

Abstract
[Rh(CO)2Cl]2 is as an effective catalyst for endo-selective cyclizations and cascades of epoxy-(E)-enoate alcohols, thus enabling the synthesis of oxepanes and oxepane-containing polyethers from di- and trisubstituted epoxides. Syntheses of the ABC and EF ring systems of (−)-brevisin via all endo-diepoxide-opening cascades using this method constitute a formal total synthesis and demonstrate the utility of this methodology in the context of the synthesis of marine ladder polyether natural products.
Introduction
The unique structural features, limited abundance, and potent biological activity of the marine ladder polyether family of natural products have inspired many innovative achievements in total synthesis enabled by the development of new methodology.1 Guided by the biogenesis proposed for these compounds,2 several groups have investigated the feasibility of all-endo3 epoxide-opening cascades as a potentially rapid and general approach to these polyethers.4 An ongoing challenge, these kinetically disfavored processes have been addressed in part by previous methodology developed in our laboratory.5 Enabling expeditious synthesis of polytetrahydropyran fragments, template-guided, water-promoted cascades nevertheless as yet do not appear to be amenable to the synthesis of oxepanes, 7-membered rings that represent an important motif present in every natural ladder polyether isolated to date. Herein we demonstrate a new tactic that not only constructs oxepane rings, but also provides a new means for selective initiation of epoxide-opening cascades, as embodied by a formal synthesis of (−)-brevisin.
In the same vein as the use of epoxides and allylic alcohols as sites for selective initiation of polyene cyclizations towards sterols,6 we envisioned that an appropriately substituted alkenyl epoxide7 with a suitable activator might play a similar role in epoxide-opening cascades. Previous examples of selective initiation of epoxide-opening cascades include the Holton synthesis of hemibrevetoxin B, wherein an electrophilic selenium reagent triggered nucleophilic attack by an epoxide,8 the photochemical generation of an oxocarbenium described by Floreancig and Houk,9 and bromonium formation in a epoxide-opening cascade we utilized en route to dioxepandehydrothyrsiferol.10 Finally, although the Lewis acid-promoted epoxide-opening cascades developed by McDonald did not utilize a site-selective initiation mechanism, they nevertheless demonstrated an important and unusual endo selectivity in the construction of trans-fused bis-oxepanes.11
Our design is depicted in Scheme 1 and can be summarized as follows: incorporation of an electronically tailored alkene at the distal12 epoxide would provide a specific site for complexation and activation by a transition metal. The use of transition metals to selectively activate alkenyl epoxides for nucleophilic attack has excellent and diverse precedents. Although Pd catalysis is the most well known,13 we eschewed this path because of the limited examples of oxygen nucleophiles in this context and, more importantly, the likelihood that an undesired stereochemical outcome would be observed, i.e., net retention (double inversion) at the site of epoxide opening, rather than the necessary inversion of configuration. In contrast, [Rh(CO)2Cl]2 has been shown to activate alkenyl epoxides for intermolecular opening by alcohol and amine nucleophiles with inversion of stereochemistry.14 Further development by Ha and coworkers led to cyclizations of trans-disubstituted enoate epoxy-alcohols and carbamates to provide five- and six-membered saturated heterocycles.15 Prior to our investigations, however, no examples of oxepane formation via [Rh(CO)2Cl]2 catalysis, activation of trisubstituted epoxides, or its use in initiation of epoxide-opening cascades had been reported.
Scheme 1.
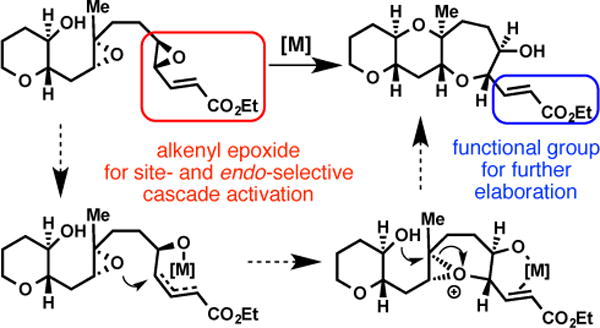
Substrate and promoter combination designed for selective initiation of all-endo epoxide-opening cascades
Important in these designs was consideration of the natural product (−)-brevisin (1), isolated by Wright and Baden in 2008 from the dinoflagellate Karenia brevis.16 The use of two cascades as described above (and shown in Scheme 2) would intercept two tricyclic intermediates (2 and 3) in the only previous total synthesis of (−)-brevisin from Tachibana and coworkers.17
Scheme 2.
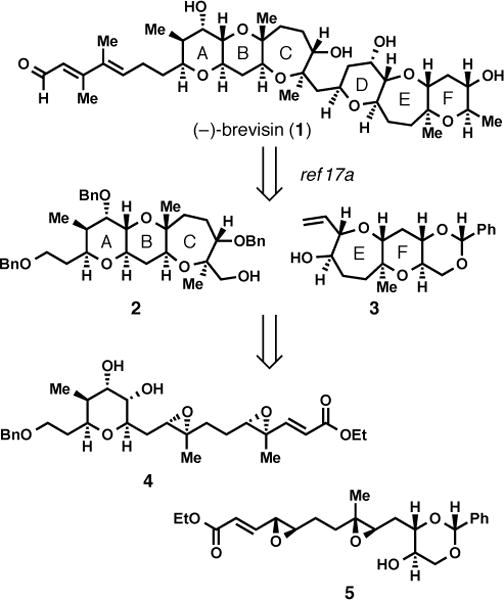
Retrosynthetic Analysis of (−)-Brevisin (1)
Results and Discussion
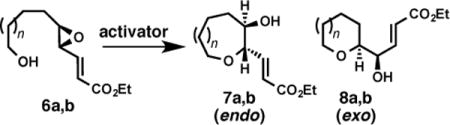
Before embarking on epoxide-opening cascades towards (−)-brevisin, we first explored the feasibility of oxepane formation from epoxy alcohols promoted by [Rh(CO)2Cl]2.. Subjecting trans-disubstituted epoxy alcohol 6b with the required enoate π-activating group18 to conditions for [Rh(CO)2Cl]2 catalysis provided the desired oxepane 7b as the only observable product (Table 1, entry 3). This combination of alkenyl epoxide and Rh catalysis provides rapid access to oxepanes from relatively simple starting materials19 via endo-cyclization with stereospecific inversion observed at the formed O–C bond.
Table 1.
Trans-disubstituted epoxy alcohol cyclizations under [Rh(CO)2Cl]2 and (±)-CSA promotion
| ||||
|---|---|---|---|---|
| entry | substrate | activator | 7/8 | yield 7 (%)a |
| 1 | 6a, n = 0 | [Rh(CO)2Cl]2b | >20 : 1 | 94 |
| 2 | 6a, n = 0 | (±)-CSAc | 1 : 1 | – |
| 3 | 6b, n = 1 | [Rh(CO)2Cl]2d | >20 : 1 | 81 |
| 4 | 6b, n = 1 | (±)-CSAe | 1 : 3 | – |
Isolated yield.
[Rh(CO)2Cl]2 (2.5 mol %), THF, rt.
(±)-CSA (10 mol %), CH2Cl2, rt.
[Rh(CO)2Cl]2 (5 mol %), THF, rt.
(±)-CSA (100 mol %), CH2Cl2, rt.
Noteworthy by contrast were the results observed under Brønsted acid catalysis. For example, subjecting epoxy alcohol 6b to (±)-CSA activation conditions afforded primarily the smaller ring (exo), THP 8b, consistent with results reported Nicolaou and coworkers (Table 1, entry 4).20 The reversal of regioselectivity by [Rh(CO)2Cl]2 relative to acid catalysis supports an alternative mechanism for epoxide activation beyond a typical Lewis acid. We found similar direct comparisons in these studies to be useful measures of the biasing ability of the enoate and [Rh(CO)2Cl]2 combination on regioselectivity.
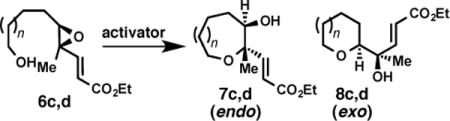
Critical to the success of this method toward the synthesis of the ABC tricycle of (−)-brevisin, distal methyl substitution was well tolerated under [Rh(CO)2Cl]2 promotion, providing complete endo selectivity for the synthesis of both tetrahydropyran (7c) and oxepane (7d) from epoxy alcohols 6c and 6d respectively (Table 2, entries 1 and 3). In comparison, promotion with (±)-CSA yielded a mixture of endo- and exo-products, albeit with a slight improvement in regioselectivity compared to disubstituted epoxides 6a and 6b. Encouraged by these one-oxepane studies, we turned our attention to epoxide-opening cascades.
Table 2.
Trisubstituted epoxy alcohol cyclizations under [Rh(CO)2Cl]2 and (±)-CSA promotion
| ||||
|---|---|---|---|---|
| entry | substrate | activator | 7/8 | yield 7 (%)a |
| 1 | 6c, n = 0 | [Rh(CO)2Cl]2b | >20: 1 | 93 |
| 2 | 6c, n = 0 | (±)-CSAc | 3.4: 1 | – |
| 3 | 6d, n = 1 | [Rh(CO)2Cl]2d | >20: 1 | 88 |
| 4 | 6d, n = 1 | (±)-CSAe | 1: 1.8 | – |
Isolated yield.
[Rh(CO)2Cl]2 (1 mol %), THF, rt.
(±)-CSA (10 mol %), CH2Cl2, rt.
[Rh(CO)2Cl]2 (2.5 mol %), THF, rt.
(±)-CSA (100 mol %), CH2Cl2, rt.
Toward the EF fragment, the route illustrated in Scheme 3 allowed for rapid access to the desired cascade precursor 5. Ozonolysis of known enoate 9,21 followed by nucleophilic addition of isopropenyl magnesium bromide and a tandem vinylation–Claisen process22 afforded aldehyde 10. Olefination of 1023 and subsequent asymmetric Shi epoxidation24 and TBAF desilylation rapidly provides cascade precursor 5 in six steps from enoate 9.25
Scheme 3. Synthesis of EF diepoxy cascade precursors and formal synthesis target.
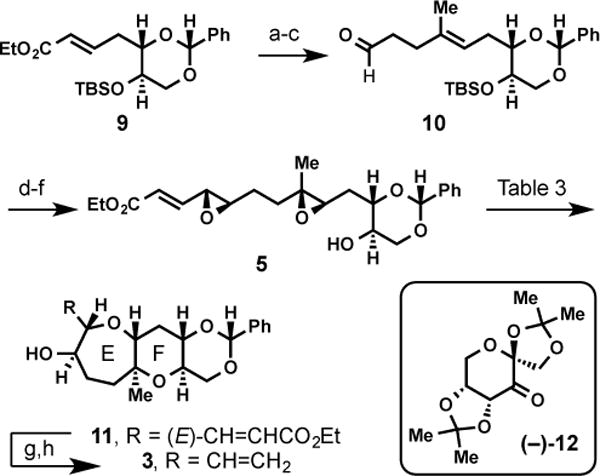
Reaction conditions: a) O3, CH2Cl2, −78 °C; then Me2S, 82%; b) H2C=C(CH3)MgBr, THF, −78 °C to 0 °C, 1:1 dr, 89%; c) Triethyleneglycol divinyl ether, (1,10-phenanthroline)Pd(OAc)2, 80 °C, 7:1 E/Z, 63%; d) LDA, (MeO)2P(O)CH2CH=CHCO2Et, −78 °C to rt, 2:1 E/Z, 84%; e) (−)-12, KHSO5, Bu4NHSO4, K2CO3, pH 10.5, DMM/CH3CN (2:1), 22%; f) TBAF, THF, rt, 69%; g) O3, CH2Cl2/MeOH (4:1), −78 °C; Ph3P, −78 °C to rt; h) Ph3PCH3Br, KOt-Bu, THF, 0 °C to rt, 84% (over 2 steps).
Investigation of cascade promoters for diepoxy alcohol 5 containing an (E)-enoate revealed [Rh(CO)2Cl]2 to be a highly chemo-, stereo-, and regioselective promoter – in the desired fashion. For example, [Rh(CO)2Cl]2 catalyzed the regioselective epoxide-opening cascade of (E)-enoate-diepoxy alcohol 5 to provide the desired product 11 in 38% isolated yield (Table 3, entry 1). Further optimization by employing 1,4-dioxane as the solvent and performing the reaction at elevated temperatures (65 °C) provided a 61% yield. Lewis or Brønsted acid activation, which we and others have employed in other all-endo epoxide-opening cascades, provided none of the desired product for enoate diepoxide 5, again highlighting the exquisite selectivity of [Rh(CO)2Cl]2 catalysis (entries 3 and 4). The ester functional group is critical to the success of the method; vinyl-diepoxide 13 under [Rh(CO)2Cl]2 catalysis provided lower yields (entry 5). Alternatively, conditions utilized by McDonald in methyl-11b or vinyl-directed11c epoxide-opening cascades led to the desired product (3), but also in a lower yield (entries 6–8). These results support the mechanistic hypothesis of Rh(I) activation of alkenyl epoxides via π-coordination and oxidative addition into the vinylic C–O bond of the epoxide, which contrasts the generally non site-selective epoxide activation with Lewis acids. Importantly, these results represent the first examples of both a cascade process and a seven-membered ring formation using this method. By ozonolysis followed by Wittig reaction, (E)-enoate 11 was converted to 3, corresponding to the EF-ring system of (−)-brevisin (1).
Table 3.
Investigation of diepoxide cascade towards EF Fragment
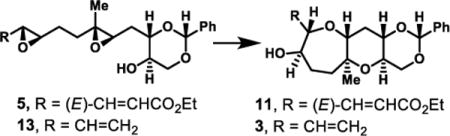
| ||||
|---|---|---|---|---|
| entry | substrate | conditions | product | yield (%) |
| 1 | 5 | [Rh(CO)2Cl]2, THF, rt | 11 | 38a |
| 2 | 5 | [Rh(CO)2Cl]2, Dioxane, 65°C | 11 | 61a |
| 3 | 5 | BF3•OEt2, CH2Cl2, −78°C | 11 | _b,c |
| 4 | 5 | CSA, CH2Cl2, rt | 11 | _b,c |
| 5 | 13 | [Rh(CO)2Cl]2, THF, rt | 3 | 21a |
| 6 | 13 | BF3•OEt2, CH2Cl2, −78°C | 3 | 23a |
| 7 | 13 | Yb(OTf)3, CH2Cl2, rt | 3 | 38b |
| 8 | 13 | Eu(OTf)3, CH2Cl2, rt | 3 | 26b |
Isolated yield.
Yield determined by 1H NMR against mesitylene standard.
– represents no observed product.
Toward the ABC fragment, synthesis of the A ring proceeded via the previously reported route to lactone 1417c (Scheme 4). This lactone was elaborated by diastereoselective dihydroxylation, and the incipient side chain installed through allyl Grignard addition. Triethylsilane reduction of the intermediate lactol, and acetylation provided the fully elaborated A ring (15).
Scheme 4. Synthetic route to ABC diepoxide-opening cascade precursor and formal synthesis target.
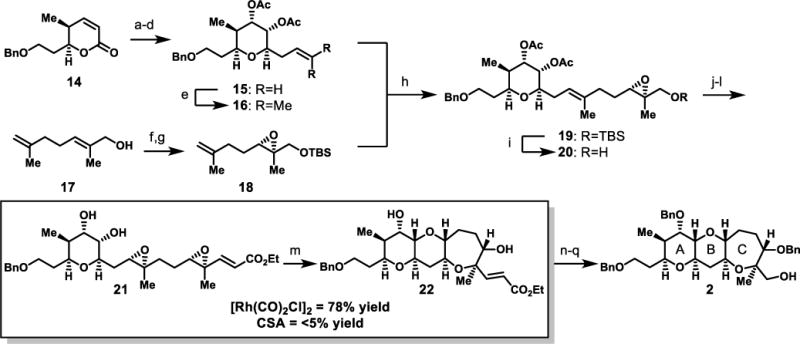
a) OsO4, NMO, Acetone/H2O (4:1), 95%; b) CH2=CHCH2MgBr, THF, −78 °C; c) Et3SiH, TMSOTf, MeCN, 44% (over 2 steps); d) Ac2O, pyr, DMAP, CH2Cl2, 87%; e) 2-methyl-2-butene, Hoveyda-Grubbs cat. (2nd generation), Benzoquinone, 87%; f) L-(+)-DET, Ti(OiPr)4, TBHP, CH2Cl2, −20 °C, 49%, 93% e.e.; g) TBSCl, Et3N, CH2Cl2, 86%; h) Hoveyda-Grubbs cat. (2nd generation), Benzoquinone, 80 °C, 78%, 2:1 E/Z; i) TBAF, THF, 84%; j) pyr•SO3, DMSO, Et3N, CH2Cl2, rt; then Ph3PCHCO2Et, rt, 91%; k) (+)-12, KHSO5, Bu4NHSO4, K2CO3, pH 10.5, DMM/CH3CN (2:1), 84%, 3:1 d.r.; l) Guanidine•HCl, NaOEt, EtOH, 77%; m) [Rh(CO)2Cl]2 (10 mol%), THF, rt, 78%; n) NaH, BnBr, TBAI, THF, 60 °C, 75%; o) K2OsO2(OH)4, Citric Acid, NMO, t-BuOH/H2O; p) Ph3BiCO3, CH2Cl2, reflux; q) NaBH4, MeOH, 0 °C, 60% (over 3 steps).
Elaboration of the allyl group of 15 was accomplished via cross metathesis with 18,26 giving trisubstituted alkene 19, albeit in only 31% yield. Motivated by reports of higher yields in cross metathesis of allyl groups with increased steric hindrance,27 alkene 16 was prepared from 15 via cross metathesis with 2-methyl-2-butene.28 In comparison, cross metathesis of 18 and trisubstituted alkene 16 provided a significantly higher yield, particularly when performed in the absence of additional solvent. Despite the modest stereoselectivity, the 2:1 E:Z mixture of alkene isomers could be separated by column chromatography. The enoate-directing group was installed by desilylation of 19, alcohol oxidation, and in-situ stabilized-Wittig olefination. Asymmetric Shi epoxidation, and acetate ethanolysis provided the diepoxide cascade precursor 21.
Exposure of diepoxide 21 to catalytic [Rh(CO)2Cl]2 at ambient temperature in THF led to full conversion and 78% yield of the desired ABC tricycle (22). Efforts directed toward lowering the catalyst loading led to inferior yields. Brønsted acid promotion with (±)-CSA did not provide any desired product, further differentiating [Rh(CO)2Cl]2 from acid promoted conditions. Completion of the formal synthesis of (−)-brevisin (1) was achieved by benzylation of both hydroxyl groups, oxidative cleavage of the pendant enoate via dihydroxylation and diol cleavage,29 and finally aldehyde reduction to provide alcohol 2, corresponding to the ABC-ring system of (−)-brevisin (1).
To help further our understanding of these successful diepoxide-opening reactions, we have put forth a mechanism for the ABC-ring cascade, shown in Scheme 5. The proposed epoxonium pathway invokes formation of a bicyclo[4.1.0] epoxonium by [Rh(CO)2Cl]2 activation of the distal epoxy-enoate, subsequent attack by the central epoxide, then rapid trapping by the A-ring hydroxyl. We favor the epoxonium pathway versus a related stepwise transformation30 as it provides a more consistent explanation of the high yield obtained under what would generally be regarded mild conditions.
Scheme 5. Proposed mechanism for Rh-promoted epoxide-opening cascade of diepoxide 22.
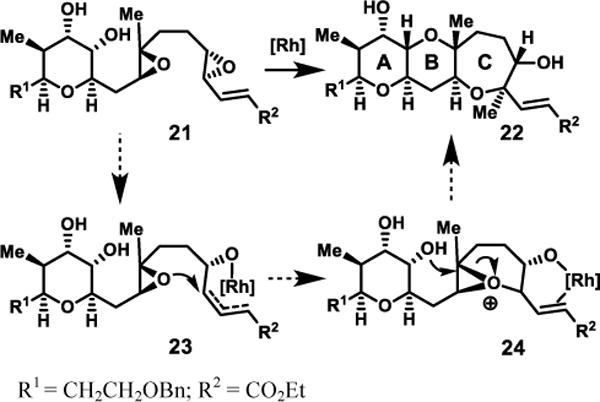
Further insight into the putative mechanism is suggested by our initial cyclization studies of epoxides with proximal methyl substitution. These substrates, which represent one of the most challenging substitution patterns to achieve high endo selectivity in epoxy alcohol cyclizations,31 have shown significant promise thus far. For example, use of the enoate and [Rh(CO)2Cl]2 overrides the near complete exo-selectivity observed with CSA for proximal methyl substitution, (Table 4, entries 1 and 2). Notably, however, measurable quantities of the exo products are observed – in contrast to the previous substrates tested (vide supra). Higher catalyst loadings and heating are required for full conversion in these cases. While the yield and selectivity are lower in the case of formation of oxepane 7f (Table 4, entry 3), this result suggests nevertheless an alternative approach to override the exo-directing methyl group in this very challenging case of oxepane formation. These results together with the diepoxide cascades further suggest that [Rh(CO)2Cl]2 is activating alkenyl epoxides via a distinct mechanism relative to Brønsted or Lewis acid catalysis.
Table 4.
Proximal-methyl (E)-trisubstituted epoxy alcohols cyclizations under [Rh(CO)2Cl]2 and (±)-CSA promotion
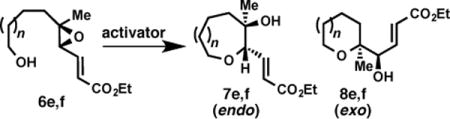
| ||||
|---|---|---|---|---|
| entry | substrate | activator | 7: 8 | yield 7 (%)a |
| 1 | 6e, n = 0 | [Rh(CO)2Cl]2b | 12: 1 | 81 |
| 2 | 6e, n = 0 | (±)-CSAc | 1: >20 | – |
| 3 | 6f, n = 1 | [Rh(CO)2Cl]2d | 4: 1 | 21 |
| 4 | 6f, n = 1 | (±)-CSAe | 1: >20 | – |
Isolated yield.
[Rh(CO)2Cl]2 (10 mol %), 1,4-Dioxane, 80 °C.
(±)-CSA (10 mol %), CH2Cl2, rt.
[Rh(CO)2Cl]2 (10 mol %), THF, 60 °C.
(±)-CSA (100 mol %), CH2Cl2, rt.
Conclusions
In summary, the combination of an enoate group and [Rh(CO)2Cl]2 catalysis is effective not only for the synthesis of 6- and 7-membered oxygen heterocycles from epoxy alcohols bearing a variety of substitution patters, but also cascades of diepoxides where control of the sequence of epoxide opening events is tantamount to success. The high yield, stereo-specificity, functional group compatibility, and endo-selectivity make this approach particularly well suited for target-directed synthesis, as highlighted by the formal synthesis of (−)-brevisin. Further application of this methodology towards other oxepane-containing natural products is an active area of research, as is investigation of other Rh(I) catalysts and alkene directing groups.
Supplementary Material
Acknowledgments
We thank the NIGMS (GM72566 and fellowship to M.G.B., F32GM095014) and the NSF (Graduate Research Fellowship to K.W.A.) for financial support of this work. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We also thank Li Li and Eric A. Standley (both MIT) for HRMS analyses, which were conducted on an instrument purchased with the assistance of NSF grant CHE-0234877. NMR spectroscopy was carried out on instruments purchased in part with funds provided by the NSF (CHE-9808061 and CHE-8915028). We are grateful to Elizabeth H. Kelley and Eric A. Standley (both MIT) for insightful discussions and input during the preparation of this manuscript.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
No competing financial interests have been declared.
References
- 1.(a) Nakata T. Chem Rev. 2005;105:4314. doi: 10.1021/cr040627q. [DOI] [PubMed] [Google Scholar]; (b) Inoue M. Chem Rev. 2005;105:4379. doi: 10.1021/cr0406108. [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Frederick MO, Aversa RJ. Angew Chem, Int Ed. 2008;47:7182. doi: 10.1002/anie.200801696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Nakanishi K. Toxicon. 1985;23:473. doi: 10.1016/0041-0101(85)90031-5. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC. Angew Chem, Int Ed Engl. 1996;35:588. [Google Scholar]
- 3.Baldwin JE. J Chem Soc, Chem Commun. 1976:734. [Google Scholar]
- 4.Vilotijevic I, Jamison TF. Angew Chem, Int Ed. 2009;48:5250. doi: 10.1002/anie.200900600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Vilotijevic I, Jamison TF. Science. 2007;317:1189. doi: 10.1126/science.1146421. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Van Dyke AR, Jamison TF. Angew Chem, Int Ed. 2009;48:4430. doi: 10.1002/anie.200900924. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Morten CJ, Jamison TF. J Am Chem Soc. 2009;131:6678. doi: 10.1021/ja9025243. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Morten CJ, Byers JA, Van Dyke AR, Vilotijevic I, Jamison TF. Chem Soc Rev. 2009;38:3175. doi: 10.1039/b816697h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abe I, Rohmer M, Prestwich GD. Chem Rev. 1993;93:2189. [Google Scholar]; (b) Van Tamelen EE. Acc Chem Res. 1975;8:152. [Google Scholar]; (c) Johnson WS, Gravestock MB, McCarry BE. J Am Chem Soc. 1971;93:4332. doi: 10.1021/ja00746a062. [DOI] [PubMed] [Google Scholar]; (d) Gravestock MB, Johnson WS, McCarry BE, Parry RJ, Ratcliffe BE. J Am Chem Soc. 1978;100:4274. [Google Scholar]; (e) Corey EJ, Luo G, Lin LS. J Am Chem Soc. 1997;119:9927. [Google Scholar]
- 7.For a recent review on vinyl epoxides in organic synthesis, see:; He J, Ling J, Chiu P. Chem Rev. 2014;114:8037. doi: 10.1021/cr400709j. [DOI] [PubMed] [Google Scholar]
- 8.Zakarian A, Batch A, Holton RA. J Am Chem Soc. 2003;125:7822. doi: 10.1021/ja029225v. [DOI] [PubMed] [Google Scholar]
- 9.Wan S, Gunaydin H, Houk KN, Floreancig PE. J Am Chem Soc. 2007;129:7915. doi: 10.1021/ja0709674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Tanuwidjaja J, Ng S-S, Jamison TF. J Am Chem Soc. 2009;131:12084. doi: 10.1021/ja9052366. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Underwood BS, Tanuwidjaja J, Ng S-S, Jamison TF. Tetrahedron. 2013;69:5205. doi: 10.1016/j.tet.2013.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) McDonald FE, Wang X, Do B, Hardcastle KI. Org Lett. 2000;2:2917. doi: 10.1021/ol0064009. [DOI] [PubMed] [Google Scholar]; (b) McDonald FE, Bravo F, Wang X, Wei X, Toganoh M, Rodríguez JR, Do B, Neiwert WA, Hardcastle KI. J Org Chem. 2002;67:2515. doi: 10.1021/jo0110092. [DOI] [PubMed] [Google Scholar]; (c) Bravo F, McDonald FE, Neiwert Wa, Hardcastle KI. Org Lett. 2004;6:4487. doi: 10.1021/ol048212e. [DOI] [PubMed] [Google Scholar]; (d) Valentine JC, McDonald FE, Neiwert WA, Hardcastle KI. J Am Chem Soc. 2005;127:4586. doi: 10.1021/ja050013i. [DOI] [PubMed] [Google Scholar]; (e) McDonald FE, Tong R, Valentine JC, Bravo F. Pure Appl Chem. 2007;79:281. [Google Scholar]
- 12.Distal is defined as the epoxide furthest from the terminating alcohol nucleophile.
- 13.Tsuji J, Kataoka H, Kobayashi Y. Tetrahedron Lett. 1981;22:2575. [Google Scholar]; (b) Trost BM, Molander GA. J Am Chem Soc. 1981;103:5969. [Google Scholar]; (c) Trost BM, Tenaglia A. Tetrahedron Lett. 1988;29:2931. [Google Scholar]; (d) Trost BM, McEachern EJ, Toste FD. J Am Chem Soc. 1998;120:12702. [Google Scholar]; (e) Trost BM, McEachern EJ. J Am Chem Soc. 1999;121:8649. [Google Scholar]; (f) Hirai A, Yu X-Q, Tonooka T, Miyashita M. Chem Commun (Cambridge, UK) 2003:2482. doi: 10.1039/b307390d. [DOI] [PubMed] [Google Scholar]; (g) Yu X-Q, Yoshimura F, Ito F, Sasaki M, Hirai A, Tanino K, Miyashita M. Angew Chem Int Ed Engl. 2008;47:750. doi: 10.1002/anie.200703889. [DOI] [PubMed] [Google Scholar]; (h) Arthuis M, Beaud R, Gandon V, Roulland E. Angew Chem Int Ed Engl. 2012;51:10510. doi: 10.1002/anie.201205479. [DOI] [PubMed] [Google Scholar]
- 14.(a) Ashworth RW, Berchtold GA. Tetrahedron Lett. 1977;18:343. [Google Scholar]; (b) Fagnou K, Lautens M. Org Lett. 2000;2:2319. doi: 10.1021/ol0060782. [DOI] [PubMed] [Google Scholar]
- 15.(a) Ha JD, Shin EY, Kang SK, Ahn JH, Choi J-K. Tetrahedron Lett. 2004;45:4193. [Google Scholar]; (b) Inoue M, Saito F, Iwatsu M, Ishihara Y, Hirama M. Tetrahedron Lett. 2007;48:2171. [Google Scholar]
- 16.(a) Satake M, Campbell A, Van Wagoner RM, Bourdelais AJ, Jacocks H, Baden DG, Wright JLC. J Org Chem. 2009;74:989. doi: 10.1021/jo802183n. [DOI] [PubMed] [Google Scholar]; (b) Van Wagoner R. J Nat Prod. 2010;73:1177. doi: 10.1021/np100159j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Kuranaga T, Ohtani N, Tsutsumi R, Baden DG, Wright JLC, Satake M, Tachibana K. Org Lett. 2011;13:696. doi: 10.1021/ol102925d. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a discussion of fragment synthesis, see:; (b) Kuranaga T, Satake M, Baden DG, Wright JLC, Tachibana K. Tetrahedron Lett. 2010;51:4673. [Google Scholar]; (c) Ohtani N, Tsutsumi R, Kuranaga T, Shirai T, Wright JL, Baden DG, Satake M, Tachibana K. Heterocycles. 2010;80:825. [Google Scholar]
- 18.“π-Activating group” refers to the group distal to furthest epoxide relative to trapping nucleophile.
- 19.See the Supporting Information for complete details of synthetic procedures and compound characterization towards epoxy alcohols 6a–6f.
- 20.Alkenes other than enoates are superior π-activating activating groups for endo-selective epoxy-alcohol cyclizations promoted by Brønsted acids. See:; (a) Nicolaou KC, Prasad CVC, Somers PK, Hwang C-K. J Am Chem Soc. 1989;111:5330. [Google Scholar]; (b) Nicolaou KC, Prasad CVC, Somers PK, Hwang C-K. J Am Chem Soc. 1989;111:5335. [Google Scholar]
- 21.Enoate 9 was prepared in three steps from commercially available 2-deoxy-D-ribose. See:; Nicolaou KC, Wallace PA, Shi S, Ouellette MA, Bunnage ME, Gunzner JL, Agrios KA, Shi G, Gartner P, Yang Z. Chem Eur J. 1999;5:618. [Google Scholar]
- 22.Wei X, Lorenz JC, Kapadia S, Saha A, Haddad N, Busacca CA, Senanayake CH. J Org Chem. 2007;72:4250. doi: 10.1021/jo062548f. [DOI] [PubMed] [Google Scholar]
- 23.The phosphonate ester was prepared in two steps by the literature method:; Mitton-Fry MJ, Cullen AJ, Sammakia T. Angew Chem, Int Ed. 2007;46:1066. doi: 10.1002/anie.200602601. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Y, Wang Q, Cornwall RG, Shi Y. Chem Rev. 2014;114:8199. doi: 10.1021/cr500064w. [DOI] [PubMed] [Google Scholar]
- 25.Diepoxide 13 was synthesized from aldehyde 10 via a related sequence in seven steps. See the Supporting Information for details.
- 26.Yang D, Xu M. Org Lett. 2001;3:1785. doi: 10.1021/ol015722p. [DOI] [PubMed] [Google Scholar]
- 27.(a) Netscher T. J Organomet Chem. 2006;691:5155. [Google Scholar]; (b) Wang ZJ, Jackson WR, Robinson AJ. Org Lett. 2013;15:3006. doi: 10.1021/ol401194h. [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee AK, Sanders DP, Grubbs RH. Org Lett. 2002;4:1939. doi: 10.1021/ol0259793. [DOI] [PubMed] [Google Scholar]
- 29.(a) Barton DHR, Kitchin JP, Lester DJ, Motherwell WB, Papoula MTB. Tetrahedron. 1981;37:73. [Google Scholar]; (b) Anaya J, Barton DHR, Gero SD, Grande M, Martin N, Tachdijian C. Angew Chem, Int Ed Engl. 1993;32:867. [Google Scholar]; (c) See the Supporting Information for a discussion regarding the optimization of this oxidative cleavage sequence.
- 30.Morten CJ, Byers JA, Jamison TF. J Am Chem Soc. 2011;133:1902. doi: 10.1021/ja1088748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Use of specific π-activating groups can also lead to endo-selective cyclizations albeit with erosion of trans-stereochemistry in the products. See:; Matsukura H, Morimoto M, Koshino H, Nakata T. Tetrahedron Lett. 1997;38:5545. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.