

Abstract
Background & Aims
The safety profiles of boceprevir and telaprevir in the treatment of chronic hepatitis C, administered in academic and community centres across the United States, were evaluated.
Methods
In 90 medical centres, patients with chronic HCV received pegylated interferon, ribavirin, and either telaprevir or boceprevir per local standard of care. Demographic, adverse event, clinical, and virological data were collected during treatment and follow-up.
Results
A total of 2084 patients (97% HCV genotype 1) received at least one dose of a protease inhibitor. At baseline, 38% of patients had cirrhosis, and 57% had received at least one prior treatment for hepatitis C. Serious adverse events occurred in 12% of patients receiving protease inhibitor therapy. Overall, 66% of patients experienced anaemia, leading to frequent ribavirin dose reductions (42%) and erythropoietin use (37%); 11% received blood transfusion. More than 90% of patients had adverse events that led to a prescription, treatment, or dosage change, and 39% of patients discontinued treatment early, most commonly because of adverse events (18%) or lack of efficacy (16%). Hepatic decompensation events occurred in 3% of all patients. Age, female gender, cirrhosis, HCV genotype 1 subtype, creatinine clearance, platelet levels, albumin levels and haemoglobin levels were independent predictors of anaemia. Five deaths occurred. Overall, 52% of all patients achieved a sustained virologic response.
Conclusions
In academic and community centres, where chronic hepatitis C patients commonly have advanced liver disease, triple therapy was associated with a high rate of adverse events and involved frequent treatment modifications and adverse event management.
Keywords: Hepatitis C, Boceprevir, Telaprevir, Anaemia, Cirrhosis, Hepatic decompensation
Introduction
Following a decade of only modest advances in the treatment of chronic hepatitis C virus (HCV), telaprevir and boceprevir, both HCV protease inhibitors, were approved by the FDA in 2011 as the first direct-acting antiviral agents (DAAs) to be used in combination with pegylated interferon-alfa (PegIFNα) and ribavirin for genotype 1 HCV infection. For treatment-naïve patients, the protease inhibitors improved the likelihood of a sustained virologic response relative to PegIFNα and ribavirin alone from 40–50% to 67–75% [1–4]. The triple therapy regimens involved frequent on-treatment monitoring for response, and the addition of the third agent created a new set of adverse events, such as rash and incremental anaemia, requiring additional resources for management [5].
The patient population in the real world may differ from that which was reflected in the phase 3 registration trials. Whereas the current patient population seen in clinic settings may include a large proportion of older patients with cirrhosis, relatively few patients with cirrhosis (all compensated) were included in the registration trials of both boceprevir and telaprevir [2,3]. Recently, a large European cohort enrolled a broad clinical patient population including those with compromised liver function and reported that the addition of a protease inhibitor to PegIFNα and ribavirin increased the likelihood of adverse events, many of which were serious and led to premature discontinuation of therapy [6]. Other studies reported that the presence of cirrhosis further increased the potential for anaemia [3–5,7] and hepatic decompensation [2–4,6]. A recent study of patients at tertiary European referral centres reported a higher proportion of patients who stopped HCV triple therapy than in registration trials [8]. The difference in discontinuation rates was attributed to the more frequent occurrences of both virological treatment failure and adverse events. In order to provide a more accurate safety profile of the clinical HCV population, we designed and executed a cohort study of the safety of triple therapy with boceprevir or telaprevir in predominantly genotype 1-infected patients, treated at a consortium of academic and community medical centres.
Materials and methods
Study population and design
HCV-TARGET is a longitudinal, observational study in chronic hepatitis C patients from a consortium of academic (n = 38) and community (n = 52) medical centres. Patients ≥18 years old were included if they were being treated or had been treated with antiviral regimens containing PegIFNα and ribavirin, in combination with telaprevir or boceprevir. Treatment was chosen and administered per local standards at the study site. The study protocol did not define specific treatment populations, regimens, dosing, or safety management guidelines. Data was captured from sequentially enrolled patients using a common database that utilized novel, standardized source data abstraction methods. A centralized team of trained coders reviewed all de-identified medical records, obtained from participating sites for data entry. Throughout treatment and during post-treatment follow-up, demographic, clinical, adverse event, and virological data were collected. Independent data monitors systematically reviewed the data entries for completeness and accuracy. All records were screened for extreme or unlikely values and verified/resolved with additional queries. Data from 337 patients from PEG-BASE-USA, an observational study of peginterferon (e.g. Pegasys®)-based direct-acting antiviral triple therapy in patients with chronic hepatitis C genotype 1 (NCT01508130), a study funded by Genentech (South San Francisco, CA, USA), were mapped to harmonize with the HCV-TARGET database and are included in the current analyses. Patients received pegylated interferon alfa-2a or pegylat-ed interferon alfa-2b and ribavirin plus either telaprevir or boceprevir in accordance with the local standard of care and U.S. labelling and were followed for the duration of their treatment and for up to 24 weeks post-treatment.
This study was approved by the local Institutional Review Boards at all participating sites. Participants provided written informed consent, or were enrolled under a waiver of consent according to local IRB policies.
Definitions
Sustained virologic response (SVR)
SVR was defined as HCV RNA below the level of quantification (BLOQ) 12 weeks after the end of HCV treatment. Patients lacking a 12-week post-treatment plasma sample were considered treatment failures.
Cirrhosis
The presence of cirrhosis was defined by biopsy and/or a combination of clinical, laboratory, and imaging criteria established a priori. Patients were determined to have cirrhosis if they had: (1) evidence of stage 4 fibrosis by liver biopsy at any time prior to therapy; or (2) evidence of stage 3 fibrosis by liver biopsy at any time prior to therapy with any of the following criteria: platelets count <140,000 per μl, presence of oesophageal varices on oesophagogastroduodenoscopy, evidence of cirrhosis and/or portal hypertension and/or of ascites by imaging studies, FibroSure test or equivalent, compatible with stage 4 fibrosis; or (3) in the absence of liver biopsy, any two of the following criteria: platelet count <140,000 per μl, presence of oesophageal varices on oesophagogastroduodenoscopy, evidence of cirrhosis and/or portal hypertension and/or ascites by imaging studies, FibroSure test or equivalent, compatible with stage 4 fibrosis.
Adverse events (AEs)
(1) Any AE that required a prescription medication for management, or resulted in a dose reduction or discontinuation of HCV treatment; or (2) any adverse event of special interest (rash, dysgeusia, anaemia, anorectal symptoms) regardless of treatment prescribed at any time during treatment; that happened at any time during treatment or up to 12 weeks after the end of treatment.
Anaemia
Defined as the presence of at least one of the following: (1) anaemia was reported as AE by healthcare practitioner; (2) haemoglobin <10 g/dl; or (3) administration of RBC growth factors; or (4) transfusion occurred.
Hepatic decompensation during therapy
Patient experienced one or more of the following AEs: (1) Hepatic decompensation was listed as an AE by the healthcare practitioner; or (2) new onset of hepatic encephalopathy; (3) new onset of spontaneous bacterial peritonitis; (4) new onset of variceal haemorrhage; (5) new onset of ascites; (6) patient received a new prescription for a medication to treat one of the above indications (1–5).
Serious adverse event (SAE)
An AE that required hospitalization or met criteria for expedited reporting per FDA form MEDWATCH 3500.
Statistical analyses
The rate of treatment completion and frequency of adverse events were calculated for the entire study population and for subpopulations of special interest. Confidence intervals of the proportions were calculated using exact binomial methods. Kaplan-Meier estimates of treatment persistence were constructed in graphical displays, using the dates of starting and stopping peginterferon to determine the duration of persistence. Multivariable logistic regression models and a stepwise variable selection algorithm were used to identify potential baseline risk factors for anaemia in the study population. The set of potential variables of interest were selected a priori based upon a consensus of clinical expertise. The model was restricted to HCV genotype 1 patients; laboratory values used in the model were baseline measurements. The estimates of the stepwise-selected variables were compared with estimates from minimally adjusted model. Because some observations were missing values for some baseline variables, the estimated odds ratios and confidence intervals of the selected risk factors of the multivariate logistic model with stepwise selection were based on the data filled in using multiple imputation method. Analyses were performed using SAS software version 9.3 (SAS Institute Inc., Cary, North Carolina) and R 3.0.2 (R Core Team, Vienna, Austria).
Results
Patient characteristics
Between May 2011 and June 2013, 2757 patients consented to participate in HCV-TARGET and 2122 started therapy prior to September 1, 2012. Of these, 2084 received at least one dose of telaprevir or boceprevir and were included in the current safety analysis (Fig. 1). Baseline characteristics for all treated patients are shown in Table 1. Seventy nine percent of patients were white and 16% were black. Median age was 56 years and 61% of patients were male. HCV genotype 1a was reported in 56% and genotype 1b in 23% of patients. Of note, an additional 18% of treated patients were genotype 1, although no further subtyping was specified. Fifty-seven percent of patients were previously treated with an interferon-containing regimen.
Fig. 1.
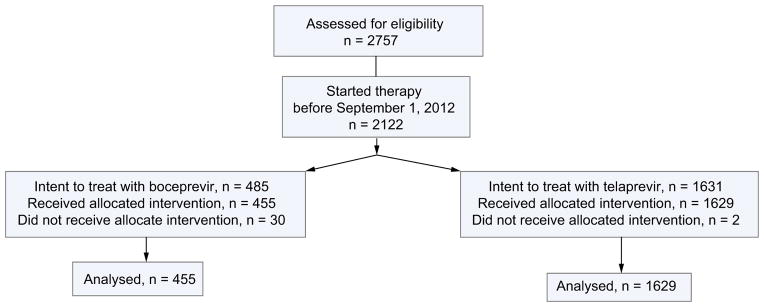
Disposition of patients from enrolment to treatment initiation.
Table 1.
Baseline characteristics of patients.
| Characteristics | Boceprevir (n = 455) | Telaprevir (n = 1629) | Total (n = 2084) |
|---|---|---|---|
| Median age (range), yr | 55 (20–77) | 56 (18–75) | 56 (18–77) |
| Male sex, n (%) | 283 (62) | 982 (60) | 1265 (61) |
| Median BMI (range), kg/m2 | 28 (18–57) | 28 (16–69) | 28 (16–69) |
| Race, n (%) | |||
| White | 364 (80) | 1281 (79) | 1645 (79) |
| Black | 66 (15) | 277 (17) | 343 (16) |
| Asian | 13 (3) | 33 (2) | 46 (2) |
| Other/missing | 12 (3) | 38 (2) | 50 (2) |
| Ethnicity Hispanic,1 n (%) | 28 (6) | 103 (6) | 131 (6) |
| Mean ALT (SD), mean IU/L | 88 (81) | 92 (78) | 91 (79) |
| Mean total bilirubin (SD), mg/dl | 0.7 (0.4) | 0.8 (0.5) | 0.8 (0.5) |
| Mean albumin (range), g/dl | 4.1 (1.4–5.1) | 4.0 (1.0–5.1) | 4.1 (1.0–5.1) |
| Mean haemoglobin (SD), g/dl | 15 (1.5) | 15 (1.4) | 15 (1.4) |
| Mean platelet count (range) (x103) per μl | 182 (33–472) | 173 (25–500) | 175 (25–500) |
| History of cirrhosis,2 n (%) | 140 (31) | 648 (40) | 788 (38) |
| History of esophageal varices,3 n (%) | 56 (12) | 213 (13) | 269 (13) |
| History of liver decompensation, n (%) | 12 (3) | 56 (3) | 68 (3) |
| Presence of diabetes, n (%) | 79 (17) | 224 (14) | 303 (15) |
| IL28B genotype, n (%) | |||
| CC | 60 (13) | 152 (9) | 212 (10) |
| CT, TT | 142 (31) | 393 (24) | 535 (26) |
| Unknown | 253 (56) | 1084 (67) | 1337 (64) |
| HCV genotype, n (%) | |||
| 1a | 254 (56) | 914 (56) | 1168 (56) |
| 1b | 105 (23) | 380 (23) | 485 (23) |
| 1, subtype unspecified | 86 (19) | 286 (18) | 372 (18) |
| Other | 10 (2) | 49 (3) | 59 (3) |
| Prior HCV treatment, n (%) | |||
| Treatment-naïve | 201 (44) | 701 (43) | 902 (43) |
| Treatment-experienced | 254 (56) | 925 (57) | 1179 (57) |
| Unknown, multiple response | 0 | 3 (<1) | 3 (<1) |
| HCV RNA ≥800,000 IU/ml, n (%) | 321 (71) | 1150 (71) | 1471 (71) |
ALT, alanine aminotransferase; HCV, hepatitis C virus.
Data available on 2064 patients.
Data available on 2045 patients.
Data available on 794 patients.
Cirrhosis was present in 38% of patients (Table 1). Among patients with cirrhosis, mean platelet count per μl was 122 × 103, compared to 208 ×103 in non-cirrhotic patients, and a mean platelet count of 96 ×103 was observed in cirrhotic patients with a history of hepatic decompensation (Supplementary Table 1). The mean albumin level was 3.9 g/dl in cirrhotic patients and their mean MELD score was 8.2 (range 6.0–21.0). Oesophageal varices were noted on prior endoscopy in 257/485 (53%) cirrhotic patients with available history of varices and 47/ 67 (70%) patients with history of hepatic decompensation (Supplementary Table 1).
Treatment completion status
Overall, 60% completed a full course of therapy, which included 56% of those treated with boceprevir and 61% of those treated with telaprevir. Adverse events and lack of efficacy were the leading causes for early discontinuation: 18% of boceprevir patients and 18% of telaprevir patients discontinued treatment due to an AE, and 20% of boceprevir patients and 16% of telaprevir patients stopped treatment due to lack of efficacy. Only 3% of patients were lost to follow-up during the treatment phase (Table 2).
Table 2.
Patient disposition, sustained virologic response, safety profile, and anaemia management.
| Boceprevir (n = 455) | Telaprevir (n = 1629) | All patients (n = 2084) | |
|---|---|---|---|
| Patient disposition during treatment, n (%) | |||
| Completed therapy | 255 (56) | 989 (61) | 1244 (60) |
| Discontinued early | 200 (44) | 621 (38) | 821 (39) |
| Adverse event | 83 (18) | 290 (18) | 373 (18) |
| Lack of efficacy | 89 (20) | 255 (16) | 344 (17) |
| Lost to follow-up | 22 (5) | 48 (3) | 70 (3) |
| Other | 6 (1) | 28 (2) | 34 (2) |
| No EOT data | 0 | 19 (1) | 19 (<1) |
| Sustained virologic response, n (%, 95% CI) | 200 (44, 39–49) | 883 (54, 52–57) | 1083 (52, 50–54) |
| Adverse events, n (%) | 422 (93) | 1561 (96) | 1983 (95) |
| Anaemia | 310 (68) | 1056 (65) | 1366 (66) |
| Rash | 155 (34) | 1026 (63) | 1181 (57) |
| Anorectal symptoms | 43 (9) | 648 (40) | 691 (33) |
| Infection | 78 (17) | 242 (15) | 320 (15) |
| Dysgeusia | 57 (13) | 65 (4) | 112 (6) |
| Decompensating event | 12 (3) | 56 (3) | 68 (3) |
| Serious adverse events, n (%) | 57 (13) | 183 (11) | 240 (12) |
| Anaemia | 18 (4) | 57 (3) | 75 (4) |
| Infection | 14 (3) | 43 (3) | 57 (3) |
| Other | 8 (2) | 29 (2) | 37 (2) |
| Decompensation | 3 (<1) | 13 (<1) | 16 (<1) |
| Rash | 0 | 8 (<1)* | 8 (<1) |
| Anorectal symptoms | 0 | 7 (<1) | 7 (<1) |
| Dysgeusia | 0 | 0 | 0 |
| Haemoglobin decline, n (%) | |||
| Haemoglobin <10 g/dl | 236 (52) | 812 (50) | 1048 (50) |
| Haemoglobin <8.5 g/dl | 69 (15) | 285 (17) | 354 (17) |
| Anaemia management, n (%) | |||
| Ribavirin dose reduction | 188 (41) | 686 (42) | 874 (42) |
| Epoetin use | 182 (40) | 580 (36) | 762 (37) |
| Blood transfusion | 44 (10) | 193 (12) | 237 (11) |
| Ribavirin interruptions | 16 (4) | 41 (3) | 57 (3) |
| Deaths, n | 0 | 5 | 5 |
SAE rash included 2 patients with DRESS.
Treatment persistence and efficacy
Mean treatment duration (based on interferon treatment first and last dates) was 209 days for telaprevir patients and 209 days for boceprevir patients. Treatment persistence estimates plotted at various time points are shown in Fig. 2. The period of greatest treatment discontinuation was around day 150 of treatment in patients treated with telaprevir and around day 90 in patients treated with boceprevir. Forty-four percent (95% CI: 39–49%) of boceprevir patients and 54% (95% CI: 52–57%) of telaprevir patients achieved a sustained virologic response (SVR) (Table 2), and overall 43% (95% CI: 40–47%) of patients with cirrhosis, treated with DAA-based therapy, were cured (Supplementary Table 1).
Fig. 2.
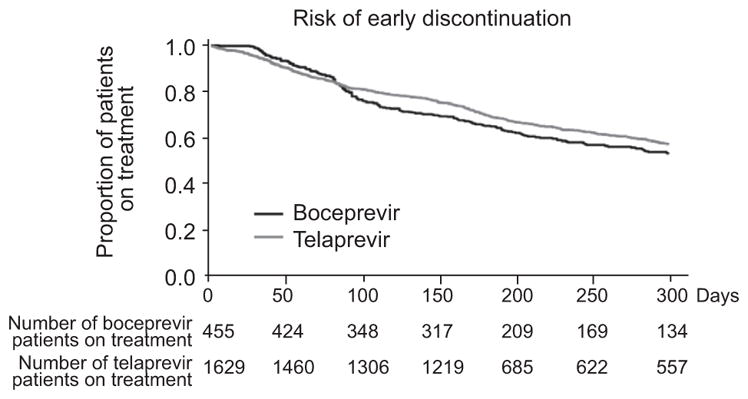
Treatment persistence.
Safety
Most patients (95%) experienced an AE (Table 2). Anaemia was observed in two thirds of patients, regardless of the DAA used and was the adverse event that most frequently required clinical intervention. Haemoglobin decrease to a nadir of <10 g/dl occurred in about half of patients treated with either DAA (boceprevir or telaprevir), severe anaemia (defined as haemoglobin <8.5 g/dl) developed in about 17%. Plots for changes in haemoglobin during the first 24 weeks of therapy are shown in Fig. 3. Anaemia was most frequently managed by ribavirin dose reductions (41% boceprevir and 42% telaprevir), although erythropoiesis-stimulating agent use was also common (40% boceprevir and 36% telaprevir). Blood transfusions were required in 10% of boceprevir-treated patients and in 12% of those treated with telaprevir (Table 2). Among patients with HCV genotype 1, Multivariable logistic regression analysis was used to identify variables associated with anaemia, defined as haemoglobin <10 g/dl. The following variables were considered as potential baseline predictors of anaemia: creatinine clearance, platelets, haemoglobin, albumin total, bilirubin, HCV RNA level, gender, age, presence of cirrhosis, diabetes status, previous treatment experience, HCV subtype, and IL28B. Odds ratios adjusted for age and sex and their corresponding 95% confidence intervals are reported in Table 3. Age, female gender, the presence of cirrhosis, HCV subtype b, lower platelet levels (≤100,000/mm3), lower, albumin (<3.5 g/dl) and haemoglobin, and lower creatinine clearance were independent predictors of anaemia in the overall population. The odds of anaemia for women were 3 times the odds for men when controlling for age.
Fig. 3. Haemoglobin levels.

(A) Patients with haemoglobin levels ≥10 g/dl. (B) Patients with haemoglobin levels ≥8.5 g/dl.
Table 3.
Predictors of anaemia (haemoglobin <10 g/dl) among HCV genotype 1 patients adjusted for gender and age.
| Risk factor | Boceprevir patients (n = 455)
|
Telaprevir patients (n = 1629)
|
All patients (n = 2084)
|
|||
|---|---|---|---|---|---|---|
| OR | 95% CI | OR | 95% CI | OR | 95% CI | |
| Female gender2 | 3.55 | (2.33, 5.42) | 2.95 | (2.37, 3.68)* | 3.05 | (2.52, 3.71)* |
|
| ||||||
| Cirrhosis | 1.11 | (0.72, 1.73) | 1.36 | (1.09, 1.69) | 1.29 | (1.06, 1.57) |
|
| ||||||
| Total bilirubin (mg/dl) | 0.98 | (0.66, 1.46) | 1.13 | (0.92, 1.38) | 1.09 | (0.91, 1.31) |
|
| ||||||
| Age3 | 1.05 | (1.03, 1.07) | 1.06 | (1.04, 1.07)* | 1.06 | (1.04, 1.07)* |
|
| ||||||
| HCV RNA (lnsIU) | 1.02 | (0.93, 1.12) | 1.00 | (0.94, 1.06) | 1.00 | (0.95, 1.05) |
|
| ||||||
| Creatinine clearance1 (ml/min) | 0.99 | (0.98, 0.99)* | 0.99 | (0.99, 0.99)* | 0.99 | (0.99, 0.99)* |
|
| ||||||
| Diabetes | 1.20 | (0.71, 2.03) | 0.89 | (0.66, 1.20) | 0.97 | (0.75, 1.26) |
|
| ||||||
| IL28B genotype CC | 0.81 | (0.41, 1.57)* | 1.04 | (0.70, 1.55) | 0.97 | (0.69, 1.36) |
|
| ||||||
| Treatment experience | 0.91 | (0.59, 1.39) | 0.84 | (0.68, 1.05) | 0.85 | (0.71, 1.03) |
|
| ||||||
| Albumin (<3.5 g/dl) | 1.23 | (0.58, 2.61) | 1.6 | (1.12, 2.29) | 1.52 | (1.1, 2.1) |
|
| ||||||
| HCV genotype 1a | 0.70 | (0.43, 1.14) | 0.74 | (0.57, 0.96)* | 0.73 | (0.58, 0.91)* |
|
| ||||||
| Haemoglobin | 0.55 | (0.45, 0.66)* | 0.61 | (0.55, 0.67)* | 0.60 | (0.55, 0.65)* |
|
| ||||||
| Platelets (≤100,000 mm3 n/μl) | 1.54 | (0.87, 2.75) | 1.81 | (1.37, 2.39)* | 1.74 | (1.35, 2.23)* |
Rows were selected by a stepwise variable selection method.
estimated using Cockcroft-Gault formula.
adjusted for age.
adjusted for gender.
Among other adverse events designated of special interest, skin rash was reported most frequently by those treated with telaprevir (63%) compared to boceprevir (34%). Most rashes were mild, although serious rash was reported in 8 (<1%) of patients treated with telaprevir, including 2 with drug reaction with eosinophilia and systemic symptoms (DRESS), leading to premature treatment discontinuation. Anorectal symptoms were reported in 40% of those treated with telaprevir and 9% of those treated with boceprevir. Dysgeusia was noted in 4% and 13% of those treated with telaprevir and boceprevir, respectively. Three percent of patients, receiving either DAA, suffered a liver decompensation event.
An SAE occurred in 13% of patients on boceprevir and in 11% on telaprevir (Table 2). The most common reasons for SAE were anaemia (4%), infection (3%), and other causes (2%). Five patients, treated with a telaprevir-based regimen, died during the study (1 due to sepsis/liver failure, 2 due to myocardial infarction, and 2 unspecified).
Impact of cirrhosis
Among the enrolled patients, 76 (4%) had a history of decompensation prior to the start of therapy as defined by the past medical history or the use of concomitant medications at baseline that were indicative of hepatic decompensation, including lactulose, rifaximin, spironolactone, and furosemide. For beta blockers to be considered as indicative of hepatic decompensation it required an associated indication of bleeding from varices (not just prophylaxis). Among patients with a history of decompensation 33% experienced SAEs, 83% suffered from anaemia, and 24% had a decompensation event. In these patients with a history of decompensation, the rate of premature treatment discontinuation was 59%, mostly due to AEs, compared to 47% of well-compensated cirrhotic patients. Logistic regression analyses noted that a lower baseline serum albumin level was associated with decompensation in both the telaprevir and boceprevir-treated populations when controlling for age and sex and that bilirubin level, platelet count, baseline HCV RNA levels and mean MELD score were predictive of liver decompensation in telaprevir patients, while the haemoglobin level was predictive of liver decompensation in those who received boceprevir (data not shown).
Discussion
This report describes the safety profiles of boceprevir and telaprevir in the largest and most diverse population treated for hepatitis C with these agents in the United States. We analysed the experiences of more than 2000 patients who were among the first U.S. patients to receive triple therapy at more than 100 medical centres. Our results demonstrate that patients treated in clinical practice with these agents were older and had more advanced disease than the registration trial participants, and were more likely to sustain significant adverse events. Overall SVR rates were slightly lower than those in registration trials.
More than 90% of patients had adverse events that led to a prescription, treatment, or dosage change; and more than one third of patients discontinued treatment early. Anaemia occurred in two thirds of patients and was the most clinically significant adverse effect. Despite a high rate of ribavirin dose reductions to manage anaemia, U.S. clinicians often used epoetin, an agent that was not permitted in the telaprevir registration trials, to treat anaemia. Indeed, nearly 40% of patients overall received epoetin alfa and a surprising 11% of patients required blood transfusions for treatment of severe anaemia. The rates of anaemia and treatment discontinuations in this analysis were far higher than reported for the pivotal registration studies of both boceprevir and telaprevir [2,3,7,10,11]. Interestingly, skin rash, more frequent among patients treated with telaprevir, had received much attention prior to the launch of telaprevir, although in the present study it was mostly of mild severity and was infrequently associated with DRESS or premature treatment discontinuation. This is likely due to increased vigilance and close monitoring, which prompted early intervention and minimized the progression of potentially serious skin reactions.
The reduced tolerability seen in this analysis is likely due to differences in patient populations in clinical studies compared to clinical practice. Relative to patients included in the registration trials, with enrolment starting in 2008, the patients in our analysis, starting in 2011, were older, with a median age approximately 5 years higher and an upper range in their 70s vs. 60s [2,3,7,10,11] in the registration trials. More importantly, the patients in the present analysis received treatment at a more advanced stage of liver disease, with 38% having cirrhosis at baseline vs. the more typical 6–26% in the registration studies [2,3,7,10,11]. Further, more than half of the patients in our analysis had received prior therapy for chronic HCV, thus indicating (a) an unmet need of a large proportion of prior non-responders to interferon-based therapies and (b) patient and clinician willingness to pursue additional and more intensive antiviral therapy than previously undertaken. The findings underscore the observation that the actual treated cohort in clinical practice represents a progressively older and histologically more advanced population than that reported in the pivotal registration trials.
Despite the large number of patients with cirrhosis, the rate of serious adverse events observed in our population (11% of patients receiving telaprevir and 13% receiving boceprevir) is very similar to that observed in the telaprevir (12%) and boceprevir (12%) registration trials in patients who had previously been treated [7,10], an observation that could be attributable to the attentive management of AEs. Liver disease was more severe among those with cirrhosis in the present HCV-TARGET analysis, as evidenced by lower platelet counts, a higher frequency of oesophageal varices, and the inclusion of numerous patients with evidence of prior decompensation, compared to two recent European reports of triple therapy in patients with advanced liver disease [8,12]. Indeed, the group of patients with a prior history of decompensation had the highest rates of AEs, SAEs, and premature treatment discontinuation. Of note, cure rates remained high even in those patients.
In the prospective analysis of 674 French patients with compensated cirrhosis in the CUPIC study, triple therapy was associated with a high incidence of serious adverse events (40%), as well as severe complications, such as severe infection or hepatic decompensation in 6.4%, and death in 1% of patients [6]. Severe anaemia occurred in 29%, although only 16% of patients had a ribavirin dose reduction, a rate lower than that reported in the present study where ribavirin dose reduction was the preferred intervention [6]. Erythrocyte stimulating agents were used in 51% of patients in the French study and 12% required blood transfusions. Deaths and severe complications were associated with lower platelet counts (≤100,000/μl) and lower albumin (<3.5 g/ dl) where the observed risk was 44% if patients had both features [9]. Comparable observations have been made from another European expanded access program that treated over 1500 patients with bridging fibrosis or compensated cirrhosis with protease inhibitor based therapy [8].
Although the standard of care for the treatment of HCV genotype 1 has shifted away from telaprevir and boceprevir containing regimens, the current study nevertheless has important implications for clinicians, particularly in regions outside of the U.S. where triple therapy with boceprevir or telaprevir, in combination with peginterferon and ribavirin, will remain the mainstay of therapy for the foreseeable future due to challenges with access to the most advanced therapies. This information is also of importance to health care policy researchers who will be comparing the effectiveness of multiple regimens across different populations.
The results of the present analysis indicate that HCV patients in clinical practice settings experience a greater potential for toxicity with boceprevir- or telaprevir-based triple therapy than patients who were selected for enrolment in the HCV drug development trials. The post marketing triple therapy experience in the U.S. mirrors the recent European experiences [6,8] and highlights a major disparity between the highly controlled HCV clinical trial experience and the real world reality of an aging, cirrhotic and medically more vulnerable global HCV community. While there remains a lack of data for novel and less toxic therapies in patients with more advanced liver disease and co-morbidities, the present report underscores the need for a more inclusive patient population to be represented in future HCV clinical trials.
Supplementary Material
Acknowledgments
Financial support
HCV-TARGET is an investigator-initiated study, jointly sponsored by The University of Florida, Gainesville, FL (PI: D.R. Nelson), and The University of North Carolina at Chapel Hill, Chapel Hill, NC (PI: M.W. Fried). It was funded in part by Vertex Pharmaceuticals, Inc., Merck & Co., Kadmon Corporation, and Genentech and in part by CTSA UF UL1TR000064 and UNC1UL1TR001111. M.W. Fried is funded in part by the NIH Mid-Career Mentoring Award K24 DK066144.
We thank the staff of the data coordinating center at UNC Chapel Hill: Lucy Akushevich, Kenneth Bergquist, John Baron, Paul Stewart, Dianne Mattingly, and Wendy Robertson; the staff of clinical coordinating center at UF Gainesville: Lauren Morelli, Anthe Hoffman, Dona-Marie Mintz, Lasheaka McClellan, Angela Bauer, Patrick Horne, Rennie Mills; the nurses and patients who were involved in this study; and Valérie Philippon, Ph.D. for her assistance in the preparation of the manuscript, with funding from University of Florida.
Abbreviations
- HCV
hepatitis C virus
- DAAs
direct-acting antiviral agents
- PegIFNα
pegylated interferon alfa
- AE
adverse event
- SAE
serious adverse event
- SVR
sustained virologic response
- DRESS
drug reaction with eosinophilia and systemic symptoms
Footnotes
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.jhep.2014.08.052.
Conflict of interest
S.C. Gordon has received grant/research support from Abbott Pharmaceuticals, Bristol-Myers Squibb, Exalenz BioScience, Gilead Pharmaceuticals, Intercept Pharmaceuticals, Merck, and Vertex Pharmaceuticals; has received financial compensation for consultancy from AbbVie, Amgen, CVS Caremark, Gilead Pharmaceuticals, and Merck and received royalties from Up-To-Date. A.J. Muir has received grant/research support from AbbVie, Achillion, Bristol Myers-Squibb, Gilead, Vertex and Merck and has received financial compensation for consultancy from AbbVie, Achillion, Bristol Myers-Squibb, Gilead, Vertex and Merck. J.K. Lim has received grant/research support from Abbott, Achillion, Boehringer Ingelheim, Bristol-Myers Squibb, Janssen, Genentech, Gilead, and Vertex and has received financial compensation for consultancy from Boehringer Ingelheim, Bristol-Myers Squibb, Gilead, Janssen, and Merck. B. Pearlman has received grant/research support from Boehringer Ingelheim, Merck, Bristol-Myers Squibb, Johnson&Johnson/Tibotec/Janssen, Gilead, and AbbVie. C.K. Argo has received grant/research support from Roche-Genentech. A. Ramani has received grant/research support from Forest Pharmaceuticals and has received financial compensation for consultancy and/or lecture activities from Gilead Sciences, Forest Pharmaceuticals, and Merck. B. Maliakkal has received has received financial compensation for lecture activities from Genentech, Merck and Vertex. I. Alam has received research/grant support from AbbVie and Genentech, and has received financial compensation for consultancy and/or lecture activities from Gilead, Janssen, AbbVie, Genentech, and Takeda. D.R. Nelson has received grant/research support from Abbott, Achillion, Boehringer-Ingelheim, Bristol-Myers Squibb, Janssen, Kadmon, Genentech, Gilead, Merck, and Vertex. M.W. Fried has received research grants from AbbVie, BMS, Genentech, Gilead, Janssen, Merck, and Vertex has received financial compensation for consultancy and/or lecture activities from AbbVie, BMS, Genentech, Gilead, GlaxoSmithKline, Janssen, Merck, and Vertex. K.R. Reddy has received research/grant support from Gilead, Vertex, Janssen, BMS, AbbVie, and Merck and has received financial compensation for consultancy and/or lecture activities from Gilead, Janssen, Vertex, Bristol-Myers Squibb, Genentech-Roche, Merck, and AbbVie.
References
- 1.Kwo PY, Lawitz EJ, McCone J, Schiff ER, Vierling JM, Pound D, et al. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet. 2010;376:705–716. doi: 10.1016/S0140-6736(10)60934-8. [DOI] [PubMed] [Google Scholar]
- 2.Poordad F, McCone J, Jr, Bacon BR, Bruno S, Manns MP, Sulkowski MS, et al. Boceprevir for untreated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1195–1206. doi: 10.1056/NEJMoa1010494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobson IM, McHutchison JG, Dusheiko G, Di Bisceglie AM, Reddy KR, Bzowej NH, et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N Engl J Med. 2011;364:2405–2416. doi: 10.1056/NEJMoa1012912. [DOI] [PubMed] [Google Scholar]
- 4.McHutchison JG, Everson GT, Gordon SC, Jacobson IM, Sulkowski M, Kauffman R, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009;360:1827–1838. doi: 10.1056/NEJMoa0806104. [DOI] [PubMed] [Google Scholar]
- 5.Barritt AS, Fried MW. Maximizing opportunities and avoiding mistakes in triple therapy for hepatitis C virus. Gastroenterology. 2012;142:1314–1323. doi: 10.1053/j.gastro.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hézode C, Fontaine H, Dorival C, Larrey D, Zoulim F, Canva V, et al. Triple therapy in treatment-experienced patients with HCV-cirrhosis in a multi-centre cohort of the French Early Access Programme (ANRS CO20-CUPIC) – NCT01514890. J Hepatol. 2013;59:434–441. doi: 10.1016/j.jhep.2013.04.035. [DOI] [PubMed] [Google Scholar]
- 7.Bacon BR, Gordon SC, Lawitz E, Marcellin P, Vierling JM, Zeuzem S, et al. Boceprevir for previously treated chronic HCV genotype 1 infection. N Engl J Med. 2011;364:1207–1217. doi: 10.1056/NEJMoa1009482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colombo M, Fernández I, Abdurakhmanov D, Ferreira PA, Strasser SI, Urbanek P, et al. Safety and on-treatment efficacy of telaprevir: the early access programme for patients with advanced hepatitis C. Gut. 2014;63:1150–1158. doi: 10.1136/gutjnl-2013-305667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maasoumy B, Port K, Markova AA, Serrano BC, Rogalska-Taranta M, Sollik L, et al. Eligibility and safety of triple therapy for hepatitis C: lessons learned from the first experience in a real world setting. PLoS One. 2013;8:e55285. doi: 10.1371/journal.pone.0055285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeuzem S, Andreone P, Pol S, Lawitz E, Diago M, Roberts S, et al. Telaprevir for retreatment of HCV infection. N Engl J Med. 2011;364:2417–2428. doi: 10.1056/NEJMoa1013086. [DOI] [PubMed] [Google Scholar]
- 11.Sherman KE, Flamm SL, Afdhal NH, Nelson DR, Sulkowski MS, Everson GT, et al. Response-guided telaprevir combination treatment for hepatitis C virus infection. N Engl J Med. 2011;365:1014–1024. doi: 10.1056/NEJMoa1014463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Afdhal NH, Reau N, Everson GT, Morelli G, Lok AS, Kenneth E, et al. Safety and efficacy of telaprevir (TVR) or boceprevir (BOC) in Patients with cirrhosis: interim results of a longitudinal, observational study. Hepatology. 2013;58:1103A–1104A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.