

Abstract
RecQ helicases are an important family of genome surveillance proteins conserved from bacteria to humans. Each of the five human RecQ helicases plays critical roles in genome maintenance and stability, and the RecQ protein family members are often referred to as guardians of the genome. The importance of these proteins in cellular homeostasis is underscored by the fact that defects in BLM, WRN, and RECQL4 are linked to distinct heritable human disease syndromes. Each human RecQ helicase has a unique set of protein-interacting partners, and these interactions dictate its specialized functions in genome maintenance, including DNA repair, recombination, replication, and transcription. Human RecQ helicases also interact with each other, and these interactions have significant impact on enzyme function. Future research goals in this field include a better understanding of the division of labor among the human RecQ helicases and learning how human RecQ helicases collaborate and cooperate to enhance genome stability.
Keywords: RECQL1, BLM, WRN, RECQL4, RECQL5, genome stability
INTRODUCTION
Helicases are ubiquitous enzymes that play critical roles in DNA replication, recombination, repair, and transcription. A recent genome-wide analysis predicted 95 genes encoding distinct helicases in the human genome (1). Most mammals possess five RecQ helicases: RECQL1, BLM, WRN, RECQL4, and RECQL5. Defects in human BLM, WRN, or RECQL4 cause monogenic disease syndromes; all RecQ helicases play critical roles in genome maintenance and are often referred to as guardians of the genome (2, 3). Although bacteria and yeast were initially thought to express only one RecQ helicase, recent studies suggest that additional homologs are present (4–7).
Most members of the DNA helicase superfamily use energy derived from ATP hydrolysis to unwind double-stranded DNA (dsDNA), including complex DNA secondary structures, and many helicases also remodel protein–DNA complexes. RecQ helicases bind to and translocate along single-stranded DNA (ssDNA) in a 3′ → 5′ direction, and ssDNA stimulates their dsDNA-unwinding activity. RecQ helicases are structure-specific DNA-binding proteins. Some promote branch migration or disrupt Holliday junctions (HJs), and all human RecQ helicases catalyze ssDNA annealing [recently reviewed by Wu (8)].
RecQ helicases share three highly conserved protein domains (9), the core helicase domain (composed of HD1 and HD2), the RecQ C-terminal (RQC) domain, and the helicase and RNase D–like C-terminal (HRDC) domain (Figure 1a, b). In addition, they possess unique protein regions and motifs that direct subcellular localization (Figure 1a). Previously, it was thought that RECQL4 lacked the RQC domain, but a recent bioinformatics analysis suggests that it may have an RQC domain (10). RECQL5 lacks part of the RQC domain and, similar to RECQL1 and RECQL4, lacks the HRDC domain (11). Unique protein segments confer distinctive functions or properties to each homolog, including specific protein–protein interactions, oligomerization signals, and nuclear or mitochondrial localization (Figures 1a and 2).
Figure 1.

RecQ helicase protein family. (a) The domain structure of RecQ helicases from Escherichia coli, Saccharomyces cerevisiae, and Homo sapiens. Homologous domains are represented with solid-color boxes outlined with solid lines; the tentatively assigned zinc and RQC domains of RECQL4 are represented by rectangles outlined with dotted lines. (b) The helicase domains of human RECQL1, based on Protein Data Bank (PDB) identifier 2WWY (24). The structure includes two RecA-like domains, helicase domains 1 [HD1 (red )] and 2 [HD2 (blue)], and an ATP-binding cleft. The conserved helicase motifs are in yellow, and the aromatic loop is in pink. (c) The truncated RECQL1 bound to DNA is based on PDB 2WWY. The RQC domain is in purple, the β-hairpin is in green, and the DNA is in black. Abbreviation: aa, amino acid.
Figure 2.

Protein–protein interactions among the RecQ helicases. The vertical panel to the left lists six DNA metabolic pathways and the RecQ helicases that participate in those pathways. Each RecQ helicase is represented as a colored hexagon. The interacting proteins are shown next to each RecQ helicase. Proteins that interact with four or five RecQ helicases are represented by light blue ovals, and proteins that interact with two or more RecQ helicases are represented by white ovals. Abbreviations: BER, base excision repair; DSBR, double-strand break repair; mRNA, messenger RNA; Topo, topoisomerase.
This article presents a comprehensive review and discussion of recent literature on human RecQ helicases, emphasizing advances toward understanding the RecQ helicase mechanisms of action and the unique and/or redundant roles each human RecQ helicase plays in DNA repair, recombination, and replication. As discussed below, critical unresolved questions in the field are whether and how the unique and overlapping roles of the five human RecQ helicases are coordinated in DNA metabolism.
CONSERVED RecQ HELICASE DOMAINS
Core Helicase Domain
There are six distinct helicase protein superfamilies (SF1–6), and their members share conserved sequence motifs as well as structural and mechanistic features (12, 13). Human RecQ helicases belong to the SF2 helicase superfamily (see the conserved motifs in the helicase core domains in Figure 1b). The RecQ proteins display strong correlations between the conserved motifs and functional DNA helicase activity, such that they unwind a wide variety of DNA substrates, many of which resemble DNA repair intermediates. Table 1 summarizes the substrate specificities of Escherichia coli and human RecQ helicases.
Table 1.
Comparison between the relative helicase activity of Escherichia coli RecQ and that of the five human RecQ helicasesa






RecQ C-Terminal Domain
In most cases, the minimal helicase functional unit includes the helicase and RQC domains (14–16). The RQC structure was solved by X-ray crystallography; it features a zinc-binding motif, a helix-hairpin-helix, winged-helix (WH) domain, and a β-hairpin motif (17–19). The RQC domain specifically mediates binding to G quadruplex (G4) DNA (20) and stabilizes binding to other DNA structures. The purified WH domain from WRN (15, 19, 21, 22) and BLM (20) shows strong DNA-binding activity. The crystal structure of the RQC domain of WRN [Protein Data Bank (PDB) identifier 3AAF] shows that the WH domain makes contacts with the phosphate backbone on the 5′ ssDNA overhang (19). Consistent with these results, mutagenesis of conserved Arg987 and Arg993 in the WH of WRN inactivates DNA-binding and helicase activity (19, 23). In the RECQL1–DNA cocrystal (PDB 2WWY) (24), the WH domain binds dsDNA, and the β-hairpin motif binds near the junction between dsDNA and ssDNA (Figure 1c). The β-hairpin motif in the RQC domain of RECQL1 regulates dimer formation and couples ATPase activity to DNA unwinding (18, 25). Notably, oligomerization of RECQL1 regulates the opposing catalytic activities of helicase and strand annealing (SA) (see the sidebar titled Structure and Oligomeric Status of RecQ Helicases). Thus, the RQC domain directs binding to ssDNA–dsDNA junctions, and the β-hairpin couples ATP hydrolysis to DNA unwinding.
STRUCTURE AND OLIGOMERIC STATUS OF RecQ HELICASES.
The structure and oligomeric status of the RecQ helicases are exciting areas of analysis. RecQ helicases are ssDNA-stimulated ATPases; however, the mechanism for the stimulation is not well understood. These proteins contain a highly conserved sequence, the aromatic loop (24, 295), immediately following the Walker B motif (important for ATP hydrolysis), which is a true signature sequence for the family. The cocrystal structure shows that the aromatic loop lies in close proximity to DNA (Figure 1c), and mutagenesis of the loop in E. coli or human RECQL1 suggests this sequence regulates dimerization and that it may be the DNA-binding sensor that couples ATPase to DNA binding (25, 295). In general, helicase subunit oligomerization is another unanswered fundamental question. Interestingly, BLM and WRN form hexamers and trimers (57, 296), and RECQL1 monomers and dimers have been observed (reviewed in Reference 297). RECQL4 exists primarily as a dimer (298), and RECQL5 is a monomer (11). Recent studies suggest that the oligomeric status regulates the catalytic activities of RECQL1, allowing it to toggle between DNA unwinding, DNA strand annealing, and HJ branch migration/disruption (299). Thus, oligomerization represents a mechanism whereby the helicases could potentially be differentially regulated.
Helicase and RNase D–Like C-Terminal Domain
The HRDC domain is found in WRN and BLM but not in other human RecQ enzymes, suggesting that it plays a specialized role. Consistent with this finding, the HRDC domain in yeast Sgs1 and E. coli RecQ promotes stable DNA binding (26, 27) but is dispensable for ATPase and helicase activities (27, 28). In WRN and BLM, the HRDC domain plays a role in recruitment to laser-induced dsDNA breaks (DSBs) (29, 30), and methyl methanesulfonate– and mitomycin C–induced DNA damage (31). Thus, the HRDC domain promotes the localization of WRN and BLM to specific DNA lesions.
THE RecQ PROTEIN FAMILY
RecQ homologs interact with a wide variety of protein partners and play multiple important roles in DNA metabolism (Figure 2). Some partners are unique, but many are common to more than one RecQ protein. Defects in human BLM, WRN, and RECQL4 cause rare recessive autosomal diseases for which there are currently no effective therapies. Mutations in BLM (15q26.1) cause Bloom syndrome (BS) [Online Mendelian Inheritance in Man (OMIM) code number 604610]; mutations in WRN (8p12) cause Werner syndrome (WS) (OMIM 604611); and mutations in RECQL4 (8q24.3) cause Rothmund–Thomson syndrome (RTS) (OMIM 268400), RA-PADILINO syndrome (OMIM 266280), and Baller–Gerold syndrome (BGS) (OMIM 218600). Each syndrome has unique clinical and developmental patterns, suggesting that each human RecQ helicase has some specialized biological function, even though all five human RecQ helicases are thought to widely contribute to genomic stability.
RECQL1
RECQL1 is the smallest (72 kDa) and most abundant human RecQ helicase (recently reviewed in Reference 36). Immunohistochemical studies show that human RECQL1 is widely expressed in the nucleus (32) and that messenger RNA encoding murine Recql1 is ubiquitous, with highest expression in testis (33). Mice lacking Recql1 are phenotypically normal; however, mouse embryonic fibroblasts demonstrate chromosome instability, including aneuploidy, spontaneous chromosomal breakage, frequent translocations, hypersensitivity to ionizing radiation, DNA damage, and elevated spontaneous sister chromatid exchanges (SCEs) (33). Many diverse studies show that defects in or depletion of RECQL1 impairs cell growth, causes genome instability, or induces mitotic catastrophe (33–37). RECQL1 is proposed to have a unique role during replication origin firing and nascent DNA synthesis (38), and it is critical for replication fork restart (39). RECQL1 is highly expressed in a variety of human solid tumors (40), but there are no known diseases associated with defects in RECQL1.
BLM
Expression of BLM increases in the S and G2/M phases (41), and it is localized to promyelocytic leukemia bodies and telomeres in the nucleus (42, 43). BLM is rapidly recruited to damaged DNA (42, 44). It is an essential gene in the mouse (45–47), given that defects in or depletion of murine Blm is linked to an increase in apoptosis during embryogenesis and to severe anemia, chromosomal instability, and a high incidence of cancer in adult mice (45, 46, 48, 49). Cell lines derived from Blm-defective mice and patient-derived human BS cells demonstrate a hyperrecombination phenotype.
The most prominent clinical feature of BS is dwarfism, which is due to pre- and postnatal growth retardation (Supplemental Table 1; follow the Supplemental Material link from the Annual Reviews home page at http://www.annualreviews.org) (50). BS patients prematurely develop cancer, diabetes, and chronic progressive lung disease. The types and frequencies of cancers observed in BS patients are similar to those in the general population; however, BS patients develop cancer early, and they develop multiple primary tumors (51). Cancer is the leading cause of death, often in childhood, among BS patients (51).
WRN
In undamaged human cells, WRN localizes predominantly to the nucleolus, but after DNA damage, it rapidly mobilizes to other nuclear regions (29, 52–54). Immunochemical staining of S-phase cells reveals a punctate pan-nuclear distribution of WRN, suggesting that WRN may be associated with nascent replicating DNA and telomeres (55).
WRN is unique among RecQ helicases in possessing an N-terminal 3′ → 5′ exonuclease (56), similar to E. coli DnaQ-like replication–associated bacterial exonucleases (57). Forked dsDNA and bubble structures with 3′-recessed ends are the preferred DNA substrates of the WRN exonuclease (58–60), which is unable to bypass certain oxidative and bulky DNA lesions (61–65). Although the in vivo role of the WRN exonuclease in DNA metabolism is not yet known, it is possible that the WRN exonuclease proofreads repair tracts synthesized by translesion DNA polymerases (see the section titled RecQ Helicases in DNA Replication). The collaboration between the WRN exonuclease and helicase is another important area for future research.
Wrn-knockout mice have no overt pathology and fail to recapitulate features of clinical WS (66, 67). However, other Wrn-null mice are insulin resistant and prone to diabetes and obesity when fed a high-fat and sugary diet (68), and telomere shortening and premature aging are observed in double-mutant mice with null alleles of Wrn and Terc, the RNA components of telomerase (69). The latter result suggests that long telomeres in wild-type mice might mask the effect of the Wrn-null mutation on genome stability.
Recently, the clinical features of WS patients were reviewed in detail and therefore are not discussed in detail here, except to note that cancer and atherosclerosis are the leading causes of death, with susceptibility to specific types of rare cancer as well as multiple primary or sequential neoplasms (70, 71). The clinical features of BS, WS, and RECQL4 syndrome are summarized in Supplemental Table 1.
RECQL4
RECQL4 partitions between the nucleus and cytosol in a manner that varies in different cell types and tissues (72). RECQL4, WRN, and BLM are thought to play roles in telomeric DNA maintenance, but RECQL4 is unique in localizing to mitochondria (73–75). RECQL4 expression peaks during S phase and is highest in the thymus and testis (41).
Three mouse models for Recql4 have been characterized in which specific exons were deleted as follows: Δ exons 5–8, Δ exons 9–13 (the entire helicase domain), and Δ exon 13 (the last exon in the helicase domain). Mice with deleted Δ exons 5–8 die as embryos (76); 95% of the mice with deleted Δ exon 13 die perinatally (77), and the “escapers” fail to thrive. Mice with deleted Δ exons 9–13 have palate and limb defects, as well as poikiloderma-like features, and are susceptible to cancer in the absence of APC (the tumor-suppressor adenomatosis polyposis coli gene) (78). Recql4-deficient mice lacking Δ exons 9–13 have a normal life span.
Mutations in RECQL4 cause type II RTS (79), a disease characterized by hypo- and hyperpigmentation, punctate atrophy, and telangiectasia (three symptoms known collectively as poikiloderma), as well as congenital skeletal abnormalities (reviewed in Reference 80). The leading cause of death for patients with RECQL4 mutations is cancer (L.L. Wang, personal communication), whereas life expectancy in cancer-free RTS patients is normal (80). The characteristic clinical feature of BGS is congenital coronal craniosynostosis (81). Poikiloderma is not observed in RECQL4-defective patients with RAPADILINO syndrome, but these patients are susceptible to rare cancers (82). RTS is more prevalent than BGS or RAPADILINO syndrome. See Supplemental Table 1 for a comparison between the RECQL4-related disorders.
Mutations causing RTS, BGS, and RAPADILINO syndrome are found in all RECQL4 protein domains. However, of the missense mutations available for analysis, those that compromise RECQL4 structurally segregate with RTS, whereas those that impair helicase activity segregate with RAPADILINO syndrome (83, 84). Additional mutational analysis of RECQL4 is warranted. For further information about RECQL4’s protein interaction partners, see Figure 2 and Reference 85.
RECQL5
There are three alternatively spliced forms of human RECQL5: RECQL5α, RECQL5β, and RECQL5γ (86). RECQL5β (referred to here as RECQL5) is the most abundant isoform. RECQL5 is ubiquitously expressed in a cell-cycle phase– and cell type–independent manner, contains a nuclear localization signal, and is an ATP-dependent DNA helicase (Figure 1a) (11, 41, 86, 87). RECQL5’s protein interaction partners were described in a recent review (88), and some are shown in Figure 2. Most notably, RECQL5 binds both initiating and elongating forms of RNA polymerase II and inhibits transcription (89, 90).
RQC-5-deficient Caenorhabditis elegans develop normally but show a 37% reduction in life span and increased sensitivity to ionizing radiation (91). Recql5-knockout mice also develop normally but display cancer susceptibility (92). Cancer-free Recql5-deficient mice have a normal life span. Recql5-deficient mouse embryonic fibroblasts accumulate endogenous DNA damage, possibly due to faulty DSB repair by homologous recombination (HR), and display dramatically increased frequencies of gross chromosomal aberrations after exposure to camptothecin (CPT). Thus, model organisms lacking RECQL5 confirm that RECQL5 is not essential for embryonic development but demonstrate that it is important for tumor suppression, and suggest that the functions of RECQL5 are evolutionarily conserved.
RecQ HELICASES IN DNA REPAIR
RecQ helicases demonstrate specific and preferred binding to DNA substrates that resemble DNA repair intermediates (Table 1), suggesting that they play a role in DNA repair. Consistent with this finding, RecQ helicases interact with and modulate the activity of proteins that play critical roles in DNA repair. Studies in mouse models confirm this putative role and suggest additional roles in recombinational DNA repair and DNA replication. RecQ helicases are currently thought to influence base excision repair (BER) directly and indirectly play a significant role in DSB repair, but have a less clear influence on nucleotide excision repair (NER) and mismatch repair. These roles are discussed at length below, with an emphasis on how each RecQ impinges on distinct DNA repair pathways in unique and/or overlapping ways. Figure 3 summarizes this discussion.
Figure 3.

RecQ helicases in base excision repair (BER) and double-strand break repair (DSBR). (a) Summary of the BER pathway. Interactions between RecQ helicases and BER proteins are depicted by a green arrow (for activation) or a red bar (for inhibition). Asterisks after PARP1 and XRCC1 indicate that these genes are specifically downregulated by RECQL5 loss. The brown RecQ hexagon indicates that multiple RecQs interact with these proteins. (b) Summary of DSBR pathways. (i ) Nonhomologous end joining (NHEJ). (ii ) Homologous recombination (HR). (iii ) Alternative (Alt)-NHEJ. The involvement of RecQ helicases in DSBR is depicted. Each RecQ helicase is represented as a colored hexagon. The colored hexagons are consistent with the coloring in Figure 2. Other key proteins are shown as white ovals. DNA molecules are shown as blue/red or light blue/red duplexes. Dashed lines denote nascent DNA synthesis. Abbreviations: dHJ, double Holliday junction; dRP, deoxyribose phosphate; IR, ionizing radiation; MPG, methylpurine DNA glycosylase; P, 3′-phosphate; PCNA, proliferating cell nuclear antigen; Pol, DNA polymerase; RFC, replication factor C; SDSA, synthesis-dependent strand annealing; Topo, topoisomerase; UA, 3′-α,β unsaturated aldehyde; UNG, uracil DNA glycosylase.
Base Excision Repair
BER removes oxidative, abasic, and other monofunctional base modifications from DNA. Base lesions are recognized by a glycosylase that removes the damaged nitrogenous base while leaving the sugar–phosphate backbone intact, creating an apurinic/apyrimidinic (AP) site. Mammalian cells express multiple DNA glycosylases with overlapping DNA lesion specificity. AP endonuclease 1 (APE1) is the next enzyme in the BER pathway; it cleaves AP sites, producing a single-strand break intermediate, which can be utilized by DNA polymerase β (Pol β) for DNA repair synthesis, and is followed by ligation of nicked DNA by a DNA ligase (93).
WRN, but not other human RecQ helicases, strongly stimulates NEIL1 glycosylase (94), which incises formamidopyrimidines, 5-hydroxyuracil, and to a lesser extent, 8-oxoG. APE1 is inhibited by WRN (95) but stimulated by RECQL4 (96) in vitro. APE1 is upregulated in RTS cells (96) and is overexpressed in some sarcomas, cancers that are common to RTS and WS (97). Pol β is significantly stimulated by helicase-proficient, but not by helicase-defective, WRN (98). Pol β strand displacement synthesis is stimulated by RECQL4 (96) but not by RECQL5 (99). DNA Pol β lacks 3′ proofreading exonuclease activity, so when DNA Pol β and WRN work in a coordinated manner, WRN exonuclease activity can play a role in correcting DNA synthesis errors, contribute to postreplication repair, and increase DNA synthesis fidelity during BER (100).
WRN physically and functionally interacts with proteins involved in short-patch BER, in which one base is replaced, and long-patch BER, in which up to eight bases are replaced (Figure 3a). The replicative protein flap endonuclease 1 (FEN1) plays a role in long-patch BER and interacts with most of the human RecQs (Figure 2) (96, 99, 101–103). Cooperation between FEN1 and the RecQ helicases, and/or stimulation of FEN1 by one of the enzymes, may also promote postreplication DNA repair (104).
WRN, RECQL1, and RECQL4 interact with and are modulated by poly(ADP-ribose) polymerase 1 (PARP1). PARP1 adds poly(ADP-ribose) moieties to chromatin-binding proteins, thereby modulating chromatin structure and function (105, 106). WRN-deficient cells fail to activate PARP1 in response to oxidative and alkylation DNA damage, indicating that the WRN–PARP1 interaction is biologically relevant and significant (107). However, PARP1 is hyperactivated in RECQL1-depleted cells exposed to oxidative stress and in BLM-, WRN-, and RECQL5-depleted cells that are not exposed to exogenous stress (108–110). Interestingly, WRN and RECQL1 bind to and are inhibited by poly(ADP-ribose) (39, 108, 111). The C-terminal region of RECQL4 is a PARP1 substrate, and PARP1 inhibitors alter the cellular localization of RECQL4 (112). The complex interactions between PARP1 and RecQ helicases warrant further study, especially in light of the critical role played by PARP1 in cancer susceptibility and recent reports of PARP1 as a cancer chemotherapeutic target (113).
RECQL5, but not the other RecQ helicases, is recruited to laser-induced ssDNA breaks in vivo, and RECQL5-depleted cells are sensitive to and accumulate oxidative DNA damage (110). RECQL5 does not stimulate APE1 or Pol β (99), but it may positively regulate transcription of PARP1 and XRCC1 (110). Thus, RecQ helicases may modulate BER indirectly by regulating transcription of BER-related genes (96, 110).
Nucleotide Excision Repair
NER removes bulky lesions from DNA, such as UV-induced pyrimidine dimers and carcinogen adducts (for a review, see Reference 114). Limited evidence suggests that RecQ helicases might play a role in NER. For example, the NER protein XPG (xeroderma pigmentosum complementation group G) stimulates WRN helicase (115), and the XPA protein interacts with RECQL4 (116). However, RecQ helicase–deficient cells are not hypersensitive to UV irradiation, and there is no direct evidence that the RecQ helicases alter NER functionally in vivo. Thus, the significance of the observed interactions remains unclear.
DNA Double-Strand Break Repair
DNA DSBs can cause transient or permanent cell-cycle arrest, mutagenesis, gross chromosomal rearrangements, and ultimately cell death or tumorigenesis. Human cells can repair DSBs by nonhomologous end joining (NHEJ), alternative nonhomologous end joining (Alt-NHEJ), or HR (Figure 3b) (117, 118). HR-dependent DSB repair is based on a sister chromatid template, is error free, and occurs only during the late S and G2 phases (119). NHEJ and Alt-NHEJ occur throughout the cell cycle and are error prone (reviewed in Reference 117). Alt-NHEJ is active mainly when NHEJ is impaired (117). As described below, all five human RecQ helicases play putative roles in one or more subpathways of DSB repair.
Nonhomologous end joining
Nonproliferating human cells repair DSBs by NHEJ in a reaction that requires 53BP1, the Ku70/Ku80 heterodimer (Ku), DNA-PKcs, and XLF/XRCC4/LIG4 (120, 121). When necessary, nonligatable ends are processed in an intermediate step by Artemis or a DNA polymerase–associated exonuclease. 53BP1 promotes NHEJ by suppressing DSB 5′-end resection (122, 123). Figures 2 and 3b summarize the interactions between RecQ helicases and NHEJ proteins. For example, WRN interacts with Ku and is a substrate of the DNA-PKcs kinase (124–127). Ku has no effect on WRN helicase or ATPase activities but strongly stimulates the WRN exonuclease (124). In contrast, DNA-PKcs stimulates the WRN helicase but not the WRN exonuclease (128); however, the WRN exonuclease is also stimulated by XRCC4/LIG4 (129). WS cells are mildly sensitive to ionizing radiation–induced DSBs (126), but in the context of in vitro DSB repair, they are competent in ligating linear plasmid DNA (130). However, WS cells exhibit elevated frequencies of deletions in chromosomal DNA (131, 132), and likewise, more deletion mutations are recovered from in vitro plasmid ligation assays (57, 133).
RECQL1 may also play a role in NHEJ (134) because Recql1-deficient mice (33) and human cells (34) are defective in DSB repair. RECQL1 interacts with Ku, and extracts from RECQL1-null cells display wild-type levels of plasmid end-joining activity and reduced Ku-DNA-binding activity (134). Whether WRN and RECQL1 play compensatory or redundant roles in NHEJ at the Ku step is unknown.
Alternative nonhomologous end joining
The precise mechanism of Alt-NHEJ is poorly understood; however, there is often microhomology between the DSB break point and the DNA repair template (117). PARP1 may act as a DNA damage recognition protein during Alt-NHEJ (135, 136), followed by end resection by MRE11, CtIP, and EXO1 (137–140) and ligation by LigIII/XRCC1 (135, 136, 141). WRN, BLM, and RECQL1 interact with EXO1 (136, 142–145) and MRE11/RAD50/NBS1 (MRN) (146–148). WRN also interacts with LigIIIα (149). Although preliminary, these findings are consistent with the possibility that RecQ helicases modulate Alt-NHEJ.
When 53BP1-defective human cells were treated with a small-molecule WRN inhibitor or a hypomorphic Blm allele (Blm3/3) was expressed, an I-SceI intrachromosomal break was repaired normally by Alt-NHEJ, but the efficiency of B cell class-switch recombination, which requires Alt-NHEJ, increased (140, 150). To explain these results, the investigators proposed that 53BP1 and phosphorylated histone H2AX (γ-H2AX) compete with proresection enzymes, WRN and BLM, for DSBs and that the circumstances that limit DNA resection in B cells promote Alt-NHEJ and class-switch recombination (140).
Homologous recombination–dependent double-strand break repair
HR is a conserved cellular process that occurs in the late S and G2 phases of the cell cycle. It repairs DNA DSBs when a homologous template is available. HR is involved in DSB repair at stalled replication forks during S phase, promotes repair at DNA lesions during mitosis, and can promote correct chromosomal pairing or exchange during meiosis. Some of the proteins required for HR-mediated DSB repair include CtIP, MRN, RPA, DNA2, EXO1, and RAD51 (118). Notably, RPA, a human ssDNA binding protein that is essential for DNA replication and repair, interacts with all human RecQ proteins and stimulates their helicase functions (Figures 2 and 3b) (11, 151–154).
DNA end resection is an exonucleolytic process that generates ssDNA protruding tails at a DNA DSB. In human cells, the first step toward resection is the binding of CtIP and MRN at the DSB (148, 155). CtIP stimulates the MRN exonuclease, and MRN recruits two additional nucleases, EXO1 and DNA2. RPA also plays a role by stimulating DNA unwinding using BLM and enforcing 5′–3′ resection polarity by DNA2 (148). Alternatively, EXO1, instead of MRN, can catalyze excision at the DSB. BLM increases the affinity of EXO1 for DNA ends independently of its helicase, whereas MRN recruits and enhances the processivity of EXO1 (148). In vitro, only BLM, but not WRN, RECQL4, or RECQL5, stimulates DNA end resection by DNA2 (148); however, RECQL1, BLM, and WRN stimulate EXO1 (142, 144, 145). Even though WRN has inherent exonuclease activity, the 3′–5′ polarity is counterproductive and thus would inhibit recombination. Therefore, it appears that BLM, via its interactions with both EXO1 and DNA2, plays a dominant role in promoting DSB resection.
In yeast, these complexes are conserved because Sgs1 (RecQ homolog in Saccharomyces cerevisiae), Dna2, and RPA (156), or Sgs1, Exo1, and Sae2, catalyze DNA end resection (143, 157, 158). Interestingly, in Xenopus, xWRN, xDNA2, and RPA are implicated in DNA end resection (159, 160).
Evidence suggests that Sgs1, BLM, and RECQL5 act as antirecombination factors because they inhibit an early step in HR-mediated DSB repair. They do so by disrupting the RAD51 nucleoprotein filament, which is essential for the homology search and D-loop formation (92, 161–163). WRN, RECQL1, and RECQL4 also colocalize with RAD51; however, WRN and RECQL1 cannot disrupt RAD51 filaments (92, 162), whereas RECQL4 has not been tested. Thus, BLM and RECQL5 may be partially complementary to one another at this step. An analysis of SCEs in BLM and RECQL5 single- and double-deficient chicken and mouse cells supports the idea that RECQL5 may play additional roles in suppressing SCEs when BLM function is impaired (164, 165).
Although WRN does not have the same antirecombinogenic activity as BLM, it plays a role in DSB pathway choice and suppresses recombination (166, 167). Ectopic expression of an HJ resolvase in WS cells enhances viability and proliferation, suggesting that there is defective HR in these cells (168, 169). How WRN facilitates this action is unclear, but it interacts with several HR proteins, including MRN (146), BRCA1 (170), RAD52 (53), RAD51, and RAD54 (Figure 2) (171).
RECQL4 is also recruited to laser-induced DSBs, and RECQL4-deficient cells are moderately sensitive to ionizing radiation (54, 172). RECQL4 interacts with RAD51 in cells exposed to etoposide (173), an inhibitor of topo-isomerase II (Topo II) that generates DSBs. Although these are preliminary findings, the data support a role for RECQL4 in DSB repair.
Homology-dependent branch migration and DNA synthesis–dependent strand annealing (SDSA) generate HJs or double HJs (dHJs), and RecQ helicases (RECQL1/ WRN/BLM) interact with and can branch migrate HJs and dHJs or dissolve them (BLM/Topo/RMI1/RMI2), as discussed in more detail below (174–179). Alternatively, HJs can be resolved endonucleolytically by nucleases (GEN1, MUS81/EME1, or SLX1/SLX4) to generate crossover or noncrossover products (180–184). Interestingly, in the absence of WRN, MUS81/EME1 promotes HR-dependent DSB repair by a poorly understood process that leads to chromosomal aberrations and appears to be error prone (167). SDSA is another alternative pathway for noncrossover resolution of recombination intermediates and a pathway for generating and extending D-loops. All RecQ helicases possess DNA SA activity (reviewed in Reference 8). More specifically, WRN enhances RAD52-mediated SA (53), and BLM stimulates DNA polymerase η–dependent SDSA (162) and is specifically required for SDSA in Drosophila (185). Because all RecQs possess SA activity, they could functionally complement one another at this step.
Thus, RecQ proteins interact with key enzymes or are themselves critical for proper DSB repair (Figures 2 and 3b). This observation could become clinically significant if synthetic lethal relationships involving RecQ helicases and DSB repair pathway components are identified or if therapeutic agents that complement or correct an inherited defect in one of the RecQ helicases are determined.
RecQ HELICASES IN DNA REPLICATION
Genome Replication
RecQ helicases participate in all phases of DNA replication (including initiation, Okazaki fragment processing, leading- and lagging-strand elongation, and resolution). Other replication-associated events in which RecQ helicases may play roles include interactions with RPA, translesion DNA synthesis, replication fork restart, telomere replication, and intra-S-phase DNA damage signaling. Although the experimental and molecular details are not yet understood, specific roles for each RecQ helicase in DNA replication have been postulated (Figure 4).
Figure 4.

RecQ helicases in DNA replication. Putative roles of RecQ helicases during initiation of DNA replication, Okazaki fragment processing, leading- and lagging-strand elongation, fork restart, and regression are indicated. The orange star and triangle denote polymerase blocking DNA damage. In addition, RECQL5 may specifically inhibit transcription to prevent replication and transcription fork collisions. Abbreviations: RQ1, RECQL1 helicase; RQ4, RECQL4 helicase; RQ5, RECQL5 helicase.
RECQL4 and RECQL1 may play unique roles during initiation of DNA replication, but RECQL1’s role is not well characterized (38), whereas RECQL4’s role in initiation is better characterized in several species. The N-terminal region of xRTS, the Xenopus homolog of RECQL4, shares homology with Sld2, a yeast replication initiation protein (186, 187), and is essential for loading DNA Pol α onto replication origins (186). Similarly, RECQL4 plays a role during initiation of DNA replication in Drosophila and chicken DT40 cells (188–190), where it associates with replication origins during G1/S phase (38) and interacts with replisome factors MCM10, MCM2-7 helicase, CDC45, and GINS (190, 191). MCM10 directly regulates the RECQL4 DNA helicase, and this interaction may be modulated by cyclin-dependent kinase phosphorylation (190). Consistent with these findings, homozygous deletion of Recql4 is lethal during embryogenesis in mice and flies (76, 192).
FEN1 interacts with BLM, WRN, RECQL4, and RECQL5 (Figure 2), suggesting that RecQ helicases may play a role during maturation of Okazaki fragments (102, 193). However, whether RecQ helicases play complementary or redundant roles during lagging-strand DNA replication, or how they are coordinated, remains unknown.
Early research on cells from WS and BS patients suggested that replication fork movement might be slower in defective BLM and WRN cells than in controls because S phase lasts longer in these cells (194, 195). Later studies using fiber tracking analysis confirmed that defects in BLM or WRN decrease the rate of replication fork progression (196, 197). One explanation for this observation is that WRN and BLM resolve or remove DNA structures that impede fork movement (60, 104, 198), such as hairpins and G4 DNA (199, 200). Slower fork movement has also been observed in RECQL1-deficient (but not RECQL4-deficient) cells (38).
Chromosomal fragile sites (CFSs) are chromosomal regions that are prone to breakage, probably because they are inherently difficult to replicate (201, 202). WRN (203, 204) and BLM (205) strongly influence the stability of CFSs in normal cells (206). WRN specifically increases the ability of Pol δ to replicate through common fragile sites, such as FRA16D (207). RECQL1 also localizes to FRA16D and FRA3B in response to replicative stress (208) and may play a similar role in CFS DNA replication.
WRN can also stimulate bypass of DNA damage by translesion polymerases (209). WRN interacts with and stimulates translesion polymerases Pol η, Pol κ, Pol ι, and REV1 (210–212). Additionally, the WRN exonuclease can act as a proofreader for Pol η (211) and the replicative polymerase Pol δ (213). BLM stimulates primer extension by Pol η, but whether it promotes lesion bypass by Pol η is unknown.
RecQ helicases are thought to play important roles during replication restart after fork collapse. For example, defects in WRN, BLM, and RECQL1 increase the fragility of hot spots after replication fork stalling (203, 205), and single-molecule DNA fiber studies reveal that DNA synthesis tracks are shorter in hydroxyurea-treated WRN-defective cells than in BLM-depleted or control cells (196). At the biochemical level, WRN strongly prefers substrates that mimic stalled replication forks (214–216), suggesting that WRN and BLM can convert a regressed fork via branch migration and/or degradation to a normal fork structure (Figure 4) (52, 217, 218).
Depletion of RECQL1 alters cell-cycle progression and induces replication-associated DSBs during S phase (219). Stable depletion of RECQL1 also renders cells sensitive to DNA damaging agents that directly or indirectly block replication fork progression (35, 38, 40, 219); activates phospho-CHK1; and induces accumulation of phospho-RPA, indicating replicative stress (219). RECQL1, like BLM, RECQL5, and WRN, promotes strand exchange on synthetic stalled replication fork–like structures (219, 220). RECQL1 and WRN, but not RECQL4 or RECQL5, unwind the leading strand of the replication fork. RPA modulates RECQL1 and WRN to favor unwinding over strand exchange and reannealing (219). Thus, RECQL1 and WRN behave similarly in vivo and in vitro in relation to replication fork–like structures with or without a leading-strand gap.
A unique role has been proposed for RECQL1 during replication restart after DNA damage (39), and this role is regulated by PARP1 (39, 221). In vitro RECQL1 helicase activity remodels chicken foot–like DNA substrates into replication fork–like molecules but fails to regress forks, and PARP1 inhibits this reaction (39). Single-molecule DNA fiber analysis revealed that RECQL1-deficient cells exposed to low-dose CPT fail to restart replication (39). Therefore, the current model proposes that PARP1 protects CPT-induced stalled forks from RECQL1 to limit premature fork restart and that RECQL1 plays a unique and essential role in fork restart (39). These results have significant clinical implications because CPT and other Topo inhibitors are widely used chemotherapeutic agents whose efficacy might be enhanced by combination with a small-molecule inhibitor of RECQL1.
In summary, depending on the type of DNA damage, BLM and WRN may stabilize replication forks, whereas RECQL1 and RECQL4 may play unique roles in replication fork restart or initiation. The precise roles of RECQL4 and RECQL5 in replication fork collapse or restart after DNA damage are unresolved and represent important areas for future research. RECQL5 might acquire a more important role in DNA replication as a backup in WRN-deficient cells (37). However, RECQL5 is also thought to inhibit transcription, thereby preventing fork collapse due to head-on collision between the transcription and replication machineries in a more proactive or preventive role (90; reviewed in References 88 and 222).
Resolution of Replication Intermediates and DNA Decatenation
During and after DNA replication, the parental and nascent DNA strands are linked together due to recombination between two molecules. The process of untangling the DNA strands, known as decatenation, usually occurs during the late S and G2/M phases of the cell cycle. BLM directly suppresses aberrant recombination at stalled forks; therefore, in BLM-deficient cells there is a high degree of SCEs (223). This phenomenon is, in part, attributed to BLM’s unique role in dissolution of late recombination intermediates, which is mediated by a protein complex, referred to as the BLM dissolvasome or BTR complex, composed of BLM, Topo IIIα, and RMI1 and -2, (recently reviewed in References 3 and 224). This complex plays an important role in promoting dissolution, noncrossover outcomes from dHJs (see review in Reference 225). In yeast, the Sgs1/Topo IIIα/RMI1 protein complex has a similar function (3, 226).
RECQL5 physically interacts with Topo IIα and specifically stimulates its DNA decatenation activity (227). Stable depletion of RECQL5 inhibits cell proliferation and induces a G2/M checkpoint. BLM also interacts with Topo IIα, which is important in preventing chromosomal breakage (228). These observations indicate that RecQ helicases play significant roles in the resolution of late replication intermediates and DNA decatenation.
Replication and Repair of Telomeres
Telomeres are protein–DNA structures at chromosome ends that preserve genome stability, survival, and proliferation at the cellular level and prevent degenerative diseases and cancer at the organismal level (229, 230). Human telomeres consist of ~10 kb of TTAGGG tandem repeats, a 3′ ssDNA overhang, and a six-member shelterin protein complex (231, 232). Normal cell division causes the progressive loss of telomeric repeats, which are compensated for by telomerase; however, most human cells lack telomerase activity (233, 234). Critically short telomeres are recognized as chromosome breaks and trigger cell senescence or chromosome end-to-end fusions (235, 236). Telomere dysfunction contributes to the WS and BS pathologies in mouse models (69, 237). Telomeres are more sensitive to replicative stress than is the bulk genome (238, 239), which may partly explain the need for RecQ helicases in telomere replication.
WRN, BLM, and RECQL4 interact with shelterin proteins TRF1, TRF2, and POT1 (Figure 5) (73, 240–245). The shelterin proteins TRF1, TRF2, and POT1 generally stimulate unwinding of telomeric D-loops and forks by RecQ helicases (55, 73, 176, 240, 241, 244). WRN helicase and exonuclease activities cooperate to dissociate D-loops (55). TRF2 stimulates (242) and POT1 inhibits the WRN exonuclease on substrates containing a telomeric ssDNA overhang (246).
Figure 5.
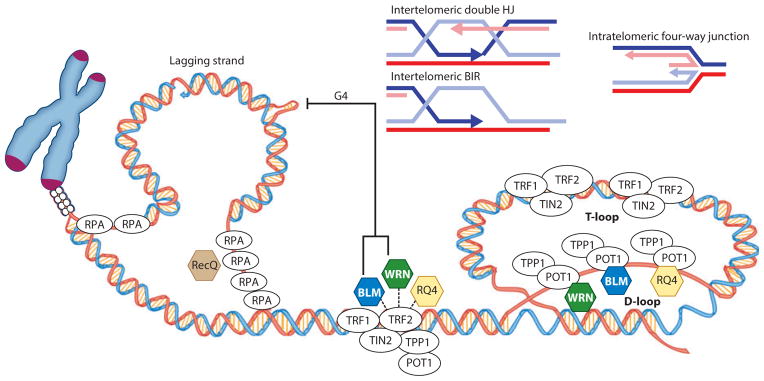
RecQ helicases (BLM, WRN, and RECQL4) in telomere maintenance and repair. Interactions with shelterin proteins (TRF1, TRF2, and POT1) and resolution of secondary structures that are potential barriers to telomere replication are shown. RecQs may resolve inter- and intratelomeric substrates, G quadruplexes (G4s), and telomeric T-loops and D-loops. Abbreviations: BIR, break-induced replication; HJ, Holliday junction; RPA, replication protein A; RQ4, RECQL4; TIN2, TRF1-interacting nuclear factor 2; TPP1, POT1-interacting telomere end–binding protein.
WRN-, BLM-, and RECQL4-deficient cells have distinct properties, but all are defective in telomere replication. WRN and RECQL4 localize to some telomeres during S phase (73, 247), and replication stress enhances telomeric localization of WRN (248). In contrast, BLM localizes to a subset of telomeres in either late S or G2/M phase (43, 241). Thus, RecQ helicases may be differentially enriched at the telomere in a cell cycle–dependent manner, reflecting their different roles in telomere maintenance.
BLM and WRN play nonredundant roles in suppressing sister telomere loss (STL) and critical telomere shortening (43). In WRN-deficient cells, STL is more frequent in telomeres replicated from the G-rich lagging strand (247, 249), and WRN prevents large deletions during telomere replication (132). These data suggest that lagging-strand TTAGGG repeats are resistant to replication in the absence of WRN, possibly because G4s form (Figure 5). However, other evidence suggests that G4s cause telomere fragility rather than telomere loss, and telomere fragility was not reported in WRN-deficient cells but was observed in RECQL4- and BLM-deficient cells (73, 238). Fragile telomeres manifest as multiple distinct telomeric spots at a single chromatid end (often a doublet) and result from treatments that cause breaks at CFSs (238). However, WRN and BLM unwind G4s, and RECQL4 does not (154, 250). Therefore, although BLM may unwind G4s and reduce telomere fragility, RECQL4 or WRN may disrupt other structures that cause telomere fragility or STL, respectively. The division of labor may relate to substrate specificity.
Regressed replication forks and D-loops can also interfere with telomere replication. Electron microscopy studies revealed that stalled replication forks at telomeric repeats spontaneously regress into four-way junctions (Figure 5) (251). Both WRN and BLM promote the formation and subsequent disruption of regressed replication forks (250, 252), whereas unwinding of four-way junctions by RECQL4 has not been observed (154). By contrast, all three helicases can disrupt telomeric D-loops (55, 73, 177, 179, 253). Intratelomeric D-loops form naturally when the 3′ ssDNA telomeric overhang wraps around and invades the duplex telomeric sequence, forming a large T-loop that is stabilized by the D-loop (Figure 5) (254, 255). Failure to resolve this structure can cause T-loop cleavage and dramatic telomere shortening, which may explain why cells express several helicases that can dismantle the telomeric D-loop (243, 256, 257). Interestingly, not all telomeres are affected equally in BLM- and WRN-deficient cells, but loss of both proteins considerably increases the number of STLs (43).
RecQ helicases may also disrupt intertelomeric D-loops generated by aberrant strand invasions of the 3′ telomeric overhang. This would prevent inappropriate recombination at telomeres and is consistent with cellular observations that WRN, BLM, or RECQL4 deficiency increases SCEs at telomeres (Figure 5) (73, 258). HR is suppressed at telomeres in normal cells but is elevated in cancer cells that use alternative lengthening of telomeres (ALT) to prevent telomere attrition during cell division (259). Evidence indicates that WRN and BLM dissociate intermediates of the ALT pathway (260–262). Thus, RecQ helicases exhibit a division of labor at telomeres by disrupting intermediate structures to (a) prevent telomeric HR in normal cells or (b) facilitate completion of the ALT pathway in cancer cells.
The roles of RECQL1 and RECQL5 in telomere maintenance are poorly characterized. However, RECQL5 does not unwind telomeric D-loops (73). Recql1-knockout mice have normal length telomeres (33). Notably, simultaneous knockout of C. elegans rtel-1 and recql5 causes synthetic lethality due to elevated recombination (263). Rtel-1 is the C. elegans homolog of human RTEL1, a helicase that plays a significant role in regulating telomere length (256, 264), which implies that RECQL5 may have an uncharacterized role in telomere maintenance in human cells.
RecQ Helicases, BRCA1, and the Fanconi’s Anemia Pathway
RecQ helicases may also engage in cross talk with BRCA1 and the Fanconi’s anemia (FA) core complex. Both WRN and BRCA1 are thought to participate in repair of interstrand cross-links (170), the primary targets of the FA pathway. BLM also forms foci that colocalize with the proliferating cell nuclear antigen and the BRCA1-associated genome surveillance complex (265). FANCM, a member of this complex, is required for formation of BLM nuclear foci after replication stalling, and it helps prevent chromosome instability (266). FANCD2 and FANCJ interact with BLM and colocalize in DNA damage–induced nuclear foci, and BLM stability correlates positively with expression of FANCD2 and FANCJ (267, 268). Further characterization of the RecQ proteins and the FA pathway is an important area for additional research.
S-Phase Checkpoint
The intra-S-phase checkpoint is activated to maintain DNA replication fork stability, promote fork recovery, and facilitate fork restart. Ataxia-telangiectasia- and RAD3-related protein kinase (ATR) and ataxia-telangiectasia-mutated protein kinase (ATM) play critical but distinct roles in this process. Depending on the type of damage, both are required to recover from replication-induced DSBs (269). RecQ helicases play a limited role in the intra-S-phase checkpoint (270). However, the checkpoint kinases ATM and ATR are not fully activated in WRN-deficient cells exposed to CPT (271, 272), which leads to replication fork collapse (271) and DSBs that are subsequently repaired by recombination (167).
Depletion of RAD9, which disrupts RAD911, eliminates recruitment of WRN to CPT- and hydroxyurea-induced DNA damage and prevents ATR/ATM-dependent serine and tyrosine phosphorylation of WRN (273). The current model is that WRN participates in the intra-S-phase checkpoint in response to replication-dependent DSBs (274, 275); the RAD911 complex recruits WRN, and ATR phosphorylation of WRN stabilizes WRN at stalled replication forks. Finally, ATM may dissociate WRN from DSBs to allow binding and initiation of RAD51-dependent recombination repair (270). Studies also show that BLM plays a role in recovery from S-phase arrest in response to hydroxyurea and that ATR plays a role in the regulation of BLM function (276). Posttranslational modifications, such as phosphorylation, are known to modulate the catalytic activities of the RecQ proteins (see the sidebar titled Posttranslational Modifications of WRN).
RECQL1-depleted cells activate the intra-S-phase checkpoint normally (219), and RECQL5-depleted cells accumulate in late S phase (227). Thus, these RecQ helicases are not required for intra-S-phase arrest. RECQL4 may play a role in the intra-S-phase checkpoint because RECQL4-deficient or -defective cells fail to arrest in S phase after UV, hydroxyurea, or ionizing radiation treatment (172, 277). However, the molecular details and/or mechanisms are not yet known.
TOPOISOMERASES AND RecQ HELICASES
BLM interacts with and reciprocally stimulates Topo I on RNA–DNA hybrids (278). BLM stimulates ribosomal transcription, and the BLM–Topo I interaction may modulate transcription of nucleolar ribosomal DNA (278). WRN also interacts with Topo I and stimulates Topo I–catalyzed relaxation of negatively supercoiled DNA (279). Furthermore, it protects against the deleterious effects of Topo I inhibitors (197, 279–281). WRN stimulates ribosomal transcription, but whether this action is mediated by an interaction with Topo I is unknown (282). Collectively, there are multiple Topo–RecQ connections, which may contribute to the faithful segregation of chromosomes.
POSTTRANSLATIONAL MODIFICATIONS OF WRN.
Multiple posttranslational modifications (PTMs) regulate the catalytic activities, cellular localization, and protein–protein interactions of RecQ helicases. For example, WRN is phosphorylated by ATM, ATR, DNA-PKcs, and c-Abl tyrosine kinase (271, 272, 275, 300). Suppression of ATR-mediated phosphorylation in vivo alters the localization of WRN to nuclear foci and its colocalization with RPA, and can lead to breakage of stalled replication forks (275). Serine/threonine phosphorylation of WRN by DNA-PKcs and tyrosine phosphorylation by c-Abl kinase, in vitro, inhibit the WRN exonuclease and helicase (126, 300). Oxidation also inhibits both WRN activities (301). WRN is also acetylated in vivo and in vitro by p300, a lysine acetyltransferase, and acetylation stimulates the helicase and exonuclease, possibly enhancing the role of WRN in BER (302). Acetylation of WRN by p300 causes translocation from nucleolar to nuclear foci (303). WRN also associates with SUMO-1 (304), but the functional consequence of SUMO modification of WRN remains to be established. Clearly, understanding PTMs for each of the RecQs is an important line of research.
RecQ HELICASES AS THERAPEUTIC TARGETS
RecQ helicases are dysregulated in cancer (283); therefore, they represent novel therapeutic targets. Recently, small-molecule inhibitors of both WRN and BLM have been identified (284, 285). Furthermore, when cells defective in the FA pathway were treated with a WRN inhibitor, the response to mitomycin C was abrogated (286). As described above, RecQ helicases play a critical role in the response to DNA damage; thus, combination therapies that target one of the RecQ helicases might induce synthetic lethality. For example, certain oncogenes such as Myc, which are frequently mutated and overexpressed in cancers, are synthetically lethal when WRN is inactivated due to incipient replication catastrophe (287). Although RecQ-targeted drugs may be useful for therapy, they are also of interest as tools for cell-based studies.
INTERACTIONS BETWEEN RecQ HELICASES
Early studies in human cells reported, with some surprise, the discovery of five distinct RecQ helicase–related enzymes, initiating an ongoing debate about whether the activities and functions of the five human RecQ helicases are complementary or redundant. After extensive research, this question remains unresolved, although some insights have been gained (Figures 2 and 6). Most importantly, significant functional interactions between the RecQ helicases have been documented. Although WRN and BLM share similar substrate specificity (250), BLM inhibits the WRN exonuclease (288), and RECQL4 specifically stimulates BLM (289). BLM promotes retention of RECQL4 at DSBs in vivo (289), and cells defective in BLM and RECQL4 proliferate slowly with elevated SCEs. Cells that are defective in WRN and RECQL5 demonstrate synthetic lethality (37). In vivo RECQL5 dissociates slowly from DSBs in WS and BS fibro-blasts. However, RECQL5 specifically stimulates WRN but not BLM, and RECQL5 and WRN cooperate during repair and restart of synthetic stalled replication fork–like structures (37). These results suggest that RECQL5 and WRN play cooperative and complementary roles. Additional research on the interactions between RecQ helicases is needed, and these interactions remain of great interest in the field.
Figure 6.
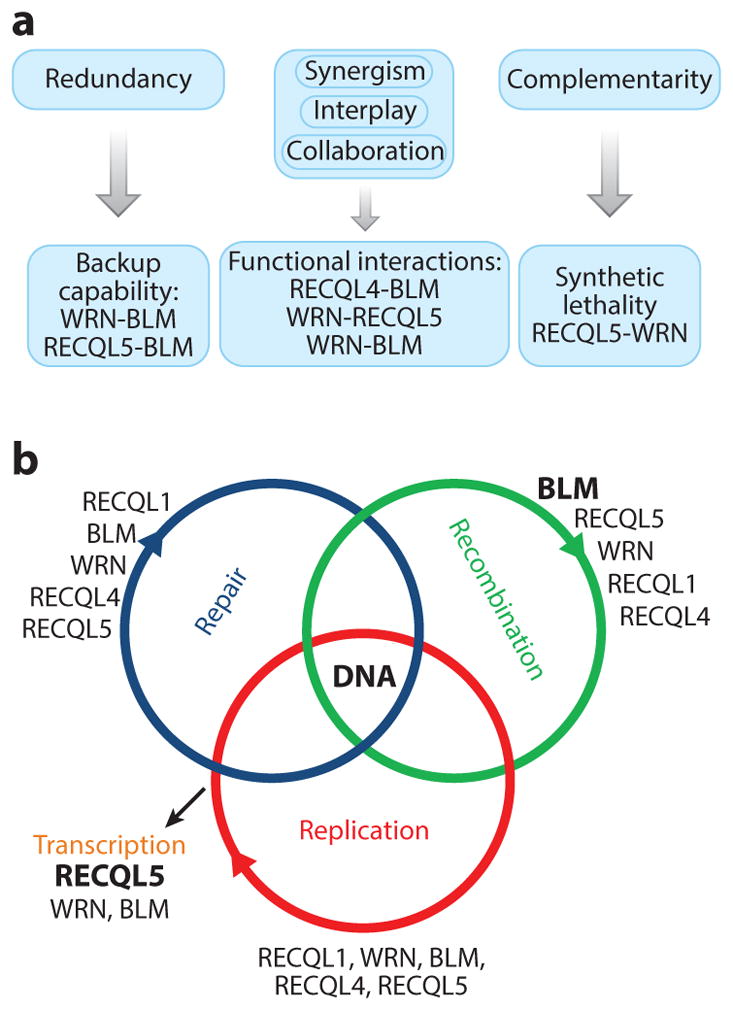
Interactions between RecQ helicases. (a) Human RecQ helicases can play both overlapping and nonredundant roles. They can cooperate with each other functionally and complement each other in certain DNA metabolic pathways. (b) The predominant role of each RecQ helicase in each DNA metabolic pathway is indicated. Larger, bold font equates to greater predominance for the role of a specific enzyme in a specific pathway.
CONCLUDING REMARKS
RecQ helicases play important roles in DNA repair, recombination, and replication that are mediated primarily through interactions with proteins that regulate or play critical roles in these pathways (Figure 2). Recently, it has become clear that RecQ helicases also play direct roles in these DNA metabolic pathways. The biological importance of RecQ helicases is well established; however, whether they can be exploited as therapeutic targets, or whether methods can be developed to treat or prevent illness in patients who carry modified alleles of BLM, WRN, or RECQL4, is not yet clear.
Supplementary Material
SUMMARY POINTS.
Each of the five human RecQ helicases can unwind forked dsDNA and anneal ssDNA, and RecQ helicases play critical roles in all aspects of DNA metabolism, including DNA repair, recombination, replication, and transcription.
There is a division of labor among the human RecQ helicases, and each has at least one specialized biological function, but collectively they all contribute widely to genome stability.
Human RecQ helicases interact with each other, and their roles in different DNA metabolic pathways can be both overlapping and complementary, creating a functional complexity that is a challenge to decipher.
Mutations in BLM, WRN, and RECQL4 are associated with heritable disease syndromes and susceptibility to cancer.
Because RecQ helicases play diverse roles in the response to DNA damage, they have potential as novel cancer chemotherapeutic targets.
FUTURE ISSUES.
Additional analyses of the structure–function relationships among human RecQ helicases are warranted because they could yield insight into their shared and unique properties. For example, there is no recognizable winged helix in RECQL5, so does it interact with DNA differently than do the other family members?
Additional novel and disease-linked mutant alleles of BLM, WRN, and RECQL4 should be characterized to better understand the molecular basis of the clinical manifestations of BS, WS, BGS, RTS, and RAPADILINO syndrome. Curiously, WRN deficiency is associated with an aging-specific phenotype, and it is important to understand how the separate activities of its helicase and exonuclease cooperate in biological pathways.
The role of the RecQ helicases in the context of the cellular DNA damage response network should continue to be investigated because of the critical role played by the DNA damage response in preserving genome stability.
The RecQ proteins, and their interaction partners, are posttranslationally modified by phosphorylation, acetylation, and SUMO-ylation. These and other PTMs are critically important regulators of the enzymes’ functions, and additional research on this subject is needed.
Small-molecule inhibitors and enhancers for each RecQ helicase should be developed because (a) such compounds could potentially enhance the efficacy of chemotherapeutic agents already in clinical use; (b) they might also prove beneficial in other clinical contexts; and (c) specific protein inhibitors and activators will be useful in studying RecQ helicase mechanisms and functions.
Acknowledgments
We thank Miriam Sander (Page One Editorial Services, Boulder, Colorado) for her contribution to this review as scientific writer/editor. We thank Drs. Robert Brosh, Tomasz Kulikowicz, and Raghavendra Shamanna for their comments. We also thank Thomas Wynn for his excellent artwork. We apologize to our colleagues whose work is not cited in the review due to space limitations. This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute on Aging. Support was also provided by the Scaife Foundations through the Center for Nucleic Acid Technology to P.L.O.
Glossary
- Translocate
refers to the directional movement of helicases in a 5′ → 3′ or 3′ → 5′ direction along DNA
- RQC domain
RecQ C-terminal domain
- HRDC domain
helicase and RNase D–like C-terminal domain
- G quadruplex (G4)
a four-stranded nucleic acid structure stabilized by stacked non–Watson–Crick Hoogsteen base pairs between four planar-orientated guanosine nucleotides
- SCE
sister chromatid exchange
- HR
homologous recombination
- CPT
camptothecin
- BER
base excision repair
- NER
nucleotide excision repair
- NHEJ
nonhomologous end joining
- Alt-NHEJ
alternative nonhomologous end joining
- Nucleoprotein filament
a critical intermediate in HR in which eukaryotic recombinase RAD51 coats ssDNA and initiates a homology search
- Synthetic lethality
cell death resulting from inactivation or inhibition of two gene products that are nonlethal when inactivated individually
- Shelterin
a six-protein complex that localizes to the terminal TTAGGG repeats of mammalian telomeres
- T-loop
a structure in which telomeric ssDNA at the chromosome terminus invades and hybridizes to the complementary strand of adjacent telomeric dsDNA
- Intra-S-phase checkpoint
a mechanism that aborts or slows replication and cell-cycle progression through S phase in cells with DNA damage
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Umate P, Tuteja N, Tuteja R. Genome-wide comprehensive analysis of human helicases. Commun Integr Biol. 2011;4:118–37. doi: 10.4161/cib.4.1.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends Biochem Sci. 2008;33:609–20. doi: 10.1016/j.tibs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Larsen NB, Hickson ID. RecQ helicases: conserved guardians of genomic integrity. Adv Exp Med Biol. 2013;767:161–84. doi: 10.1007/978-1-4614-5037-5_8. [DOI] [PubMed] [Google Scholar]
- 4.Mandell JG, Goodrich KJ, Bahler J, Cech TR. Expression of a RecQ helicase homolog affects progression through crisis in fission yeast lacking telomerase. J Biol Chem. 2005;280:5249–57. doi: 10.1074/jbc.M412756200. [DOI] [PubMed] [Google Scholar]
- 5.Barea F, Tessaro S, Bonatto D. In silico analyses of a new group of fungal and plant RecQ4-homologous proteins. Comput Biol Chem. 2008;32:349–58. doi: 10.1016/j.compbiolchem.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 6.Groocock LM, Prudden J, Perry JP, Boddy MN. The RecQ4 orthologue Hrq1 is critical for DNA interstrand cross-link repair and genome stability in fission yeast. Mol Cell Biol. 2012;32:276–87. doi: 10.1128/MCB.06184-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi DH, Lee R, Kwon SH, Bae SH. Hrq1 functions independently of Sgs1 to preserve genome integrity in Saccharomyces cerevisiae. J Microbiol. 2013;51:105–12. doi: 10.1007/s12275-013-3048-2. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y. Unwinding and rewinding: double faces of helicase? J Nucleic Acids. 2012;2012:140601. doi: 10.1155/2012/140601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gorbalenya AE, Koonin EV. Helicases: amino acid sequence comparisons and structure–function relationships. Curr Opin Struct Biol. 1993;3:419–29. [Google Scholar]
- 10.Marino F, Vindigni A, Onesti S. Bioinformatic analysis of RecQ4 helicases reveals the presence of a RQC domain and a Zn knuckle. Biophys Chem. 2013;177/178:34–39. doi: 10.1016/j.bpc.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Garcia PL, Liu Y, Jiricny J, West SC, Janscak P. Human RECQ5β, a protein with DNA helicase and strand-annealing activities in a single polypeptide. EMBO J. 2004;23:2882–91. doi: 10.1038/sj.emboj.7600301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 13.Fairman-Williams ME, Guenther UP, Jankowsky E. SF1 and SF2 helicases: family matters. Curr Opin Struct Biol. 2010;20:313–24. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Janscak P, Garcia PL, Hamburger F, Makuta Y, Shiraishi K, et al. Characterization and mutational analysis of the RecQ core of the Bloom syndrome protein. J Mol Biol. 2003;330:29–42. doi: 10.1016/s0022-2836(03)00534-5. [DOI] [PubMed] [Google Scholar]
- 15.Lee JW, Kusumoto R, Doherty KM, Lin GX, Zeng W, et al. Modulation of Werner syndrome protein function by a single mutation in the conserved RecQ domain. J Biol Chem. 2005;280:39627–36. doi: 10.1074/jbc.M506112200. [DOI] [PubMed] [Google Scholar]
- 16.Ren H, Dou SX, Zhang XD, Wang PY, Kanagaraj R, et al. The zinc-binding motif of human RECQ5β suppresses the intrinsic strand-annealing activity of its DExH helicase domain and is essential for the helicase activity of the enzyme. Biochem J. 2008;412:425–33. doi: 10.1042/BJ20071150. [DOI] [PubMed] [Google Scholar]
- 17.Bernstein DA, Zittel MC, Keck JL. High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 2003;22:4910–21. doi: 10.1093/emboj/cdg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pike AC, Shrestha B, Popuri V, Burgess-Brown N, Muzzolini L, et al. Structure of the human RECQ1 helicase reveals a putative strand-separation pin. Proc Natl Acad Sci USA. 2009;106:1039–44. doi: 10.1073/pnas.0806908106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitano K, Kim SY, Hakoshima T. Structural basis for DNA strand separation by the unconventional winged-helix domain of RecQ helicase WRN. Structure. 2010;18:177–87. doi: 10.1016/j.str.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Huber MD, Duquette ML, Shiels JC, Maizels N. A conserved G4 DNA binding domain in RecQ family helicases. J Mol Biol. 2006;358:1071–80. doi: 10.1016/j.jmb.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 21.von Kobbe C, Thoma NH, Czyzewski BK, Pavletich NP, Bohr VA. Werner syndrome protein contains three structure-specific DNA binding domains. J Biol Chem. 2003;278:52997–3006. doi: 10.1074/jbc.M308338200. [DOI] [PubMed] [Google Scholar]
- 22.Kitano K, Yoshihara N, Hakoshima T. Crystal structure of the HRDC domain of human Werner syndrome protein, WRN. J Biol Chem. 2007;282:2717–28. doi: 10.1074/jbc.M610142200. [DOI] [PubMed] [Google Scholar]
- 23.Tadokoro T, Kulikowicz T, Dawut L, Croteau DL, Bohr VA. DNA binding residues in the RQC domain of Werner protein are critical for its catalytic activities. Aging. 2012;4:417–30. doi: 10.18632/aging.100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vindigni A, Marino F, Gileadi O. Probing the structural basis of RecQ helicase function. Biophys Chem. 2010;149:67–77. doi: 10.1016/j.bpc.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 25.Lucic B, Zhang Y, King O, Mendoza-Maldonado R, Berti M, et al. A prominent β-hairpin structure in the winged-helix domain of RECQ1 is required for DNA unwinding and oligomer formation. Nucleic Acids Res. 2011;39:1703–17. doi: 10.1093/nar/gkq1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Z, Macias MJ, Bottomley MJ, Stier G, Linge JP, et al. The three-dimensional structure of the HRDC domain and implications for the Werner and Bloom syndrome proteins. Structure. 1999;7:1557–66. doi: 10.1016/s0969-2126(00)88346-x. [DOI] [PubMed] [Google Scholar]
- 27.Bernstein DA, Keck JL. Domain mapping of Escherichia coli RecQ defines the roles of conserved N- and C-terminal regions in the RecQ family. Nucleic Acids Res. 2003;31:2778–85. doi: 10.1093/nar/gkg376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bennett RJ, Sharp JA, Wang JC. Purification and characterization of the Sgs1 DNA helicase activity of Saccharomyces cerevisiae. J Biol Chem. 1998;273:9644–50. doi: 10.1074/jbc.273.16.9644. [DOI] [PubMed] [Google Scholar]
- 29.Lan L, Nakajima S, Komatsu K, Nussenzweig A, Shimamoto A, et al. Accumulation of Werner protein at DNA double-strand breaks in human cells. J Cell Sci. 2005;118:4153–62. doi: 10.1242/jcs.02544. [DOI] [PubMed] [Google Scholar]
- 30.Karmakar P, Seki M, Kanamori M, Hashiguchi K, Ohtsuki M, et al. BLM is an early responder to DNA double-strand breaks. Biochem Biophys Res Commun. 2006;348:62–69. doi: 10.1016/j.bbrc.2006.07.037. [DOI] [PubMed] [Google Scholar]
- 31.Samanta S, Karmakar P. Recruitment of HRDC domain of WRN and BLM to the sites of DNA damage induced by mitomycin C and methyl methanesulfonate. Cell Biol Int. 2012;36:873–81. doi: 10.1042/CBI20110510. [DOI] [PubMed] [Google Scholar]
- 32.Puranam KL, Blackshear PJ. Cloning and characterization of RECQL, a potential human homologue of the Escherichia coli DNA helicase RecQ. J Biol Chem. 1994;269:29838–45. [PubMed] [Google Scholar]
- 33.Sharma S, Stumpo DJ, Balajee AS, Bock CB, Lansdorp PM, et al. RECQL, a member of the RecQ family of DNA helicases, suppresses chromosomal instability. Mol Cell Biol. 2007;27:1784–94. doi: 10.1128/MCB.01620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma S, Brosh RM., Jr Human RECQ1 is a DNA damage responsive protein required for genotoxic stress resistance and suppression of sister chromatid exchanges. PLoS ONE. 2007;2:e1297. doi: 10.1371/journal.pone.0001297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Futami K, Kumagai E, Makino H, Goto H, Takagi M, et al. Induction of mitotic cell death in cancer cells by small interference RNA suppressing the expression of RecQL1 helicase. Cancer Sci. 2008;99:71–80. doi: 10.1111/j.1349-7006.2007.00647.x. [DOI] [PubMed] [Google Scholar]
- 36.Sharma S, Brosh RM., Jr Unique and important consequences of RECQ1 deficiency in mammalian cells. Cell Cycle. 2008;7:989–1000. doi: 10.4161/cc.7.8.5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Popuri V, Huang J, Ramamoorthy M, Tadokoro T, Croteau DL, Bohr VA. RECQL5 plays co-operative and complementary roles with WRN syndrome helicase. Nucleic Acids Res. 2013;41:881–99. doi: 10.1093/nar/gks1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thangavel S, Mendoza-Maldonado R, Tissino E, Sidorova JM, Yin J, et al. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2010;30:1382–96. doi: 10.1128/MCB.01290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berti M, Chaudhuri AR, Thangavel S, Gomathinayagam S, Kenig S, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013;20:347–54. doi: 10.1038/nsmb.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mendoza-Maldonado R, Faoro V, Bajpai S, Berti M, Odreman F, et al. The human RECQ1 helicase is highly expressed in glioblastoma and plays an important role in tumor cell proliferation. Mol Cancer. 2011;10:83. doi: 10.1186/1476-4598-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kitao S, Ohsugi I, Ichikawa K, Goto M, Furuichi Y, Shimamoto A. Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics. 1998;54:443–52. doi: 10.1006/geno.1998.5595. [DOI] [PubMed] [Google Scholar]
- 42.Bischof O, Kim SH, Irving J, Beresten S, Ellis NA, Campisi J. Regulation and localization of the Bloom syndrome protein in response to DNA damage. J Cell Biol. 2001;153:367–80. doi: 10.1083/jcb.153.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barefield C, Karlseder J. The BLM helicase contributes to telomere maintenance through processing of late-replicating intermediate structures. Nucleic Acids Res. 2012;40:7358–67. doi: 10.1093/nar/gks407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eladad S, Ye TZ, Hu P, Leversha M, Beresten S, et al. Intra-nuclear trafficking of the BLM helicase to DNA damage-induced foci is regulated by SUMO modification. Hum Mol Genet. 2005;14:1351–65. doi: 10.1093/hmg/ddi145. [DOI] [PubMed] [Google Scholar]
- 45.Chester N, Kuo F, Kozak C, O’Hara CD, Leder P. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 1998;12:3382–93. doi: 10.1101/gad.12.21.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo G, Santoro IM, McDaniel LD, Nishijima I, Mills M, et al. Cancer predisposition caused by elevated mitotic recombination in Bloom mice. Nat Genet. 2000;26:424–29. doi: 10.1038/82548. [DOI] [PubMed] [Google Scholar]
- 47.Goss KH, Risinger MA, Kordich JJ, Sanz MM, Straughen JE, et al. Enhanced tumor formation in mice heterozygous for Blm mutation. Science. 2002;297:2051–53. doi: 10.1126/science.1074340. [DOI] [PubMed] [Google Scholar]
- 48.McDaniel LD, Chester N, Watson M, Borowsky AD, Leder P, Schultz RA. Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA Repair. 2003;2:1387–404. doi: 10.1016/j.dnarep.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 49.Chester N, Babbe H, Pinkas J, Manning C, Leder P. Mutation of the murine Bloom’s syndrome gene produces global genome destabilization. Mol Cell Biol. 2006;26:6713–26. doi: 10.1128/MCB.00296-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanz MM, German J. Bloom’s syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews at GeneTests: Medical Genetics Information Resource database. Seattle: Univ Wash; 2013. Updated March 28 2013 http://www.ncbi.nlm.nih.gov/books/NBK1398/ [Google Scholar]
- 51.German J. Bloom’s syndrome. XX The first 100 cancers. Cancer Genet Cytogenet. 1997;93:100–6. doi: 10.1016/s0165-4608(96)00336-6. [DOI] [PubMed] [Google Scholar]
- 52.Constantinou A, Tarsounas M, Karow JK, Brosh RM, Bohr VA, et al. Werner’s syndrome protein (WRN) migrates Holliday junctions and co-localizes with RPA upon replication arrest. EMBO Rep. 2000;1:80–84. doi: 10.1093/embo-reports/kvd004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baynton K, Otterlei M, Bjørås M, von Kobbe C, Bohr VA, Seeberg E. WRN interacts physically and functionally with the recombination mediator protein RAD52. J Biol Chem. 2003;278:36476–86. doi: 10.1074/jbc.M303885200. [DOI] [PubMed] [Google Scholar]
- 54.Singh DK, Karmakar P, Aamann M, Schurman SH, May A, et al. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell. 2010;9:358–71. doi: 10.1111/j.1474-9726.2010.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004;14:763–74. doi: 10.1016/j.molcel.2004.05.023. [DOI] [PubMed] [Google Scholar]
- 56.Huang S, Li B, Gray MD, Oshima J, Mian IS, Campisi J. The premature ageing syndrome protein, WRN, is a 3′ 5′ exonuclease. Nat Genet. 1998;20:114–16. doi: 10.1038/2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Perry JJ, Yannone SM, Holden LG, Hitomi C, Asaithamby A, et al. WRN exonuclease structure and molecular mechanism imply an editing role in DNA end processing. Nat Struct Mol Biol. 2006;13:414–22. doi: 10.1038/nsmb1088. [DOI] [PubMed] [Google Scholar]
- 58.Huang S, Beresten S, Li B, Oshima J, Ellis NA, Campisi J. Characterization of the human and mouse WRN 3′ 5′ exonuclease. Nucleic Acids Res. 2000;28:2396–405. doi: 10.1093/nar/28.12.2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen JC, Loeb LA. Werner syndrome exonuclease catalyzes structure-dependent degradation of DNA. Nucleic Acids Res. 2000;28:3260–68. doi: 10.1093/nar/28.17.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ozgenc A, Loeb LA. Current advances in unraveling the function of the Werner syndrome protein. Mutat Res. 2005;577:237–51. doi: 10.1016/j.mrfmmm.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 61.Machwe A, Ganunis R, Bohr VA, Orren DK. Selective blockage of the 3′ 5′ exonuclease activity of WRN protein by certain oxidative modifications and bulky lesions in DNA. Nucleic Acids Res. 2000;28:2762–70. doi: 10.1093/nar/28.14.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Driscoll HC, Matson SW, Sayer JM, Kroth H, Jerina DM, Brosh RM., Jr Inhibition of Werner syndrome helicase activity by benzo[c]phenanthrene diol epoxide dA adducts in DNA is both strand- and stereoisomer-dependent. J Biol Chem. 2003;278:41126–35. doi: 10.1074/jbc.M304798200. [DOI] [PubMed] [Google Scholar]
- 63.Choudhary S, Doherty KM, Handy CJ, Sayer JM, Yagi H, et al. Inhibition of Werner syndrome helicase activity by benzo[a]pyrene diol epoxide adducts can be overcome by replication protein A. J Biol Chem. 2006;281:6000–9. doi: 10.1074/jbc.M510122200. [DOI] [PubMed] [Google Scholar]
- 64.Harrigan JA, Fan J, Momand J, Perrino FW, Bohr VA, Wilson DM., 3rd WRN exonuclease activity is blocked by DNA termini harboring 3′ obstructive groups. Mech Ageing Dev. 2007;128:259–66. doi: 10.1016/j.mad.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bukowy Z, Harrigan JA, Ramsden DA, Tudek B, Bohr VA, Stevnsner T. WRN exonuclease activity is blocked by specific oxidatively induced base lesions positioned in either DNA strand. Nucleic Acids Res. 2008;36:4975–87. doi: 10.1093/nar/gkn468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci USA. 1998;95:13097–102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lombard DB, Beard C, Johnson B, Marciniak RA, Dausman J, et al. Mutations in the WRN gene in mice accelerate mortality in a p53-null background. Mol Cell Biol. 2000;20:3286–91. doi: 10.1128/mcb.20.9.3286-3291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moore G, Knoblaugh S, Gollahon K, Rabinovitch P, Ladiges W. Hyperinsulinemia and insulin resistance in Wrn null mice fed a diabetogenic diet. Mech Ageing Dev. 2008;129:201–6. doi: 10.1016/j.mad.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36:877–82. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 70.Goto M, Ishikawa Y, Sugimoto M, Furuichi Y. Werner syndrome: a changing pattern of clinical manifestations in Japan (1917–2008) Biosci Trends. 2013;7:13–22. [PubMed] [Google Scholar]
- 71.Lauper JM, Krause A, Vaughan TL, Monnat RJ., Jr Spectrum and risk of neoplasia in Werner syndrome: a systematic review. PLoS ONE. 2013;8:e59709. doi: 10.1371/journal.pone.0059709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yin J, Kwon YT, Varshavsky A, Wang W. RECQL4, mutated in the Rothmund-Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Hum Mol Genet. 2004;13:2421–30. doi: 10.1093/hmg/ddh269. [DOI] [PubMed] [Google Scholar]
- 73.Ghosh AK, Rossi ML, Singh DK, Dunn C, Ramamoorthy M, et al. RECQL4, the protein mutated in Rothmund–Thomson syndrome, functions in telomere maintenance. J Biol Chem. 2012;287:196–209. doi: 10.1074/jbc.M111.295063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Croteau DL, Rossi ML, Canugovi C, Tian J, Sykora P, et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell. 2012;11:456–66. doi: 10.1111/j.1474-9726.2012.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De S, Kumari J, Mudgal R, Modi P, Gupta S, et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J Cell Sci. 2012;125:2509–22. doi: 10.1242/jcs.101501. [DOI] [PubMed] [Google Scholar]
- 76.Ichikawa K, Noda T, Furuichi Y. Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund-Thomson syndrome caused by the mutation of DNA helicases. Folia Pharmacol Jpn. 2002;119:219–26. doi: 10.1254/fpj.119.219. (in Japanese) [DOI] [PubMed] [Google Scholar]
- 77.Hoki Y, Araki R, Fujimori A, Ohhata T, Koseki H, et al. Growth retardation and skin abnormalities of the Recql4-deficient mouse. Hum Mol Genet. 2003;12:2293–99. doi: 10.1093/hmg/ddg254. [DOI] [PubMed] [Google Scholar]
- 78.Mann MB, Hodges CA, Barnes E, Vogel H, Hassold TJ, Luo G. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund–Thomson syndrome. Hum Mol Genet. 2005;14:813–25. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- 79.Wang LL, Gannavarapu A, Kozinetz CA, Levy ML, Lewis RA, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund–Thomson syndrome. J Natl Cancer Inst. 2003;95:669–74. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- 80.Larizza L, Roversi G, Volpi L. Rothmund–Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Van Maldergem L. Bhaaller–Gerold syndrome. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews at GeneTests: Medical Genetics Information Resource database. Seattle: Univ Wash; 2013. Updated June 7, 2011 http://www.ncbi.nlm.nih.gov/books/NBK1204/ [Google Scholar]
- 82.Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–58. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Croteau DL, Rossi ML, Ross J, Dawut L, Dunn C, et al. RAPADILINO RECQL4 mutant protein lacks helicase and ATPase activity. Biochim Biophys Acta. 2012;1822:1727–34. doi: 10.1016/j.bbadis.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jensen MB, Dunn CA, Keijzers G, Kulikowicz T, Rasmussen LJ, et al. The helicase and ATPase activities of RECQL4 are compromised by mutations reported in three human patients. Aging. 2012;4:790–802. doi: 10.18632/aging.100506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Croteau DL, Singh DK, Hoh Ferrarelli L, Lu H, Bohr VA. RECQL4 in genomic instability and aging. Trends Genet. 2012;28:624–31. doi: 10.1016/j.tig.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shimamoto A, Nishikawa K, Kitao S, Furuichi Y. Human RecQ5β, a large isomer of RecQ5 DNA helicase, localizes in the nucleoplasm and interacts with topoisomerases 3α and 3β. Nucleic Acids Res. 2000;28:1647–55. doi: 10.1093/nar/28.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kawabe T, Tsuyama N, Kitao S, Nishikawa K, Shimamoto A, et al. Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene. 2000;19:4764–72. doi: 10.1038/sj.onc.1203841. [DOI] [PubMed] [Google Scholar]
- 88.Popuri V, Tadokoro T, Croteau DL, Bohr VA. Human RECQL5: guarding the crossroads of DNA replication and transcription and providing backup capability. Crit Rev Biochem Mol Biol. 2013;48:289–99. doi: 10.3109/10409238.2013.792770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aygün O, Xu X, Liu Y, Takahashi H, Kong SE, et al. Direct inhibition of RNA polymerase II transcription by RECQL5. J Biol Chem. 2009;284:23197–203. doi: 10.1074/jbc.M109.015750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Islam MN, Fox D, III, Guo R, Enomoto T, Wang W. RecQL5 promotes genome stabilization through two parallel mechanisms—interacting with RNA polymerase II and acting as a helicase. Mol Cell Biol. 2010;30:2460–72. doi: 10.1128/MCB.01583-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeong YS, Kang Y, Lim KH, Lee MH, Lee J, Koo HS. Deficiency of Caenorhabditis elegans RecQ5 homologue reduces life span and increases sensitivity to ionizing radiation. DNA Repair. 2003;2:1309–19. doi: 10.1016/j.dnarep.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 92.Hu Y, Raynard S, Sehorn MG, Lu X, Bussen W, et al. RECQL5/Recql5 helicase regulates homologous recombination and suppresses tumor formation via disruption of Rad51 presynaptic filaments. Genes Dev. 2007;21:3073–84. doi: 10.1101/gad.1609107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wilson DM, III, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair. 2007;6:544–59. doi: 10.1016/j.dnarep.2006.10.017. [DOI] [PubMed] [Google Scholar]
- 94.Das A, Boldogh I, Lee JW, Harrigan JA, Hegde ML, et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J Biol Chem. 2007;282:26591–602. doi: 10.1074/jbc.M703343200. [DOI] [PubMed] [Google Scholar]
- 95.Ahn B, Harrigan JA, Indig FE, Wilson DM, III, Bohr VA. Regulation of WRN helicase activity in human base excision repair. J Biol Chem. 2004;279:53465–74. doi: 10.1074/jbc.M409624200. [DOI] [PubMed] [Google Scholar]
- 96.Schurman SH, Hedayati M, Wang Z, Singh DK, Speina E, et al. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum Mol Genet. 2009;18:3470–83. doi: 10.1093/hmg/ddp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang D, Luo M, Kelley MR. Human apurinic endonuclease 1 (APE1) expression and prognostic significance in osteosarcoma: enhanced sensitivity of osteosarcoma to DNA damaging agents using silencing RNA APE1 expression inhibition. Mol Cancer Ther. 2004;3:679–86. [PubMed] [Google Scholar]
- 98.Harrigan JA, Opresko PL, von Kobbe C, Kedar PS, Prasad R, et al. The Werner syndrome protein stimulates DNA polymerase β strand displacement synthesis via its helicase activity. J Biol Chem. 2003;278:22686–95. doi: 10.1074/jbc.M213103200. [DOI] [PubMed] [Google Scholar]
- 99.Speina E, Dawut L, Hedayati M, Wang Z, May A, et al. Human RECQL5β stimulates flap endonuclease 1. Nucleic Acids Res. 2010;38:2904–16. doi: 10.1093/nar/gkp1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harrigan JA, Wilson DM, III, Prasad R, Opresko PL, Beck G, et al. The Werner syndrome protein operates in base excision repair and cooperates with DNA polymerase beta. Nucleic Acids Res. 2006;34:745–54. doi: 10.1093/nar/gkj475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brosh RM, Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, et al. Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J. 2001;20:5791–801. doi: 10.1093/emboj/20.20.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Sharma S, Sommers JA, Wu L, Bohr VA, Hickson ID, Brosh RM., Jr Stimulation of flap endonuclease-1 by the Bloom’s syndrome protein. J Biol Chem. 2004;279:9847–56. doi: 10.1074/jbc.M309898200. [DOI] [PubMed] [Google Scholar]
- 103.Sharma S, Sommers JA, Gary RK, Friedrich-Heineken E, Hubscher U, Brosh RM., Jr The interaction site of flap endonuclease 1 with WRN helicase suggests a coordination of WRN and PCNA. Nucleic Acids Res. 2005;33:6769–81. doi: 10.1093/nar/gki1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sharma S, Otterlei M, Sommers JA, Driscoll HC, Dianov GL, et al. WRN helicase and FEN-1 form a complex upon replication arrest and together process branch migrating DNA structures associated with the replication fork. Mol Biol Cell. 2004;15:734–50. doi: 10.1091/mbc.E03-08-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sousa FG, Matuo R, Soares DG, Escargueil AE, Henriques JA, et al. PARPs and the DNA damage response. Carcinogenesis. 2012;33:1433–40. doi: 10.1093/carcin/bgs132. [DOI] [PubMed] [Google Scholar]
- 106.Thomas C, Tulin AV. Poly-ADP-ribose polymerase: machinery for nuclear processes. Mol Asp Med. 2013;34:1124–37. doi: 10.1016/j.mam.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.von Kobbe C, Harrigan JA, May A, Opresko PL, Dawut L, et al. Central role for the Werner syndrome protein/poly(ADP-ribose) polymerase 1 complex in the poly(ADP-ribosyl)ation pathway after DNA damage. Mol Cell Biol. 2003;23:8601–13. doi: 10.1128/MCB.23.23.8601-8613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sharma S, Phatak P, Stortchevoi A, Jasin M, Larocque JR. RECQ1 plays a distinct role in cellular response to oxidative DNA damage. DNA Repair. 2012;11:537–49. doi: 10.1016/j.dnarep.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gottipati P, Vischioni B, Schultz N, Solomons J, Bryant HE, et al. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010;70:5389–98. doi: 10.1158/0008-5472.CAN-09-4716. [DOI] [PubMed] [Google Scholar]
- 110.Tadokoro T, Ramamoorthy M, Popuri V, May A, Tian J, et al. Human RECQL5 participates in the removal of endogenous DNA damage. Mol Biol Cell. 2012;23:4273–85. doi: 10.1091/mbc.E12-02-0110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Popp O, Veith S, Fahrer J, Bohr VA, Burkle A, Mangerich A. Site-specific noncovalent interaction of the biopolymer poly(ADP-ribose) with the Werner syndrome protein regulates protein functions. ACS Chem Biol. 2013;8:179–88. doi: 10.1021/cb300363g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM. The Rothmund–Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res. 2006;312:3443–57. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 113.Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31:955–60. doi: 10.1093/carcin/bgq064. [DOI] [PubMed] [Google Scholar]
- 114.Kamileri I, Karakasilioti I, Garinis GA. Nucleotide excision repair: new tricks with old bricks. Trends Genet. 2012;28:566–73. doi: 10.1016/j.tig.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 115.Trego KS, Chernikova SB, Davalos AR, Perry JJ, Finger LD, et al. The DNA repair endonuclease XPG interacts directly and functionally with the WRN helicase defective in Werner syndrome. Cell Cycle. 2011;10:1998–2007. doi: 10.4161/cc.10.12.15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fan W, Luo J. RecQ4 facilitates UV light–induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA) J Biol Chem. 2008;283:29037–44. doi: 10.1074/jbc.M801928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 118.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, et al. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J. 1998;17:5497–508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rooney S, Chaudhuri J, Alt FW. The role of the non-homologous end-joining pathway in lymphocyte development. Immunol Rev. 2004;200:115–31. doi: 10.1111/j.0105-2896.2004.00165.x. [DOI] [PubMed] [Google Scholar]
- 121.Fattah F, Lee EH, Weisensel N, Wang Y, Lichter N, Hendrickson EA. Ku regulates the non-homologous end joining pathway choice of DNA double-strand break repair in human somatic cells. PLoS Genet. 2010;6:e1000855. doi: 10.1371/journal.pgen.1000855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med. 2010;207:855–65. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bunting SF, Callen E, Wong N, Chen HT, Polato F, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cooper MP, Machwe A, Orren DK, Brosh RM, Ramsden D, Bohr VA. Ku complex interacts with and stimulates the Werner protein. Genes Dev. 2000;14:907–12. [PMC free article] [PubMed] [Google Scholar]
- 125.Li B, Comai L. Functional interaction between Ku and the Werner syndrome protein in DNA end processing. J Biol Chem. 2000;275:28349–52. doi: 10.1074/jbc.C000289200. [DOI] [PubMed] [Google Scholar]
- 126.Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, et al. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. J Biol Chem. 2001;276:38242–48. doi: 10.1074/jbc.M101913200. [DOI] [PubMed] [Google Scholar]
- 127.Karmakar P, Snowden CM, Ramsden DA, Bohr VA. Ku heterodimer binds to both ends of the Werner protein and functional interaction occurs at the Werner N-terminus. Nucleic Acids Res. 2002;30:3583–91. doi: 10.1093/nar/gkf482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kusumoto-Matsuo R, Opresko PL, Ramsden D, Tahara H, Bohr VA. Cooperation of DNA-PKcs and WRN helicase in the maintenance of telomeric D-loops. Aging. 2010;2:274–84. doi: 10.18632/aging.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kusumoto R, Dawut L, Marchetti C, Wan Lee J, Vindigni A, et al. Werner protein cooperates with the XRCC4-DNA ligase IV complex in end-processing. Biochemistry. 2008;47:7548–56. doi: 10.1021/bi702325t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bohr VA, Souza Pinto N, Nyaga SG, Dianov G, Kraemer K, et al. DNA repair and mutagenesis in Werner syndrome. Environ Mol Mutagen. 2001;38:227–34. doi: 10.1002/em.1076. [DOI] [PubMed] [Google Scholar]
- 131.Oshima J, Huang S, Pae C, Campisi J, Schiestl RH. Lack of WRN results in extensive deletion at nonhomologous joining ends. Cancer Res. 2002;62:547–51. [PubMed] [Google Scholar]
- 132.Damerla RR, Knickelbein KE, Strutt S, Liu FJ, Wang H, Opresko PL. Werner syndrome protein suppresses the formation of large deletions during the replication of human telomeric sequences. Cell Cycle. 2012;11:3036–44. doi: 10.4161/cc.21399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chen L, Huang S, Lee L, Davalos A, Schiestl RH, et al. WRN, the protein deficient in Werner syndrome, plays a critical structural role in optimizing DNA repair. Aging Cell. 2003;2:191–99. doi: 10.1046/j.1474-9728.2003.00052.x. [DOI] [PubMed] [Google Scholar]
- 134.Parvathaneni S, Stortchevoi A, Sommers JA, Brosh RM, Jr, Sharma S. Human RECQ1 interacts with Ku70/80 and modulates DNA end-joining of double-strand breaks. PLoS ONE. 2013;8:e62481. doi: 10.1371/journal.pone.0062481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:55117–26. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 136.Wang M, Wu W, Wu W, Rosidi B, Zhang L, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nat Struct Mol Biol. 2009;16:808–13. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16:814–18. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct Mol Biol. 2011;18:80–84. doi: 10.1038/nsmb.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bothmer A, Rommel PC, Gazumyan A, Polato F, Reczek CR, et al. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J Exp Med. 2013;210:115–23. doi: 10.1084/jem.20121975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang H, Rosidi B, Perrault R, Wang M, Zhang L, et al. DNA ligase III as a candidate component of backup pathways of nonhomologous end joining. Cancer Res. 2005;65:4020–30. doi: 10.1158/0008-5472.CAN-04-3055. [DOI] [PubMed] [Google Scholar]
- 142.Aggarwal M, Sommers JA, Morris C, Brosh RM., Jr Delineation of WRN helicase function with EXO1 in the replicational stress response. DNA Repair. 2010;9:765–76. doi: 10.1016/j.dnarep.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–72. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC. Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair. Proc Natl Acad Sci USA. 2008;105:16906–11. doi: 10.1073/pnas.0809380105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Doherty KM, Sharma S, Uzdilla LA, Wilson TM, Cui S, et al. RECQ1 helicase interacts with human mismatch repair factors that regulate genetic recombination. J Biol Chem. 2005;280:28085–94. doi: 10.1074/jbc.M500265200. [DOI] [PubMed] [Google Scholar]
- 146.Cheng WH, von Kobbe C, Opresko PL, Arthur LM, Komatsu K, et al. Linkage between Werner syndrome protein and the Mre11 complex via Nbs1. J Biol Chem. 2004;279:21169–76. doi: 10.1074/jbc.M312770200. [DOI] [PubMed] [Google Scholar]
- 147.Cheng WH, Sakamoto S, Fox JT, Komatsu K, Carney J, Bohr VA. Werner syndrome protein associates with H2AX in a manner that depends upon Nbs1. FEBS Lett. 2005;579:1350–56. doi: 10.1016/j.febslet.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 148.Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011;25:350–62. doi: 10.1101/gad.2003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sallmyr A, Tomkinson AE, Rassool FV. Up-regulation of WRN and DNA ligase IIIα in chronic myeloid leukemia: consequences for the repair of DNA double-strand breaks. Blood. 2008;112:1413–23. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Babbe H, McMenamin J, Hobeika E, Wang J, Rodig SJ, et al. Genomic instability resulting from Blm deficiency compromises development, maintenance, and function of the B cell lineage. J Immunol. 2009;182:347–60. doi: 10.4049/jimmunol.182.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cui S, Klima R, Ochem A, Arosio D, Falaschi A, Vindigni A. Characterization of the DNA-unwinding activity of human RECQ1, a helicase specifically stimulated by human replication protein A. J Biol Chem. 2003;278:1424–32. doi: 10.1074/jbc.M209407200. [DOI] [PubMed] [Google Scholar]
- 152.Brosh RM, Jr, Li JL, Kenny MK, Karow JK, Cooper MP, et al. Replication protein A physically interacts with the Bloom’s syndrome protein and stimulates its helicase activity. J Biol Chem. 2000;275:23500–8. doi: 10.1074/jbc.M001557200. [DOI] [PubMed] [Google Scholar]
- 153.Brosh RM, Jr, Orren DK, Nehlin JO, Ravn PH, Kenny MK, et al. Functional and physical interaction between WRN helicase and human replication protein A. J Biol Chem. 1999;274:18341–50. doi: 10.1074/jbc.274.26.18341. [DOI] [PubMed] [Google Scholar]
- 154.Rossi ML, Ghosh AK, Kulikowicz T, Croteau DL, Bohr VA. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair. 2010;9:796–804. doi: 10.1016/j.dnarep.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, et al. Human CtIP promotes DNA end resection. Nature. 2007;450:509–14. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, et al. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature. 2010;467:112–16. doi: 10.1038/nature09355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–74. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–94. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liao S, Toczylowski T, Yan H. Mechanistic analysis of Xenopus EXO1’s function in 5′-strand resection at DNA double-strand breaks. Nucleic Acids Res. 2011;39:5967–77. doi: 10.1093/nar/gkr216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yan H, Toczylowski T, McCane J, Chen C, Liao S. Replication protein A promotes 5′ 3′ end processing during homology-dependent DNA double-strand break repair. J Cell Biol. 2011;192:251–61. doi: 10.1083/jcb.201005110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Wu L, Davies SL, Levitt NC, Hickson ID. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J Biol Chem. 2001;276:19375–81. doi: 10.1074/jbc.M009471200. [DOI] [PubMed] [Google Scholar]
- 162.Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the Bloom’s syndrome helicase. Genes Dev. 2007;21:3085–94. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Schwendener S, Raynard S, Paliwal S, Cheng A, Kanagaraj R, et al. Physical interaction of RECQ5 helicase with RAD51 facilitates its anti-recombinase activity. J Biol Chem. 2010;285:15739–45. doi: 10.1074/jbc.M110.110478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang W, Seki M, Narita Y, Nakagawa T, Yoshimura A, et al. Functional relation among RecQ family helicases RecQL1, RecQL5, and BLM in cell growth and sister chromatid exchange formation. Mol Cell Biol. 2003;23:3527–35. doi: 10.1128/MCB.23.10.3527-3535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hu Y, Lu X, Barnes E, Yan M, Lou H, Luo G. Recql5 and Blm RecQ DNA helicases have nonredundant roles in suppressing crossovers. Mol Cell Biol. 2005;25:3431–42. doi: 10.1128/MCB.25.9.3431-3442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Prince PR, Emond MJ, Monnat RJ., Jr Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001;15:933–38. doi: 10.1101/gad.877001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Franchitto A, Pirzio LM, Prosperi E, Sapora O, Bignami M, Pichierri P. Replication fork stalling in WRN-deficient cells is overcome by prompt activation of a MUS81-dependent pathway. J Cell Biol. 2008;183:241–52. doi: 10.1083/jcb.200803173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ., Jr Homologous recombination resolution defect in Werner syndrome. Mol Cell Biol. 2002;22:6971–78. doi: 10.1128/MCB.22.20.6971-6978.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Rodriguez-Lopez AM, Whitby MC, Borer CM, Bachler MA, Cox LS. Correction of proliferation and drug sensitivity defects in the progeroid Werner’s syndrome by Holliday junction resolution. Rejuvenation Res. 2007;10:27–40. doi: 10.1089/rej.2006.0503. [DOI] [PubMed] [Google Scholar]
- 170.Cheng WH, Kusumoto R, Opresko PL, Sui X, Huang S, et al. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. 2006;34:2751–60. doi: 10.1093/nar/gkl362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Otterlei M, Bruheim P, Ahn B, Bussen W, Karmakar P, et al. Werner syndrome protein participates in a complex with RAD51, RAD54, RAD54B and ATR in response to ICL-induced replication arrest. J Cell Sci. 2006;119:5137–46. doi: 10.1242/jcs.03291. [DOI] [PubMed] [Google Scholar]
- 172.Kohzaki M, Chiourea M, Versini G, Adachi N, Takeda S, et al. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis. 2012;33:1203–10. doi: 10.1093/carcin/bgs149. [DOI] [PubMed] [Google Scholar]
- 173.Petkovic M, Dietschy T, Freire R, Jiao R, Stagljar I. The human Rothmund–Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci. 2005;118:4261–69. doi: 10.1242/jcs.02556. [DOI] [PubMed] [Google Scholar]
- 174.LeRoy G, Carroll R, Kyin S, Seki M, Cole MD. Identification of RecQL1 as a Holliday junction processing enzyme in human cell lines. Nucleic Acids Res. 2005;33:6251–57. doi: 10.1093/nar/gki929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Bugreev DV, Brosh RM, Jr, Mazin AV. RECQ1 possesses DNA branch migration activity. J Biol Chem. 2008;283:20231–42. doi: 10.1074/jbc.M801582200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sowd G, Lei M, Opresko PL. Mechanism and substrate specificity of telomeric protein POT1 stimulation of the Werner syndrome helicase. Nucleic Acids Res. 2008;36:4242–56. doi: 10.1093/nar/gkn385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Opresko PL, Sowd G, Wang H. The Werner syndrome helicase/exonuclease processes mobile D-loops through branch migration and degradation. PLoS ONE. 2009;4:e4825. doi: 10.1371/journal.pone.0004825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Karow JK, Constantinou A, Li JL, West SC, Hickson ID. The Bloom’s syndrome gene product promotes branch migration of Holliday junctions. Proc Natl Acad Sci USA. 2000;97:6504–8. doi: 10.1073/pnas.100448097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bachrati CZ, Borts RH, Hickson ID. Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 2006;34:2269–79. doi: 10.1093/nar/gkl258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ip SC, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–61. doi: 10.1038/nature07470. [DOI] [PubMed] [Google Scholar]
- 181.Ciccia A, McDonald N, West SC. Structural and functional relationships of the XPF/MUS81 family of proteins. Annu Rev Biochem. 2008;77:259–87. doi: 10.1146/annurev.biochem.77.070306.102408. [DOI] [PubMed] [Google Scholar]
- 182.Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, et al. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009;138:78–89. doi: 10.1016/j.cell.2009.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Munoz IM, Hain K, Declais AC, Gardiner M, Toh GW, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009;35:116–27. doi: 10.1016/j.molcel.2009.06.020. [DOI] [PubMed] [Google Scholar]
- 184.Svendsen JM, Smogorzewska A, Sowa ME, O’Connell BC, Gygi SP, et al. Mammalian BTBD12/SLX4 assembles a Holliday junction resolvase and is required for DNA repair. Cell. 2009;138:63–77. doi: 10.1016/j.cell.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Adams MD, McVey M, Sekelsky JJ. Drosophila BLM in double-strand break repair by synthesis-dependent strand annealing. Science. 2003;299:265–67. doi: 10.1126/science.1077198. [DOI] [PubMed] [Google Scholar]
- 186.Matsuno K, Kumano M, Kubota Y, Hashimoto Y, Takisawa H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase α in the initiation of DNA replication. Mol Cell Biol. 2006;26:4843–52. doi: 10.1128/MCB.02267-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, et al. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund–Thomson syndrome. Cell. 2005;121:887–98. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 188.Abe T, Yoshimura A, Hosono Y, Tada S, Seki M, Enomoto T. The N-terminal region of RECQL4 lacking the helicase domain is both essential and sufficient for the viability of vertebrate cells: role of the N-terminal region of RECQL4 in cells. Biochim Biophys Acta. 2011;1813:473–79. doi: 10.1016/j.bbamcr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 189.Capp C, Wu J, Hsieh TS. Drosophila RecQ4 has a 3′–5′ DNA helicase activity that is essential for viability. J Biol Chem. 2009;284:30845–52. doi: 10.1074/jbc.M109.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Xu X, Rochette PJ, Feyissa EA, Su TV, Liu Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009;28:3005–14. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Im JS, Ki SH, Farina A, Jung DS, Hurwitz J, Lee JK. Assembly of the Cdc45–Mcm2-7–GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc Natl Acad Sci USA. 2009;106:15628–32. doi: 10.1073/pnas.0908039106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Xu Y, Lei Z, Huang H, Dui W, Liang X, et al. dRecQ4 is required for DNA synthesis and essential for cell proliferation in Drosophila. PLoS ONE. 2009;4:e6107. doi: 10.1371/journal.pone.0006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bartos JD, Wang W, Pike JE, Bambara RA. Mechanisms by which Bloom protein can disrupt recombination intermediates of Okazaki fragment maturation. J Biol Chem. 2006;281:32227–39. doi: 10.1074/jbc.M606310200. [DOI] [PubMed] [Google Scholar]
- 194.Poot M, Hoehn H, Runger TM, Martin GM. Impaired S-phase transit of Werner syndrome cells expressed in lymphoblastoid cell lines. Exp Cell Res. 1992;202:267–73. doi: 10.1016/0014-4827(92)90074-i. [DOI] [PubMed] [Google Scholar]
- 195.Hand R, German J. A retarded rate of DNA chain growth in Bloom’s syndrome. Proc Natl Acad Sci USA. 1975;72:758–62. doi: 10.1073/pnas.72.2.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sidorova JM, Kehrli K, Mao F, Monnat R., Jr Distinct functions of human RECQ helicases WRN and BLM in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair. 2013;12:128–39. doi: 10.1016/j.dnarep.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sidorova JM, Li N, Folch A, Monnat RJ., Jr The RecQ helicase WRN is required for normal replication fork progression after DNA damage or replication fork arrest. Cell Cycle. 2008;7:796–807. doi: 10.4161/cc.7.6.5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Franchitto A, Pichierri P. Werner syndrome protein and the MRE11 complex are involved in a common pathway of replication fork recovery. Cell Cycle. 2004;3:1331–39. doi: 10.4161/cc.3.10.1185. [DOI] [PubMed] [Google Scholar]
- 199.Kamath-Loeb AS, Loeb LA, Johansson E, Burgers PMJ, Fry M. Interactions between the Werner syndrome helicase and DNA polymerase δ specifically facilitate copying of tetraplex and hairpin structures of the d(CGG)n trinucleotide repeat sequence. J Biol Chem. 2001;276:16439–46. doi: 10.1074/jbc.M100253200. [DOI] [PubMed] [Google Scholar]
- 200.Selak N, Bachrati CZ, Shevelev I, Dietschy T, van Loon B, et al. The Bloom’s syndrome helicase (BLM) interacts physically and functionally with p12, the smallest subunit of human DNA polymerase δ. Nucleic Acids Res. 2008;36:5166–79. doi: 10.1093/nar/gkn498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lukusa T, Fryns JP. Human chromosome fragility. Biochim Biophys Acta. 2008;1779:3–16. doi: 10.1016/j.bbagrm.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 202.Glover TW. Common fragile sites. Cancer Lett. 2006;232:4–12. doi: 10.1016/j.canlet.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 203.Pirzio LM, Pichierri P, Bignami M, Franchitto A. Werner syndrome helicase activity is essential in maintaining fragile site stability. J Cell Biol. 2008;180:305–14. doi: 10.1083/jcb.200705126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Murfuni I, De Santis A, Federico M, Bignami M, Pichierri P, Franchitto A. Perturbed replication induced genome wide or at common fragile sites is differently managed in the absence of WRN. Carcinogenesis. 2012;33:1655–63. doi: 10.1093/carcin/bgs206. [DOI] [PubMed] [Google Scholar]
- 205.Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753–60. doi: 10.1038/ncb1882. [DOI] [PubMed] [Google Scholar]
- 206.Pellicioli A, Muzi-Falconi M. A blooming resolvase at chromosomal fragile sites. Nat Cell Biol. 2013;15:883–85. doi: 10.1038/ncb2812. [DOI] [PubMed] [Google Scholar]
- 207.Shah SN, Opresko PL, Meng X, Lee MY, Eckert KA. DNA structure and the Werner protein modulate human DNA polymerase δ–dependent replication dynamics within the common fragile site FRA16D. Nucleic Acids Res. 2010;38:1149–62. doi: 10.1093/nar/gkp1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Lu X, Parvathaneni S, Hara T, Lal A, Sharma S. Replication stress induces specific enrichment of RECQ1 at common fragile sites FRA3B and FRA16D. Mol Cancer. 2013;12:29. doi: 10.1186/1476-4598-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol. 2012;13:141–52. doi: 10.1038/nrm3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kamath-Loeb AS, Lan L, Nakajima S, Yasui A, Loeb LA. Werner syndrome protein interacts functionally with translesion DNA polymerases. Proc Natl Acad Sci USA. 2007;104:10394–99. doi: 10.1073/pnas.0702513104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Maddukuri L, Ketkar A, Eddy S, Zafar MK, Griffin WC, Eoff RL. Enhancement of human DNA polymerase activity and fidelity is dependent upon a bipartite interaction with the Werner syndrome protein. J Biol Chem. 2012;287:42312–23. doi: 10.1074/jbc.M112.410332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Phillips LG, Sale JE. The Werner’s syndrome protein collaborates with REV1 to promote replication fork progression on damaged DNA. DNA Repair. 2010;9:1064–72. doi: 10.1016/j.dnarep.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Kamath-Loeb AS, Shen JC, Schmitt MW, Loeb LA. The Werner syndrome exonuclease facilitates DNA degradation and high fidelity DNA polymerization by human DNA polymerase δ. J Biol Chem. 2012;287:12480–90. doi: 10.1074/jbc.M111.332577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Brosh RM, Jr, Waheed J, Sommers JA. Biochemical characterization of the DNA substrate specificity of Werner syndrome helicase. J Biol Chem. 2002;277:23236–45. doi: 10.1074/jbc.M111446200. [DOI] [PubMed] [Google Scholar]
- 215.Choudhary S, Sommers JA, Brosh RM., Jr Biochemical and kinetic characterization of the DNA helicase and exonuclease activities of Werner syndrome protein. J Biol Chem. 2004;279:34603–13. doi: 10.1074/jbc.M401901200. [DOI] [PubMed] [Google Scholar]
- 216.Machwe A, Lozada EM, Xiao L, Orren DK. Competition between the DNA unwinding and strand pairing activities of the Werner and Bloom syndrome proteins. BMC Mol Biol. 2006;7:1. doi: 10.1186/1471-2199-7-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Machwe A, Xiao L, Groden J, Orren DK. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–46. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- 218.Machwe A, Xiao L, Lloyd RG, Bolt E, Orren DK. Replication fork regression in vitro by the Werner syndrome protein (WRN): Holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res. 2007;35:5729–47. doi: 10.1093/nar/gkm561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Popuri V, Croteau DL, Brosh RM, Jr, Bohr VA. RECQ1 is required for cellular resistance to replication stress and catalyzes strand exchange on stalled replication fork structures. Cell Cycle. 2012;11:4252–65. doi: 10.4161/cc.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Kanagaraj R, Saydam N, Garcia PL, Zheng L, Janscak P. Human RECQ5β helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res. 2006;34:5217–31. doi: 10.1093/nar/gkl677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Chaudhuri AR, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, et al. Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat Struct Mol Biol. 2012;19:417–23. doi: 10.1038/nsmb.2258. [DOI] [PubMed] [Google Scholar]
- 222.Aygün O, Svejstrup JQ. RECQL5 helicase: connections to DNA recombination and RNA polymerase II transcription. DNA Repair. 2010;9:345–53. doi: 10.1016/j.dnarep.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 223.Ellis NA, Lennon DJ, Proytcheva M, Alhadeff B, Henderson EE, German J. Somatic intragenic recombination within the mutated locus BLM can correct the high sister-chromatid exchange phenotype of Bloom syndrome cells. Am J Hum Genet. 1995;57:1019–27. [PMC free article] [PubMed] [Google Scholar]
- 224.Chan KL, Hickson ID. New insights into the formation and resolution of ultra-fine anaphase bridges. Semin Cell Dev Biol. 2011;22:906–12. doi: 10.1016/j.semcdb.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 225.Manthei KA, Keck JL. The BLM dissolvasome in DNA replication and repair. Cell Mol Life Sci. 2013;70:4067–84. doi: 10.1007/s00018-013-1325-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Cejka P, Plank JL, Dombrowski CC, Kowalczykowski SC. Decatenation of DNA by the S. cerevisiae Sgs1-Top3-Rmi1 and RPA complex: a mechanism for disentangling chromosomes. Mol Cell. 2012;47:886–96. doi: 10.1016/j.molcel.2012.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ramamoorthy M, Tadokoro T, Rybanska I, Ghosh AK, Wersto R, et al. RECQL5 cooperates with topoisomerase II α in DNA decatenation and cell cycle progression. Nucleic Acids Res. 2012;40:1621–35. doi: 10.1093/nar/gkr844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Russell B, Bhattacharyya S, Keirsey J, Sandy A, Grierson P, et al. Chromosome breakage is regulated by the interaction of the BLM helicase and topoisomerase IIα. Cancer Res. 2011;71:561–71. doi: 10.1158/0008-5472.CAN-10-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Calado R, Young N. Telomeres in disease. F1000 Med Rep. 2012;4:8. doi: 10.3410/M4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11:171–81. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liu D, O’Connor MS, Qin J, Songyang Z. Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem. 2004;279:51338–42. doi: 10.1074/jbc.M409293200. [DOI] [PubMed] [Google Scholar]
- 232.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 233.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–60. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 234.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–52. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 235.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–98. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 236.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–97. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Du X, Shen J, Kugan N, Furth EE, Lombard DB, et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol. 2004;24:8437–46. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Suram A, Kaplunov J, Patel PL, Ruan H, Cerutti A, et al. Oncogene-induced telomere dysfunction enforces cellular senescence in human cancer precursor lesions. EMBO J. 2012;31:2839–51. doi: 10.1038/emboj.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Opresko PL, von Kobbe C, Laine JP, Harrigan J, Hickson ID, Bohr VA. Telomere-binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem. 2002;277:41110–19. doi: 10.1074/jbc.M205396200. [DOI] [PubMed] [Google Scholar]
- 241.Lillard-Wetherell K, Machwe A, Langland GT, Combs KA, Behbehani GK, et al. Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum Mol Genet. 2004;13:1919–32. doi: 10.1093/hmg/ddh193. [DOI] [PubMed] [Google Scholar]
- 242.Machwe A, Xiao L, Orren DK. TRF2 recruits the Werner syndrome (WRN) exonuclease for processing of telomeric DNA. Oncogene. 2004;23:149–56. doi: 10.1038/sj.onc.1206906. [DOI] [PubMed] [Google Scholar]
- 243.Li B, Jog SP, Reddy S, Comai L. WRN controls formation of extrachromosomal telomeric circles and is required for TRF2ΔB-mediated telomere shortening. Mol Cell Biol. 2008;28:1892–904. doi: 10.1128/MCB.01364-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Opresko PL, Mason PA, Podell ER, Lei M, Hickson ID, et al. POT1 stimulates RecQ helicases WRN and BLM to unwind telomeric DNA substrates. J Biol Chem. 2005;280:32069–80. doi: 10.1074/jbc.M505211200. [DOI] [PubMed] [Google Scholar]
- 245.Lee OH, Kim H, He Q, Baek HJ, Yang D, et al. Genome-wide YFP fluorescence complementation screen identifies new regulators for telomere signaling in human cells. Mol Cell Proteomics. 2011;10:M110001628. doi: 10.1074/mcp.M110.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Reddy S, Li B, Comai L. Processing of human telomeres by the Werner syndrome protein. Cell Cycle. 2010;9:3137–38. doi: 10.4161/cc.9.16.12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science. 2004;306:1951–53. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 248.Liu FJ, Barchowsky A, Opresko PL. The Werner syndrome protein suppresses telomeric instability caused by chromium (VI) induced DNA replication stress. PLoS ONE. 2010;5:e11152. doi: 10.1371/journal.pone.0011152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Arnoult N, Saintome C, Ourliac-Garnier I, Riou JF, Londono-Vallejo A. Human POT1 is required for efficient telomere C-rich strand replication in the absence of WRN. Genes Dev. 2009;23:2915–24. doi: 10.1101/gad.544009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Mohaghegh P, Karow JK, Brosh RM, Jr, Bohr VA, Hickson ID. The Bloom’s and Werner’s syndrome proteins are DNA structure–specific helicases. Nucleic Acids Res. 2001;29:2843–49. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Fouché N, Özgür S, Roy D, Griffith JD. Replication fork regression in repetitive DNAs. Nucleic Acids Res. 2006;34:6044–50. doi: 10.1093/nar/gkl757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Machwe A, Karale R, Xu X, Liu Y, Orren DK. The Werner and Bloom syndrome proteins help resolve replication blockage by converting (regressed) Holliday junctions to functional replication forks. Biochemistry. 2011;50:6774–88. doi: 10.1021/bi2001054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ferrarelli LK, Popuri V, Ghosh AK, Tadokoro T, Canugovi C, et al. The RECQL4 protein, deficient in Rothmund–Thomson syndrome is active on telomeric D-loops containing DNA metabolism blocking lesions. DNA Repair. 2013;12:518–28. doi: 10.1016/j.dnarep.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, et al. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–14. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 255.Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006;127:709–20. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 256.Vannier JB, Pavicic-Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell. 2012;149:795–806. doi: 10.1016/j.cell.2012.03.030. [DOI] [PubMed] [Google Scholar]
- 257.Wang RC, Smogorzewska A, de Lange T. Homologous recombination generates T-loop-sized deletions at human telomeres. Cell. 2004;119:355–68. doi: 10.1016/j.cell.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 258.Hagelstrom RT, Blagoev KB, Niedernhofer LJ, Goodwin EH, Bailey SM. Hyper telomere recombination accelerates replicative senescence and may promote premature aging. Proc Natl Acad Sci USA. 2010;107:15768–73. doi: 10.1073/pnas.1006338107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Laud PR, Multani AS, Bailey SM, Wu L, Ma J, et al. Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev. 2005;19:2560–70. doi: 10.1101/gad.1321305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Mendez-Bermudez A, Hidalgo-Bravo A, Cotton VE, Gravani A, Jeyapalan JN, Royle NJ. The roles of WRN and BLM RecQ helicases in the alternative lengthening of telomeres. Nucleic Acids Res. 2012;40:10809–20. doi: 10.1093/nar/gks862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Bhattacharyya S, Keirsey J, Russell B, Kavecansky J, Lillard-Wetherell K, et al. Telomerase-associated protein 1, HSP90, and topoisomerase IIα associate directly with the BLM helicase in immortalized cells using ALT and modulate its helicase activity using telomeric DNA substrates. J Biol Chem. 2009;284:14966–77. doi: 10.1074/jbc.M900195200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Temime-Smaali N, Guittat L, Wenner T, Bayart E, Douarre C, et al. Topoisomerase IIIα is required for normal proliferation and telomere stability in alternative lengthening of telomeres. EMBO J. 2008;27:1513–24. doi: 10.1038/emboj.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Barber LJ, Youds JL, Ward JD, McIlwraith MJ, O’Neil NJ, et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell. 2008;135:261–71. doi: 10.1016/j.cell.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Uringa EJ, Youds JL, Lisaingo K, Lansdorp PM, Boulton SJ. RTEL1: an essential helicase for telomere maintenance and the regulation of homologous recombination. Nucleic Acids Res. 2011;39:1647–55. doi: 10.1093/nar/gkq1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–39. [PMC free article] [PubMed] [Google Scholar]
- 266.Deans AJ, West SC. FANCM connects the genome instability disorders Bloom’s syndrome and Fanconi anemia. Mol Cell. 2009;36:943–53. doi: 10.1016/j.molcel.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 267.Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, et al. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 2011;30:692–705. doi: 10.1038/emboj.2010.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Chaudhury I, Sareen A, Raghunandan M, Sobeck A. FANCD2 regulates BLM complex functions independently of FANCI to promote replication fork recovery. Nucleic Acids Res. 2013;41:6444–59. doi: 10.1093/nar/gkt348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Pichierri P, Ammazzalorso F, Bignami M, Franchitto A. The Werner syndrome protein: linking the replication checkpoint response to genome stability. Aging. 2011;3:311–18. doi: 10.18632/aging.100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Cheng WH, Muftic D, Muftuoglu M, Dawut L, Morris C, et al. WRN is required for ATM activation and the S-phase checkpoint in response to interstrand cross-link-induced DNA double-strand breaks. Mol Biol Cell. 2008;19:3923–33. doi: 10.1091/mbc.E07-07-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Patro BS, Frohlich R, Bohr VA, Stevnsner T. WRN helicase regulates the ATR-CHK1-induced S-phase checkpoint pathway in response to topoisomerase-I-DNA covalent complexes. J Cell Sci. 2011;124:3967–79. doi: 10.1242/jcs.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pichierri P, Nicolai S, Cignolo L, Bignami M, Franchitto A. The RAD9-RAD1-HUS1 (9.1.1) complex interacts with WRN and is crucial to regulate its response to replication fork stalling. Oncogene. 2012;31:2809–23. doi: 10.1038/onc.2011.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Pichierri P, Rosselli F, Franchitto A. Werner’s syndrome protein is phosphorylated in an ATR/ATM-dependent manner following replication arrest and DNA damage induced during the S phase of the cell cycle. Oncogene. 2003;22:1491–500. doi: 10.1038/sj.onc.1206169. [DOI] [PubMed] [Google Scholar]
- 275.Ammazzalorso F, Pirzio LM, Bignami M, Franchitto A, Pichierri P. ATR and ATM differently regulate WRN to prevent DSBs at stalled replication forks and promote replication fork recovery. EMBO J. 2010;29:3156–69. doi: 10.1038/emboj.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Davies SL, North PS, Dart A, Lakin ND, Hickson ID. Phosphorylation of the Bloom’s syndrome helicase and its role in recovery from S-phase arrest. Mol Cell Biol. 2004;24:1279–91. doi: 10.1128/MCB.24.3.1279-1291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Park SJ, Lee YJ, Beck BD, Lee SH. A positive involvement of RecQL4 in UV-induced S-phase arrest. DNA Cell Biol. 2006;25:696–703. doi: 10.1089/dna.2006.25.696. [DOI] [PubMed] [Google Scholar]
- 278.Grierson PM, Acharya S, Groden J. Collaborating functions of BLM and DNA topoisomerase I in regulating human rDNA transcription. Mutat Res. 2013;743–744:89–96. doi: 10.1016/j.mrfmmm.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Laine JP, Opresko PL, Indig FE, Harrigan JA, von Kobbe C, Bohr VA. Werner protein stimulates topoisomerase I DNA relaxation activity. Cancer Res. 2003;63:7136–46. [PubMed] [Google Scholar]
- 280.Christmann M, Tomicic MT, Gestrich C, Roos WP, Bohr VA, Kaina B. WRN protects against topo I but not topo II inhibitors by preventing DNA break formation. DNA Repair. 2008;7:1999–2009. doi: 10.1016/j.dnarep.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Sidorova JM. Roles of the Werner syndrome RecQ helicase in DNA replication. DNA Repair. 2008;7:1776–86. doi: 10.1016/j.dnarep.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Shiratori M, Suzuki T, Itoh C, Goto M, Furuichi Y, Matsumoto T. WRN helicase accelerates the transcription of ribosomal RNA as a component of an RNA polymerase I–associated complex. Oncogene. 2002;21:2447–54. doi: 10.1038/sj.onc.1205334. [DOI] [PubMed] [Google Scholar]
- 283.Brosh RM., Jr DNA helicases involved in DNA repair and their roles in cancer. Nat Rev Cancer. 2013;13:542–58. doi: 10.1038/nrc3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Aggarwal M, Sommers JA, Shoemaker RH, Brosh RM., Jr Inhibition of helicase activity by a small molecule impairs Werner syndrome helicase (WRN) function in the cellular response to DNA damage or replication stress. Proc Natl Acad Sci USA. 2011;108:1525–30. doi: 10.1073/pnas.1006423108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Nguyen GH, Dexheimer TS, Rosenthal AS, Chu WK, Singh DK, et al. A small molecule inhibitor of the BLM helicase modulates chromosome stability in human cells. Chem Biol. 2013;20:55–62. doi: 10.1016/j.chembiol.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Aggarwal M, Banerjee T, Sommers JA, Iannascoli C, Pichierri P, et al. Werner syndrome helicase has a critical role in DNA damage responses in the absence of a functional Fanconi anemia pathway. Cancer Res. 2013;73:5497–507. doi: 10.1158/0008-5472.CAN-12-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Moser R, Toyoshima M, Robinson K, Gurley KE, Howie HL, et al. MYC-driven tumorigenesis is inhibited by WRN syndrome gene deficiency. Mol Cancer Res. 2012;10:535–45. doi: 10.1158/1541-7786.MCR-11-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.von Kobbe C, Karmakar P, Dawut L, Opresko P, Zeng X, et al. Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins. J Biol Chem. 2002;277:22035–44. doi: 10.1074/jbc.M200914200. [DOI] [PubMed] [Google Scholar]
- 289.Singh DK, Popuri V, Kulikowicz T, Shevelev I, Ghosh AK, et al. The human RecQ helicases BLM and RECQL4 cooperate to preserve genome stability. Nucleic Acids Res. 2012;40:6632–48. doi: 10.1093/nar/gks349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J. 2003;374:577–606. doi: 10.1042/BJ20030491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Sharma S, Sommers JA, Choudhary S, Faulkner JK, Cui S, et al. Biochemical analysis of the DNA unwinding and strand annealing activities catalyzed by human RECQ1. J Biol Chem. 2005;280:28072–84. doi: 10.1074/jbc.M500264200. [DOI] [PubMed] [Google Scholar]
- 292.Popuri V, Bachrati CZ, Muzzolini L, Mosedale G, Costantini S, et al. The human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J Biol Chem. 2008;283:17766–76. doi: 10.1074/jbc.M709749200. [DOI] [PubMed] [Google Scholar]
- 293.Ghosh A, Rossi ML, Aulds J, Croteau D, Bohr VA. Telomeric D-loops containing 8-oxo-2′-deoxyguanosine are preferred substrates for Werner and Bloom syndrome helicases and are bound by POT1. J Biol Chem. 2009;284:31074–84. doi: 10.1074/jbc.M109.027532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Xu X, Liu Y. Dual DNA unwinding activities of the Rothmund–Thomson syndrome protein, RECQ4. EMBO J. 2009;28:568–77. doi: 10.1038/emboj.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Zittel MC, Keck JL. Coupling DNA-binding and ATP hydrolysis in Escherichia coli RecQ: role of a highly conserved aromatic-rich sequence. Nucleic Acids Res. 2005;33:6982–91. doi: 10.1093/nar/gki999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Karow JK, Newman RH, Freemont PS, Hickson ID. Oligomeric ring structure of the Bloom’s syndrome helicase. Curr Biol. 1999;9:597–600. doi: 10.1016/s0960-9822(99)80264-4. [DOI] [PubMed] [Google Scholar]
- 297.Vindigni A, Hickson ID. RecQ helicases: multiple structures for multiple functions? HFSP J. 2009;3:153–64. doi: 10.2976/1.3079540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Suzuki T, Kohno T, Ishimi Y. DNA helicase activity in purified human RECQL4 protein. J Biochem. 2009;146:327–35. doi: 10.1093/jb/mvp074. [DOI] [PubMed] [Google Scholar]
- 299.Muzzolini L, Beuron F, Patwardhan A, Popuri V, Cui S, et al. Different quaternary structures of human RECQ1 are associated with its dual enzymatic activity. PLoS Biol. 2007;5:e20. doi: 10.1371/journal.pbio.0050020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Cheng WH, von Kobbe C, Opresko PL, Fields KM, Ren J, et al. Werner syndrome protein phosphorylation by Abl tyrosine kinase regulates its activity and distribution. Mol Cell Biol. 2003;23:6385–95. doi: 10.1128/MCB.23.18.6385-6395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Harrigan JA, Piotrowski J, Di Noto L, Levine RL, Bohr VA. Metal-catalyzed oxidation of the Werner syndrome protein causes loss of catalytic activities and impaired protein-protein interactions. J Biol Chem. 2007;282:36403–11. doi: 10.1074/jbc.M706107200. [DOI] [PubMed] [Google Scholar]
- 302.Muftuoglu M, Kusumoto R, Speina E, Beck G, Cheng WH, Bohr VA. Acetylation regulates WRN catalytic activities and affects base excision DNA repair. PLoS ONE. 2008;3:e1918. doi: 10.1371/journal.pone.0001918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Blander G, Zalle N, Daniely Y, Taplick J, Gray MD, Oren M. DNA damage-induced translocation of the Werner helicase is regulated by acetylation. J Biol Chem. 2002;277:50934–40. doi: 10.1074/jbc.M210479200. [DOI] [PubMed] [Google Scholar]
- 304.Kawabe Y, Seki M, Seki T, Wang WS, Imamura O, et al. Covalent modification of the Werner’s syndrome gene product with the ubiquitin-related protein, SUMO-1. J Biol Chem. 2000;275:20963–66. doi: 10.1074/jbc.C000273200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.