at the lipid-exposed transmembrane segment M4 of Torpedo californica acetylcholine receptor govern channel gating. Biochemistry. 1996;35:14139–14148. doi: 10.1021/bi961583l. [DOI] [PubMed] [Google Scholar]
- Lee YH, Li L, Lasalde JA, Rojas LV, McNamee M, Ortiz-Miranda SI, Pappone P. Mutations in the M4 domain of Torpedo californica acetylcholine receptor dramatically alter ion channel function. Biophys J. 1994;66:646–653. doi: 10.1016/s0006-3495(94)80838-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugovskoy AA, Maslennikov IV, Utkin YN, Tsetlin VI, Cohen JB, Arseniev AS. Spatial structure of the M3 transmembrane segment of the nicotinic acetylcholine receptor a subunit. Eur J Biochem. 1998;255:455–461. doi: 10.1046/j.1432-1327.1998.2550455.x. [DOI] [PubMed] [Google Scholar]
- Mishina M, Kurosaki T, Tobimatsu T, Morimoto Y, Noda M, Yamamoto T, Terao M, Lindstrom J, Takahashi T, Kuno M, Numa S. Expression of functional acetylcholine receptor from cloned cDNAs. Nature. 1984;307:604–608. doi: 10.1038/307604a0. [DOI] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Stowell M, Unwin N. Nicotinic acetylcholine receptor at 4.6 Å resolution: transverse tunnels in the channel wall. J Mol Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- Ortiz-Miranda SI, Lasalde JA, Pappone PA, McNamee MG. Mutations in the M4 domain of the Torpedo californica nicotinic acetylcholine receptor alter channel opening and closing. J Membrane Biol. 1997;158:17–30. doi: 10.1007/s002329900240. [DOI] [PubMed] [Google Scholar]
- Pedersen SE, Sharp SD, Liu WS, Cohen JB. Structure of the noncompetitive antagonist-binding site of the Torpedo nicotinic acetylcholine receptor [3H] meproadifen mustard reacts selectively with alpha-subunit Glu-262. J Biol Chem. 1992;267:10489–10499. [PubMed] [Google Scholar]
- Tamamizu S, Lee YH, Hung B, McNamee MG, Lasalde-Dominicci JA. Alteration in ion-channel function of the mouse nicotinic acetylcholine receptor by mutations in the M4 transmembrane domain. J Membrane Biol. 1999;170:157–164. doi: 10.1007/s002329900545. [DOI] [PubMed] [Google Scholar]
- Tamamizu S, Guzmán G, Santiago J, Rojas LV, McNamee MG, Dominicci JA. Functional effects of periodic tryptophan substitutions in the α M4 transmembrane domain of the Torpedo californica nicotinic acetylcholine receptor. Biochemistry. 2000;39:4666–4673. doi: 10.1021/bi992835w. [DOI] [PubMed] [Google Scholar]
- Unwin N. The nicotinic acetylcholine receptor at 9Å resolution. J Mol Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- Villarroel A, Herlitze S, Sakmann B. Amino acids responsible for differences in conductance between adult and fetal forms of the acetylcholine receptor. Biophys J. 1991;59:34a. [Google Scholar]
- Wang HL, Milone M, Ohno K, Shen X, Tsujino A, Batocchi AP, Tonali P, Brengman J, Engel AG, Sine SM. Acetylcholine receptor M3 domain: Stereochemical and volume contributions to channel gating. Nature Neuroscience. 1999;2:226–233. doi: 10.1038/6326. [DOI] [PubMed] [Google Scholar]
- White BH, Cohen JB. Agonist-induced changes in the structure of the acetylcholine receptor M2 regions revealed by photoin-corporation of an uncharged nicotinic noncompetitive agonist. J Biol Chem. 1992;267:15770–15783. [PubMed] [Google Scholar]