

Abstract
Vascular endothelial growth factor regulates neoplastic angiogenesis through production of endothelium-derived nitric oxide. We performed a prospective evaluation of vascular function during treatment with vandetanib, a vascular endothelial growth receptor 2 and 3 receptor tyrosine kinase inhibitor, to determine the effects of vascular endothelial growth receptor signal interruption on endothelial function in humans. Seventeen patients with stage IV breast cancer received dose escalated vandetanib in combination with low-dose oral chemotherapy. We measured blood pressure, systemic nitrate/nitrite levels, and brachial artery vascular function. In vitro analyses of cultured endothelial cells were performed to determine the effect of vandetanib on nitric oxide production, akt473 phosphorylation, and endothelial nitric oxide synthase protein content and membrane localization. Vandetanib treatment for 6 weeks significantly increased blood pressure, decreased resting brachial artery diameter, and decreased plasma systemic nitrate/nitrite levels compared to baseline. Flow-mediated vasodilation was preserved, and no change was noted in nitroglycerin-mediated vasodilation. In vitro, endothelial cell nitrite levels and akt473 phosphorylation were reduced, vascular endothelial growth receptor 2 levels did not change, but endothelial nitric oxide synthase membrane concentration doubled. Vandetanib reduces constitutive nitric oxide production and increases blood pressure, yet flow-stimulated nitric oxide bioavailability was preserved. Changes in vascular function with tyrosine kinase inhibition are complex and require further study in humans.
Keywords: Vascular Endothelial Growth Factor, VEGF receptor 2, secondary hypertension, endothelium, nitric oxide
Angiogenesis is an obligate component of the growth and progression of many solid tumors including breast cancer. (1–3). Vascular endothelial growth factor (VEGF), a potent angiogenic factor that regulates malignant angiogenesis, has emerged as a therapeutic target for this disease. Angiogenesis inhibition using the anti-VEGF antibody bevacizumab in combination with cytotoxic chemotherapy has improved response rate and prolonged progression free survival in patients with advanced cancer (4). Small molecule tyrosine kinase inhibitors of the VEGF receptor (VEGFR) are under investigation as anti-angiogenesis therapy for breast cancer (5–7).
Hypertension is a prevalent side effect of VEGF inhibitors (8–12) and VEGFR antagonists, (13–16) and quite distinct from chemotherapy-related toxicity. The incidence ranges from 15%–60%, depending on the anti-angiogenic agent, tumor type, and patient-related factors (17). Indeed, nearly all patients experience some increase in blood pressure, if not frank hypertension (18). The mechanisms underlying hypertension associated with angiogenesis inhibitors are not well defined. Several theories have been proposed, including an imbalance in neurohumoral factors and the development of vascular rarefaction. In humans treated with the VEGFR 2 inhibitor sorafenib with a mean increase in blood pressure of 9 mm Hg, there was no change in renin, aldosterone, and endothelin 1 levels, whereas catecholamine levels trended down inversely to the increase in blood pressure (15), making humoral changes less likely.
The most compelling explanation for angiogenesis inhibitor-associated hypertension is its effect on nitric oxide (NO) bioavailability described in preclinical investigations. VEGF is known to stimulate endothelial nitric oxide synthase (eNOS) leading to NO production and vasodilation (19, 20). Inhibition of VEGF signaling could reduce eNOS activity and decrease NO levels, leading to vasoconstriction, endothelial dysfunction, and subsequent hypertension. Animal data show that VEGFR inhibition leads to reduced eNOS activity and hypertension (21).
The effect of VEGF inhibitors on NO bioavailability and its role in treatment-related hypertension in humans is not well described. Accordingly, we evaluated blood pressure, markers of NO production, and in vivo vascular function before and during exposure to the VEGFR 2/3 antagonist, vandetanib, in subjects participating in a trial of vandetanib and metronomic chemotherapy in advanced metastatic breast cancer to test constitutive and flow-stimulated NO bioavailability.
Methods
Subjects
Eligible subjects with metastatic breast cancer were recruited as part of a single center investigator-initiated phase 1 trial. The subjects had exposure to no more than 4 prior chemotherapy regimens for metastatic disease, an ECOG performance status of at least 2, were post-menopausal or surgically sterile, and normal bone marrow, hepatic, and renal function. Patients were required to have preserved cardiac function (left ventricular ejection fraction > 45%), no evidence of QTc prolongation, and adequately controlled blood pressure at baseline. Patients were excluded for a requirement of more than one medication to control blood pressure to <140/90 mm Hg, therapeutic anticoagulation, or clinically significant cardiac disease.
Study Design
Three sequential cohorts, planned enrollment 8 patients each, were treated with escalating doses of daily vandetanib, an oral inhibitor of VEGFR and EGFR, (Cohort 1, 100 mg; Cohort 2, 200 mg; Cohort 3, 300 mg) with continuous metronomic chemotherapy (cyclophosphamide 50 mg PO qd, and methotrexate 2.5 mg PO bid, day 1 and 2 of each week). Vandetanib is a potent vascular endothelial growth factor receptor-2 inhibitor (VEGFR-2: inhibitory concentration (22) = 40 nM), and shows additional inhibitory activity against Rearranged during Transfection (RET) receptor (IC50 = 100 nM), Fms-related tyrosine kinase 4 (Flt-4)/VEGFR-3 (IC50 = 110 nM) and epidermal growth factor receptor (EGFR) (IC50 = 500 nM) (23, 24).
Subjects underwent vascular function testing prior to administration of vandetanib, and after six weeks of protocol therapy. As there were no differences in change in blood pressure or vascular function between cohorts, all subjects were studied as one group. All participants provided written informed consent. The protocol was approved by the Human Research Committees of the Brigham and Women’s Hospital and Dana Farber Cancer Institute.
Blood Pressure Assessments
Blood pressure measurements were performed at each vascular study using an oscillometric device after 10 minutes of quiet recumbency prior to vascular measurements. The blood pressures were measured in triplicate with the value provided the average of the three. Blood pressure was measured prior to vascular studies. All studies were performed in the morning after an overnight fast. Antihypertensive medications were held the day of study.
Assessment of Vascular Function
To assess endothelium-dependent, nitric oxide-mediated conduit artery vasodilation, brachial artery flow-mediated vasodilation (FMD) following a hyperemic stimulus was measured at baseline and after 6 weeks on protocol therapy. The FMD technique was performed according to published protocol (25). We and others have demonstrated that brachial artery vasodilation one minute after reactive hyperemia dilation is mediated by nitric oxide (26, 27).
Subjects were studied in a controlled environment in the supine position after a minimum four hour fast. After a minimum 10 minute equilibration period, baseline two-dimensional images of the brachial artery were obtained approximately 2 cm above the antecubital fossa. A blood pressure cuff, placed proximal to the imaging transducer on the upper arm, was inflated to suprasystolic pressure for five minutes. The vessel was imaged continuously for 70 seconds after release of occlusion. Longitudinal brachial artery digital images from end-diastole (peak of the QRS) were acquired with a high-resolution (7.5 MHz) linear-array vascular ultrasound scanning probe (Vivid 7, GE Medical Systems, Piscataway, NJ). Reactive hyperemia was confirmed by measuring arterial blood flow using pulse-wave Doppler scanning interrogation. After a 10 minute period of re-equilibration, baseline measurements were repeated. Endothelium-independent vasodilation was assessed by measuring brachial artery diameter under basal conditions and 3 minutes following the administration of sublingual nitroglycerin (0.4 mg). Nitroglycerin was not administered if systolic blood pressure was below 100 mm Hg. Subjects in whom nitroglycerin was not contraindicated were administered 0.4 mg nitroglycerin sublingually and the brachial artery was imaged continuously for 5 min as described above.
Blinded measurement of vessel diameter was performed using customized image analysis software (Brachial Analyzer for Research 5.7.9, Medical Imaging Applications, Iowa City, Iowa) by investigators blinded to study day and subjects (WMR & JAB). For each condition, the results from six images at end-diastole were averaged. FMD was defined as the brachial artery diameter 60 – 70 seconds after cuff deflation compared with the baseline vessel diameter. Nitroglycerin mediated dilation (NMD) was defined as the brachial artery diameter 3 minutes after administration of nitroglycerin compared with the baseline vessel diameter. To be considered interpretable, a study had to have distinct visualization of the proximal and distal intima-media arterial layers perpendicular to the ultrasound beam with less than 5% diameter variation across the field of measurement. In our laboratory, the intraobserver variability in measuring brachial diameters was 2.9% and the variability of the hyperemic response was 1.4% (28).
Volumetric flow, Q, was calculated from commercially available software, (Brachial Analyzer, Medical Imaging Applications, Iowa City, Iowa), by integrating the area under the velocity spectral waveform and dividing by the time required to arrive at a time-averaged velocity. Mean flow was calculated by multiplying the time-averaged velocity by the mean area of the lumen (as obtained from the index segment M-mode measurements). Mean shear stress was calculated according to the Hagen-Poiseuille formula τw = 4μQ/πri3, where τw is shear stress in dynes/cm2 and mean volumetric flow is Q. The viscosity of blood, μ, is assumed to be 0.035 poise. The lumen radius, ri, is in cm. Forearm vascular resistance was calculated by dividing the mean arterial pressure by mean volumetric flow (arbitrary units).
Laboratory Analyses
The effects of angiogenesis inhibitor therapy on NO and reactive oxygen species (ROS) production were examined in patients. Plasma samples were collected at baseline and following 6 weeks of treatment and frozen until use.
NO Assay
A quantitative fluorometric extracellular NO assay (Calbiochem) was used to assess NO production in patient samples following manufacturers instructions. Since the final products of NO are nitrates and nitrites, this assay uses nitrate reductase to convert nitrates to nitrites, then uses 2,3-diaminonapthalene and NaOH to convert nitrites to a fluorescent compound to measure total NO production. Plasma samples were thawed, filtered using Ultracel 10 kDa cut-off Microcons (Millipore) by centrifugation (0.5 h, 4°C), and processed in the NO assay. Plates were read using a Wallac 1420 Multilabel Victor3 microplate reader (Perkin Elmer), with excitation at 355 nm and emission at 430 nm.
DNA Damage ELISA
The marker 8-hydroxy-2′-deoxyguanosine (8-OHdG) is an oxidized nucleoside excised from damaged DNA that increases in the setting of oxidative DNA damage. Levels of 8-OHdG were quantified in patient plasma samples using the colorimetric DNA Damage ELISA (Assay Designs) as per manufacturer’s instructions. Absorbances at 450 nm were measured using a Spectra Max 190 microplate reader with SoftMax Pro software (Molecular Devices).
Nitrotyrosine ELISA
Superoxides are highly reactive and can react with NO to form peroxynitrite, which reacts with tyrosines on proteins causing tyrosine nitration (tyrosine converted to 3-nitrotyrosine). Patient plasma samples were examined using a quantitative chemiluminescent nitrotyrosine ELISA to measure tyrosine nitration as per manufacturer’s instructions (Millipore). Luminescence was measured using a Wallac 1420 Multilabel microplate reader.
In Vitro Endothelial Cell Studies
MS1 endothelial cells (EC) were cultured as we previously described (29).
NO Assay
Vandetanib or matched vehicle control (DMSO) was added to MS1 EC (n=6/group with studies done in duplicate) in phenol red-free DMEM/0.5% BSA and incubated (0.5 h, 37°C, 10% CO2). Then, 100 μM L-arginine, and 100 μM SNAP were added, cells incubated (1.5 h, 37°C, 10% CO) and the NO assay was performed as described above.
Akt Activity
The MS1 endothelial cells were cultured in 24-well plates in low glucose (1.0 g/L) DMEM (LG-DMEM) supplemented with 10% fetal calf serum/1% L-glutamine-penicillin-streptomycin (37°C, 10% CO2) and grown to near confluency. Cells were rinsed twice in LG-DMEM/0.5% BSA. In some assays, cells were serum-starved in LG-DMEM/0.5% BSA (2 h, 37°C, 10% CO2). Vandetanib (Selleck/Husker Chemicals) was dissolved in DMSO and diluted in LG-DMEM/0.5% BSA. Either 1 μM vandetanib or matched vehicle controls were added to cells and incubated (1 h, 37°C, 10% CO2). Protein lysates were prepared and phosphorylation of aktS473 was measured as we previously described (29). Films were scanned and Scion Image software was used to conduct densitometric analyses to quantify signal intensity (n=4–6/group, studies done in triplicate).
ENOS Protein Levels in Membrane and Cytosol
The MS1 endothelial cells were cultured in their normal culture media and grown to confluency. Cells were rinsed in PBS and were serum-starved in culture media diluted 1/5 with LG-DMEM (2 h, 37°C, 10% CO2). Vandetanib was dissolved in DMSO and either 1 μM vandetanib or DMSO vehicle controls were added to cells and incubated (1 h, 37°C, 10% CO2). Protein lysates were prepared and membrane and cytosol components were isolated using the Subcellular Protein Fractionation Kit (Pierce). Both membrane and cytosolic fractions were run on gels and Western blotting conducted as we described (29). ENOS (rabbit monoclonal antibody, clone 49G3 at 1/1000), GAPDH (HRP-conjugate rabbit monoclonal antibody, clone 14C10 at 1/5000), and VE-Cadherin (rabbit polyclonal antibody, clone H72 at 1/1000) proteins levels were measured, films were scanned and Scion Image software was used to conduct densitometric analyses to quantify signal intensity (n=3/group, studies done in duplicate).
Statistical Considerations
Descriptive measures are reported as mean ±SD. Experimental measures are reported as mean ±SE. Biomarkers are reported as median [inter-quartile range]. Basal forearm blood flow and diameter were compared by paired two-tailed t tests. Laboratory measures were compared by Mann Whitney U testing for two way comparisons. Association between vascular function and blood pressure changes were correlated by Pearson correlation. Statistical significance was accepted at the 95% confidence level (P<0.05). SPSS (version 16.0, SPSS Inc., Chicago, IL) software package was utilized.
Results
A total of 23 patients were enrolled into the Phase 1 trial. Nineteen subjects consented to participate in the vascular study and 2 withdrew consent midprotocol. Patient and tumor characteristics are described in Table 1 for the 17 subjects who completed the investigation. All patients enrolled received protocol therapy.
Table 1.
Baseline Characteristics
| Characteristic | N = 17 |
|---|---|
| Age (years) | 49 [29–71] |
| Cardiovascular Risk Factors | |
| Postmenopausal (%) | 100 |
| Hypertension (%) | 35 |
| Dyslipidemia (%) | 6 |
| Current smoker (%) | 0 |
| ECOG Performance Status (median (19)) | 0 [0 – 2] |
| Antihypertensive Medications (n) | |
| Beta Adrenergic Blockers | 3 |
| Lisinopril | 1 |
| Furosemide | 1 |
| Amlodipine | 1 |
| Prior Systemic Therapy | |
| Anthracycline exposure (%) | 100 |
| Trastuzumab or lapatinib (%) | 12 |
| Bevacizumab (%) | 41 |
| Cetuximab (%) | 12 |
| Cediranib (%) | 6 |
Blood Pressure and Constitutive NO Bioavailability
Consistent with prior studies of VEGF RTK inhibitors, vandetanib increased mean arterial pressure from 91 ± 8 mm Hg to 102 ± 10 mm Hg, p = 0.001 (Table 2). The increase in blood pressure was similar in both systolic and diastolic components. In contrast, no change in heart rate was noted. Also, subjects’ weight decreased significantly. Two laboratory measures of nitric oxide were used to determine NO bioavailability before and during vandetanib use. NOx levels decreased significantly from baseline following exposure to study drugs for 6 weeks (Figure 1, p=0.036), indicating reduced NO elaboration. There was a trend between the reduction of nitrite level and increase in blood pressure, (R2 = 0.21; p = 0.068). When divided by the median blood pressure increase (10 mm Hg), the subjects with an increase of the median increased MAP by 19 ± 6 mm Hg compared with the subjects with an increase of the median, who had a nonsignificant change of 1 ± 5 mm Hg, (p < 0.001). The subjects with an above the median increase in blood pressure had a 10-fold greater reduction in NOx compared to the 9 subjects whose blood pressure was below the median (−3945 ± 5530 vs. −382 ± 2492; p = 0.048). Levels of nitrotyrosine trended lower from baseline following 6 weeks of treatment (Table 2, p = 0.054). Physiological evidence of reduced nitric oxide was evaluated with brachial ultrasonography. Baseline arterial diameter was 2.74 ± .09 mm and decreased after 6 weeks of treatment to 2.57 ± 0.08 mm, p = 0.004 (Table 2). Consistent with a reduction in NOx and increased blood pressure, forearm vascular resistance increased from 5.3 ± 0.5 U to 6.4 ± 0.5 U (p = 0.015). Shear stress did not vary significantly between study conditions, 10.9 ± 1.2 vs. 11.6 ± 1.7 poise, p = 0.4.
Table 2.
Experimental Measures
| Parameter | Baseline | 6 weeks | Comparison | |
|---|---|---|---|---|
| Systolic Blood Pressure | mm Hg | 138 +/− 20 | 150 +/− 21 | p = 0.037 |
| Diastolic Blood Pressure | mm Hg | 68 +/−7 | 77 +/− 8 | p < 0.001 |
| Mean Arterial Pressure | mm Hg | 91 +/− 8 | 102 +/− 10 | p = 0.001 |
| Heart Rate | beats per minute | 74 +/− 13 | 71 +/− 10 | p = 0.28 |
| Weight | kg | 72.8 ± 17.0 | 71.3 ± 16.5 | p = 0.004 |
| Basal Brachial Artery Diameter | mm | 2.74 ± .09 | 2.57 ± .08 | p = 0.004 |
| Post-RH Brachial Artery Diameter | mm | 3.07 ± .12 | 2.93 ± .12 | p = 0.06 |
| Absolute Increase in Brachial Artery Diameter | mm | 0.33 ± .05 | 0.36 ± .06 | P = 0.36 |
| Reactive Hyperemic Stimulus | fold increase | 5.8 ± 2.9 | 6.8 ± 3.0 | p = 0.024 |
| Flow Mediated Vasodilation | % | 12.0 ± 1.7 | 13.8 ± 1.8 | p = 0.15 |
| Pre Nitroglycerin Brachial Artery Diameter | mm | 2.78 ± .1 | 2.63 ± .1 | p = 0.024 |
| Post-NTG Brachial Artery Diameter | mm | 3.38 ± .12 | 3.16 ± .13 | p = 0.04 |
| NTG Mediated Vasodilation | % | 22.5 ± 2.0 | 20.3 ± 2.3 | p = 34 |
| Nitrates | AU | 48446±1264 | 46799±1740 | p < 0.001 |
| Nitrotyrosine | AU | 259814±26139 | 246157±20501 | p = 0.027 |
| 8OHDG | AU | 1.96±0.09 | 2.0±0.15 | p = 0.5 |
Laboratory units are fluorescence or luminescence. 8OHDG = 8-Hydroxy-2′Deoxyguanosine
Figure 1.

Vandetanib Reduced Plasma Nitrite Levels in Humans. Plasma levels of nitrites were measured in patients at baseline and after receiving vandetanib plus chemotherapy for 6 weeks. Vandetanib lowered nitrite levels after 6 (**p=0.0001) weeks suggesting that circulating levels of nitric oxide were reduced.
We investigated the putative mechanism of reduced constitutive nitric oxide bioavailability in MS1 coronary endothelial cells. Incubation of vandetanib with MS1 EC significantly decreased nitrite production compared to the matched vehicle (DMSO) (Figure 2, p=0.0003). These findings demonstrate that vandetanib lowered endothelial cell NO levels. VEGF, used as a positive control, increased nitrite levels compared to matched vehicle (PBS) (p=0.02). We next evaluated the effect of vandetanib on VEGF to eNOS signaling by measuring phosphorylation of the intermediate, AKT at serine 473. Western blotting (Figure 3) shows that vandetanib also reduced phosphorylation of Akt at serine 473 in MS1 EC compared to vehicle controls (DMSO) (Figure 3, p=0.01), indicating reduced Akt activation. Oxidative stress, as a mechanism to reduce NO bioavailability, was also evaluated. Levels of 8OHDG, a product of oxidative DNA damage, were stable with treatment (Table 2) and did not vary by vandetanib dose levels. In addition, nitrotyrosine levels trended lower during treatment. This may suggest a decrease in oxidative stress or a reduction in circulating nitrates from which to nitrosylate proteins.
Figure 2.
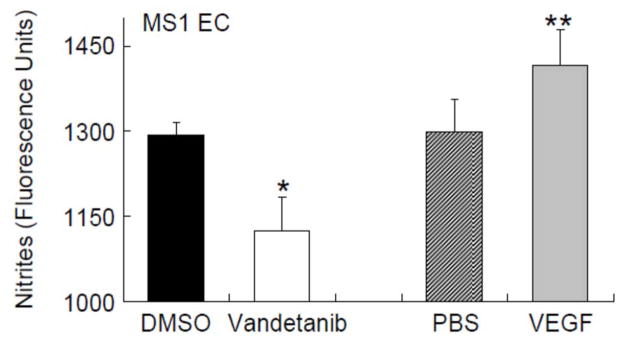
Vandetanib Reduced Extracellular Nitrite Levels in Endothelial Cells. MS1 EC were incubated with 1 μM vandetanib or matched vehicle (DMSO), 50 ng/ml VEGF, or matched vehicle (PBS) (0.5 h), and L-arginine and SNAP added (1.5 h). Vandetanib lowered nitrite levels in MS1 EC (*p=0.0003). VEGF was used a positive control and increased nitrite levels (**p=0.02). These findings indicate that vandetanib lowered endothelial cell nitric oxide levels.
Figure 3.

Vandetanib Reduced Phosphorylation of Akt in Endothelial Cells.
MS1 EC were incubated with 1 μM vandetanib or matched vehicle (DMSO) (1 h). Western blotting analysis showed that vandetanib decreased phosphorylation of akt (S473) in MS1 EC (*p=0.01) (n=6/group, studies done in triplicate). These findings show that vandetanib reduced akt activity.
Vascular Function Studies and Stimulated NO Bioavailability
We measured flow-mediated vasodilation as an index of flow-stimulated NO bioavailability. The application of a five-minute ischemic intervention created a significant reactive hyperemic stimulus during both study visits. Peak reactive hyperemic blood flow increased 5.8 ± 2.8 fold over basal flow at baseline and 6.8 ± 3.0 fold at 6 weeks (p = 0.02). In the whole cohort, flow-mediated vasodilation did not change significantly during the 6 weeks of treatment, 12.0 ± 1.7% vs. 13.8 ± 1.8%, (Figure 4, p = 0.15), even when adjusted for the variation in reactive hyperemia (p = 0.26). There was no change in nitroglycerin-mediated vasodilation during treatment. To understand the mechanism of preserved flow-mediated vasodilation despite VEGF inhibition, membrane-associated eNOS protein level and VEGFR2 protein levels were assessed. Vandetanib application increased membrane eNOS protein levels two-fold (p = 0.04, Figure 5). No changes in VEGFR2 levels were noted.
Figure 4.

Flow Mediated and Nitroglycerin-Mediated Vasodilation. Endothelium-dependent, flow-mediated vasodilation was preserved after 6 weeks of vandetanib therapy (12.0 ± 1.7% vs. 13.8 ± 1.8%, p = 0.15). Nitroglycerin-mediated, endothelium-independent vasodilation was unchanged after 6 weeks of vandetanib treatment (22.5 ± 2.0% vs. 20.0 ± 2.3%, p > 0.2).
Figure 5. Vandetanib Increases Membrane Localization of eNOS.

MS1 EC were incubated with 1 μM vandetanib or matched vehicle (DMSO). Western blotting analysis showed that vandetanib increases membrane localization of eNOS compared to control (*p =0.04) (n=4/group, studies done in triplicate). These findings show that vandetanib increased the membrane localization of eNOS compared to control.
Discussion
This study demonstrates that vandetanib, a VEGFR2/3 and RETR receptor tyrosine kinase antagonist, in combination with metronomic chemotherapy attenuates constitutive but not flow-mediated nitric oxide bioavailability in humans. Administration of this medication significantly increased mean blood pressure and decreased, in association, systemic NOx levels. These findings are bolstered by the decrease in resting conduit artery size despite the lack of change in wall shear stress and the increase in forearm vascular resistance. The mechanism of the chronic reduction in NO was tested in vitro. Vandetanib significantly reduced nitrite production and serine437 AKT phosphorylation in MS1 coronary endothelial cells. These analyses support the contention that vandetanib reduces PI3 kinase-AKT-mediated phosphorylation of eNOS activation sites and consequent attenuation in the constitutive production of NO. We also studied other potential mechanisms to explain this finding. Both eNOS and VEGFR2 total cellular and membrane fractions were evaluated in MS1 coronary endothelial cells treated with vandetanib. The treated cells showed an increase in both total and membrane-associated eNOS but no change in either parameter for VEGFR2, providing more insight into the physiological effects of vandetanib.
VEGF and Nitric Oxide
The angiogenic and vasoactive effects of VEGF occur primarily via VEGFR2 (30, 31). Supporting this finding, the incidence of hypertension seen in clinical trials appears to correlate with the potency of the kinase inhibitors to block VEGFR2 (17). VEGFR2 modulates endothelial nitric oxide synthase (eNOS) production of NO through multiple signaling pathways. Tyrosine phosphorylation of VEGFR2 initially activates phospholipase C (PLC)-γ rapidly increasing the intracellular calcium concentration, facilitating calmodulin-mediated eNOS disassociation from calveolin-1, and consequent NO production (32). Infusion of VEGF into patients induces immediate NO elaboration and hypotension (33). Later, VEGF activation of VEGFR2 stimulates the PI3-AKT-HSP90 pathway which phosphorylates eNOS serine1177, a positive regulator of the enzyme (32, 34).
Our findings of increased blood pressure, increased vascular resistance, decreased systemic NO production, and decreased basal arterial diameter, are consistent with the predicted effects of a VEGFR2 inhibitor. Indeed, the significant reduction in systemic NOx production in this paired sample with VEGFR inhibition is greater than recently reported in another study of humans treated with VEGF antagonism (35). These results and our in vitro data showing that vandetanib reduces endothelial cell Akt phosphorylation and nitrite production suggest that VEGFR2 inhibition reduces constitutive eNOS-mediated NO production. Other sources of NOx are less likely to contribute to this picture as VEGFR2 inhibition has been shown to decrease inducible NOS (36). Thus, our data are consistent with a mechanism of VEGFR2 inhibition disrupting the PI3K-Akt-HSP90 phosphorylation-dependent activation of eNOS, leading to chronically reduced NO levels and hypertension, particularly in light of other work in humans demonstrating no change in sympathetic nervous system, renin-angiotensin system, and endothelin 1 humoral activity (15).
RTKI specificity and vascular function
In contrast to our findings, in a study of the RTKI telatinib, there was an attenuation in vascular smooth muscle and possibly endothelial function (37). One explanation that may reconcile the differences in our observations with vandetanib versus telatinib is the variability of the effect of these RTK inhibitors on phosphorylation of specific tyrosine moieties and cell types. The fact that telatinib and vandetanib produced different changes in blood pressure, vascular smooth muscle function, and endothelial function over a similar time course in intact humans makes clear that the description of an agent as a VEGFR2 inhibitor reveals only its gross function. A second variance may be the non-VEGFR-related effects. Telatinib inhibits platelet derived growth factor receptor activity which has shown to be an important mechanism by which vascular smooth muscle cells increase cytosolic calcium and may explain the attenuated response to nitroglycerin seen in this study (38, 39). These results highlight the need for evaluation of each agent specifically.
Hypertension
Recently, several investigators have reported evidence of an association between treatment-induced hypertension and tumor responsiveness suggesting that vascular responsiveness to therapy is a predictor of the drug’s efficacy against the cancer (40–42). This link may reflect the fact that NO mediates vascular responsiveness as well as angiogenesis, malignant transformation, and tumorigenesis (43). These observations lead to proposals that blood pressure elevation may serve as a biomarker for efficacious VEGF signaling inhibition (15) despite the observations that neither tumor expression of VEGF nor VEGFR2, nor plasma levels of VEGF have proven useful predictors of treatment outcome (42, 44).
Our study found an average increase of mean arterial pressure of 11 mm Hg. Such changes are significant and warrant clinical attention. To illustrate, the cholesteryl ester transfer protein (CETP) inhibitor torcetrapib, increased blood pressure by 5.4 mm Hg and was associated with a 58% increase in all-cause mortality despite marked reductions in low density lipoprotein and increases in high density lipoprotein (45). We surmise that vandetanib increased blood pressure by decreasing the constitutive production of NO, manifesting as a reduction in nitrite levels in our subjects. These findings are similar to other RTKIs. We cannot exclude the possibility of an increase in the production of an endogenous vasoconstrictor, however, in patients treated with the VEGFR2 inhibitor sorafenib, despite a mean increase in blood pressure of 13 mm Hg, there was no change in catecholamines, endothelin I, urotensin II, renin, and aldosterone levels (15). Additional agent-specific mechanisms are likely present and require further study to elucidate.
Additional mechanisms of hypertension need to be considered. Vascular rarefaction has been demonstrated in response to VEGF antagonism (37) and may contribute to the cause or effects of induced hypertension. Recently, Machnik and colleagues demonstrated that inhibition of VEGFR-3 (a described property of vandetanib) augments interstitial hypertonic volume retention, decreases eNOS expression, and increases blood pressure (4). Interestingly, in this experiment, inhibition of VEGF-C mediated activation of VEGFR-3 was associated with a decrease in eNOS expression. Vandetanib is also an EGFR antagonist and RET inhibitor, but blockade of these receptors has not been associated with hypertension (46). Both VEGF and eNOS contribute importantly to natriuresis (47), so the effect of renal VEGFR-2 inhibition may also mediate changes in blood pressure. Finally, the role of the metronomic therapy (methotrexate and cyclophosphamide) remains unclear. Neither are commonly associated with hypertension, but we can not exclude any interaction. The role of these and other mechanisms need further study.
Limitations
This study would have been strengthened by inclusion of a control group, however in the setting of this phase 1 trial in advanced breast cancer, randomization and the use of placebo weren’t possible. Whether the concomitant therapies, methotrexate and cyclophosphamide, contributed to vascular function remains an open question, but they have not demonstrated this tendency in past studies of subjects with rheumatological disease (48–52). Diet is an important contributor to levels of NOx. A change in diet may have participated in changing the systemic level of NOx, however, we believe the trend noted between increasing blood pressure and decreasing NOx suggests that these two changes were related.
Perspectives
Treatment with the VEGFR2 RTK inhibitor vandetanib in combination with metronomic chemotherapy increased blood pressure, decreased constitutive NO production, and decreased conduit artery resting diameter. In vitro, vandetanib decreased Akt phosphorylation and NO production, yet increased eNOS membrane content. Further studies will be needed to elucidate the specific mechanisms underlying the vascular responses to different RTK inhibitors, the effect of specific tyrosine moiety inhibition on endothelial cell signaling, and the clinical sequelae of hypertension induced by these medications.
Acknowledgments
Sources of Funding
This research was supported by a grant from the Investigator-Sponsored Study Program of AstraZeneca and the American Diabetes Association (ADA 1-06-CD-01).
Abbreviations
- FMD
flow-mediated vasodilation
- RTKIs
receptor tyrosine kinase inhibitors
- VEGF
vascular endothelial growth factor
- NO
nitric oxide
References
- 1.Gasparini G. Angiogenesis in breast cancer: role in biology, tumor progression, and prognosis. In: Bowcock A, editor. Breast Cancer: Molecular Genetics, Pathogenesis and Therapeutics. Totowa, NJ: Humana Press; 1999. pp. 347–371. [Google Scholar]
- 2.Miller KD, Trigo JM, Wheeler C, Barge A, Rowbottom J, Sledge G, et al. A multicenter phase II trial of ZD6474, a vascular endothelial growth factor receptor-2 and epidermal growth factor receptor tyrosine kinase inhibitor, in patients with previously treated metastatic breast cancer. Clin Cancer Res. 2005;11(9):3369–3376. doi: 10.1158/1078-0432.CCR-04-1923. [DOI] [PubMed] [Google Scholar]
- 3.Kerbel RS. Tumor angiogenesis. The New England journal of medicine. 2008;358(19):2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 5.Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26(11):1810–1816. doi: 10.1200/JCO.2007.14.5375. [DOI] [PubMed] [Google Scholar]
- 6.Burstein HJ, Spigel D, Kindsvogel K, Parker LM, Bunnell CA, Partridge AH, et al. Metronomic chemotherapy with and without bevacizumab for advanced breast cancer: a randomized phase II study. Breast Cancer Res Treat. 2005:Abstract 4. [Google Scholar]
- 7.Colleoni M, Rocca A, Sandri MT, Zorzino L, Masci G, Nole F, et al. Low-dose oral methotrexate and cyclophosphamide in metastatic breast cancer: antitumor activity and correlation with vascular endothelial growth factor levels. Ann Oncol. 2002;13(1):73–80. doi: 10.1093/annonc/mdf013. [DOI] [PubMed] [Google Scholar]
- 8.Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. The New England journal of medicine. 2003;349(5):427–434. doi: 10.1056/NEJMoa021491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. The New England journal of medicine. 2007;357(26):2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 10.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. The New England journal of medicine. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 11.Herbst RS, Johnson DH, Mininberg E, Carbone DP, Henderson T, Kim ES, et al. Phase I/II trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the HER-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23(11):2544–2555. doi: 10.1200/JCO.2005.02.477. [DOI] [PubMed] [Google Scholar]
- 12.Grothey A, Sugrue MM, Purdie DM, Dong W, Sargent D, Hedrick E, et al. Bevacizumab beyond first progression is associated with prolonged overall survival in metastatic colorectal cancer: results from a large observational cohort study (BRiTE) J Clin Oncol. 2008;26(33):5326–5334. doi: 10.1200/JCO.2008.16.3212. [DOI] [PubMed] [Google Scholar]
- 13.Escudier B, Szczylik C, Eisen T, Stadler WM, Schwartz B, Shan M, et al. Randomized Phase III trial of the Raf kinase and VEGFR inhibitor sorafenib (BAY 43-9006) in patients with advanced renal cell carcinoma (RCC) J Clin Oncol. 2005;23(16S):4510. [Google Scholar]
- 14.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Phase III randomized trial of sunitinib malate (SU11248) versus interferon-alfa (IFN-α) as first-line systemic therapy for patients with metastatic renal cell carcinoma (mRCC) J Clin Oncol. 2006;24(18S):LBA3. [Google Scholar]
- 15.Veronese ML, Mosenkis A, Flaherty KT, Gallagher M, Stevenson JP, Townsend RR, et al. Mechanisms of hypertension associated with BAY 43-9006. J Clin Oncol. 2006;24(9):1363–1369. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- 16.Wu S, Chen JJ, Kudelka A, Lu J, Zhu X. Incidence and risk of hypertension with sorafenib in patients with cancer: a systematic review and meta-analysis. Lancet Oncol. 2008;9(2):117–123. doi: 10.1016/S1470-2045(08)70003-2. [DOI] [PubMed] [Google Scholar]
- 17.Bhargava P. VEGF kinase inhibitors: how do they cause hypertension? Am J Physiol Regul Integr Comp Physiol. 2009;297(1):R1–5. doi: 10.1152/ajpregu.90502.2008. [DOI] [PubMed] [Google Scholar]
- 18.Humphreys BD, Atkins MB. Rapid development of hypertension by sorafenib: toxicity or target? Clin Cancer Res. 2009;15(19):5947–5949. doi: 10.1158/1078-0432.CCR-09-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hood JD, Meininger CJ, Ziche M, Granger HJ. VEGF upregulates ecNOS message, protein, and NO production in human endothelial cells. The American journal of physiology. 1998;274(3 Pt 2):H1054–1058. doi: 10.1152/ajpheart.1998.274.3.H1054. [DOI] [PubMed] [Google Scholar]
- 20.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nature reviews. 2006;7(5):359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 21.Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. 2009;54(3):652–658. doi: 10.1161/HYPERTENSIONAHA.109.129973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dahring TK, Lu GH, Hamby JM, Batley BL, Kraker AJ, Panek RL. Inhibition of growth factor-mediated tyrosine phosphorylation in vascular smooth muscle by PD 089828, a new synthetic protein tyrosine kinase inhibitor. J Pharmacol Exp Ther. 1997;281(3):1446–1456. [PubMed] [Google Scholar]
- 23.Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res. 2002;62(16):4645–4655. [PubMed] [Google Scholar]
- 24.Carlomagno F, Vitagliano D, Guida T, Ciardiello F, Tortora G, Vecchio G, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002;62(24):7284–7290. [PubMed] [Google Scholar]
- 25.Corretti MC, Anderson TJ, Benjamin EJ, Celermajer D, Charbonneau F, Creager MA, et al. Guidelines for the ultrasound assessment of endothelial-dependent flow-mediated vasodilation of the brachial artery: a report of the International Brachial Artery Reactivity Task Force. J Am Coll Cardiol. 2002;39(2):257–265. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 26.Lieberman EH, Gerhard MD, Uehata A, Selwyn AP, Ganz P, Yeung AC, et al. Flow-induced vasodilation of the human brachial artery is impaired in patients <40 years of age with coronary artery disease. The American journal of cardiology. 1996;78(11):1210–1214. doi: 10.1016/s0002-9149(96)00597-8. [DOI] [PubMed] [Google Scholar]
- 27.Owens CD, Wake N, Conte MS, Gerhard-Herman M, Beckman JA. In vivo human lower extremity saphenous vein bypass grafts manifest flow mediated vasodilation. J Vasc Surg. 2009 doi: 10.1016/j.jvs.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uehata A, Lieberman EH, Gerhard MD, Anderson TJ, Ganz P, Polak JF, et al. Noninvasive assessment of endothelium-dependent flow-mediated dilation of the brachial artery. Vascular medicine (London, England) 1997;2(2):87–92. doi: 10.1177/1358863X9700200203. [DOI] [PubMed] [Google Scholar]
- 29.Dallabrida SM, Ismail N, Oberle JR, Himes BE, Rupnick MA. Angiopoietin-1 promotes cardiac and skeletal myocyte survival through integrins. Circulation research. 2005;96(4):e8–24. doi: 10.1161/01.RES.0000158285.57191.60. [DOI] [PubMed] [Google Scholar]
- 30.Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO, et al. Predominant role of endothelial nitric oxide synthase in vascular endothelial growth factor-induced angiogenesis and vascular permeability. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(5):2604–2609. doi: 10.1073/pnas.041359198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B, Ogasawara AK, Yang R, Wei W, He GW, Zioncheck TF, et al. KDR (VEGF receptor 2) is the major mediator for the hypotensive effect of VEGF. Hypertension. 2002;39(6):1095–1100. doi: 10.1161/01.hyp.0000018588.56950.7a. [DOI] [PubMed] [Google Scholar]
- 32.Gelinas DS, Bernatchez PN, Rollin S, Bazan NG, Sirois MG. Immediate and delayed VEGF-mediated NO synthesis in endothelial cells: role of PI3K, PKC and PLC pathways. British journal of pharmacology. 2002;137(7):1021–1030. doi: 10.1038/sj.bjp.0704956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Henry TD, Annex BH, McKendall GR, Azrin MA, Lopez JJ, Giordano FJ, et al. The VIVA trial: Vascular endothelial growth factor in Ischemia for Vascular Angiogenesis. Circulation. 2003;107(10):1359–1365. doi: 10.1161/01.cir.0000061911.47710.8a. [DOI] [PubMed] [Google Scholar]
- 34.Tanimoto T, Jin ZG, Berk BC. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS) J Biol Chem. 2002;277(45):42997–43001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- 35.Robinson ES, Khankin EV, Choueiri TK, Dhawan MS, Rogers MJ, Karumanchi SA, et al. Suppression of the Nitric Oxide Pathway in Metastatic Renal Cell Carcinoma Patients Receiving Vascular Endothelial Growth Factor-Signaling Inhibitors. Hypertension. 2010 doi: 10.1161/HYPERTENSIONAHA.110.160481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Echeverria V, Burgess S, Gamble-George J, Zeitlin R, Lin X, Cao C, et al. Sorafenib inhibits nuclear factor kappa B, decreases inducible nitric oxide synthase and cyclooxygenase-2 expression, and restores working memory in APPswe mice. Neuroscience. 2009;162(4):1220–1231. doi: 10.1016/j.neuroscience.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 37.Steeghs N, Gelderblom H, Roodt JO, Christensen O, Rajagopalan P, Hovens M, et al. Hypertension and rarefaction during treatment with telatinib, a small molecule angiogenesis inhibitor. Clin Cancer Res. 2008;14(11):3470–3476. doi: 10.1158/1078-0432.CCR-07-5050. [DOI] [PubMed] [Google Scholar]
- 38.Chandra A, Angle N. Vascular endothelial growth factor stimulates a novel calcium-signaling pathway in vascular smooth muscle cells. Surgery. 2005;138(4):780–787. doi: 10.1016/j.surg.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Sachinidis A, Skach RA, Seul C, Ko Y, Hescheler J, Ahn HY, et al. Inhibition of the PDGF beta-receptor tyrosine phosphorylation and its downstream intracellular signal transduction pathway in rat and human vascular smooth muscle cells by different catechins. Faseb J. 2002;16(8):893–895. doi: 10.1096/fj.01-0799fje. [DOI] [PubMed] [Google Scholar]
- 40.Rixe O, Billemont B, Izzedine H. Hypertension as a predictive factor of Sunitinib activity. Ann Oncol. 2007;18(6):1117. doi: 10.1093/annonc/mdm184. [DOI] [PubMed] [Google Scholar]
- 41.Scartozzi M, Galizia E, Chiorrini S, Giampieri R, Berardi R, Pierantoni C, et al. Arterial hypertension correlates with clinical outcome in colorectal cancer patients treated with first-line bevacizumab. Ann Oncol. 2009;20(2):227–230. doi: 10.1093/annonc/mdn637. [DOI] [PubMed] [Google Scholar]
- 42.Schneider BP, Wang M, Radovich M, Sledge GW, Badve S, Thor A, et al. Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J Clin Oncol. 2008;26(28):4672–4678. doi: 10.1200/JCO.2008.16.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fukumura D, Kashiwagi S, Jain RK. The role of nitric oxide in tumour progression. Nat Rev Cancer. 2006;6(7):521–534. doi: 10.1038/nrc1910. [DOI] [PubMed] [Google Scholar]
- 44.Garcia AA, Hirte H, Fleming G, Yang D, Tsao-Wei DD, Roman L, et al. Phase II clinical trial of bevacizumab and low-dose metronomic oral cyclophosphamide in recurrent ovarian cancer: a trial of the California, Chicago, and Princess Margaret Hospital phase II consortia. J Clin Oncol. 2008;26(1):76–82. doi: 10.1200/JCO.2007.12.1939. [DOI] [PubMed] [Google Scholar]
- 45.Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, et al. Effects of torcetrapib in patients at high risk for coronary events. The New England journal of medicine. 2007;357(21):2109–2122. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 46.Eng C. Toxic effects and their management: daily clinical challenges in the treatment of colorectal cancer. Nat Rev Clin Oncol. 2009;6(4):207–218. doi: 10.1038/nrclinonc.2009.16. [DOI] [PubMed] [Google Scholar]
- 47.Gu JW, Manning RD, Jr, Young E, Shparago M, Sartin B, Bailey AP. Vascular endothelial growth factor receptor inhibitor enhances dietary salt-induced hypertension in Sprague-Dawley rats. Am J Physiol Regul Integr Comp Physiol. 2009;297(1):R142–148. doi: 10.1152/ajpregu.90972.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bosello S, Santoliquido A, Zoli A, Di Campli C, Flore R, Tondi P, et al. TNF-alpha blockade induces a reversible but transient effect on endothelial dysfunction in patients with long-standing severe rheumatoid arthritis. Clin Rheumatol. 2008;27(7):833–839. doi: 10.1007/s10067-007-0803-y. [DOI] [PubMed] [Google Scholar]
- 49.Kappers MH, van Esch JH, Sluiter W, Sleijfer S, Danser AH, van den Meiracker AH. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 56(4):675–681. doi: 10.1161/HYPERTENSIONAHA.109.149690. [DOI] [PubMed] [Google Scholar]
- 50.Banerjee S, Mehta S, Haque I, Sengupta K, Dhar K, Kambhampati S, et al. VEGF-A165 induces human aortic smooth muscle cell migration by activating neuropilin-1-VEGFR1-PI3K axis. Biochemistry. 2008;47(11):3345–3351. doi: 10.1021/bi8000352. [DOI] [PubMed] [Google Scholar]
- 51.Di Meglio F, Nurzynska D, Castaldo C, Arcucci A, De Santo L, de Feo M, et al. In vitro cultured progenitors and precursors of cardiac cell lineages from human normal and post-ischemic hearts. Eur J Histochem. 2007;51(4):275–282. [PubMed] [Google Scholar]
- 52.Kattman SJ, Huber TL, Keller GM. Multipotent flk-1+ cardiovascular progenitor cells give rise to the cardiomyocyte, endothelial, and vascular smooth muscle lineages. Dev Cell. 2006;11(5):723–732. doi: 10.1016/j.devcel.2006.10.002. [DOI] [PubMed] [Google Scholar]