

Abstract
Natural products are a continual source of inspiration for chemists, particularly for organic chemists engaged in reaction development and methodology. In the early stages of our research program, we were drawn to macrocyclic natural products containing allylic alcohol moieties, such as (−)-terpestacin (1, Figure 1). We envisioned, in an ideal case, an intramolecular reductive coupling (a field still in its infancy at the time) could be developed to join an alkyne and an aldehyde to yield this allylic alcohol, simultaneously closing the macrocycle. For this reason we began studying reductive coupling as a tool for C–C bond formation. Additionally, as our program developed, it became clear that a number of other natural products, such as amphidinolide T1 (2), which, although they do not contain allylic alcohols, could be produced in an analogous fashion after modification of the allylic alcohol formed from such a macrocyclization.
As our investigation into the reductive coupling of alkynes and aldehydes continued, it quickly became clear that many other nucleophilic π-components, such as allenes and alkenes, and other electrophilic π-components, such as ketones and imines, could be competent substrates in these reactions. Additionally, σ-components, such as epoxides, are also able to participate in nickel-catalyzed reductive couplings. This Account describes our journey from inspiration by natural products to the creation of new methods to synthesize these products, the realization of their synthesis, and back again to inventing and improving methodologies with broad applications in organic synthesis.
REDUCTIVE COUPLING REACTIONS OF ALKYNES
Reductive Coupling of Alkynes and Aldehydes
Nickel(0) complexes have long been privileged catalysts for reactions of alkynes and alkenes, including oligomerization, cyclization, and polymerization.1 In the late 1990s, new strategies for nickel-catalyzed coupling of two π-components to produce a new σ-bond between them and a new σ-bond with an organometallic reducing agent were being developed, a field later known as reductive coupling.2 Although intramolecular reductive cyclizations were well precedented, the first intermolecular reductive three-component coupling was published by Montgomery and coworkers in 1997, and involved reaction of alkynes and aldehydes to give allylic alcohols (3, Scheme 1a).3 However, while the intramolecular coupling reaction conditions could be adjusted to afford either alkylative (C–R bond from reducing agent) or reductive (C–H bond from reducing agent) products, intermolecular couplings with alkylzinc reagents provided only alkylative products.
SCHEME 1.

Intermolecular a) alkylative and b) reductive alkyne–aldehyde coupling reactions.
Upon examination of the limitations of the current state of the field, we believed that the value of allylic alcohol products arising from an intermolecular reductive coupling (bypassing the traditional need for stoichiometric alkenyl metal reagents) meant that such a transformation would be synthetically and mechanistically interesting. Ultimately, we were able to achieve this goal with the use of alkyl boron reagents and phosphine ligands (Scheme 1b).4 Allylic alcohols were thus produced with high yields and regioselectivities for both aromatic (4) and aliphatic aldehydes (5).
The mechanism for reductive coupling, which was studied using DFT calculations in an enlightening and fruitful collaboration with Ken Houk, is shown in Scheme 2.5 The concerted oxidative cyclization step to form key five-membered nickelacycle 7 explains why only syn-addition across the alkyne is always observed. This step is generally considered to be rate-determining and also determines the regioselectivity of the resulting alkene products by the formation of the new C–C bond. The difference between the reductive and alkylative pathways lies in the preference for nickel–alkyl species 8 to undergo β–hydride elimination to form 9 rather than reductive elimination to form 10. The phosphine ligands employed in our chemistry promote the former. Later, Montgomery showed that reductive couplings could also be accomplished using N-heterocyclic carbene (NHC) ligands with trialkylsilanes as reducing agents.6
SCHEME 2.

Mechanism of reductive and alkylative alkyne–aldehyde coupling.
Since allylic alcohols are useful synthetic intermediates and a widespread motif in natural products, we immediately set out to render these reductive couplings enantioselective. Serendipitously, the very first chiral phosphine ligand that we tested was outstanding and for several years remained optimal: (+)-(neomenthyl)diphenylphosphine (NMDPP) (Scheme 3).7 Excellent yields, regioselectivities, and enantioselectivities were achieved for alkyne substrates containing at least one aromatic substituent. The high selectivity is hypothesized to originate from the required arrangement of the components in a highly-ordered transition state, as depicted in Scheme 3. Later, we synthesized a novel class of P-chiral monodentate ferrocenyl phosphines that were able to accomplish the same coupling using dialkyl alkynes with moderate regio- and enantioselectivity.8
SCHEME 3.

Enantioselective alkyne–aldehyde reductive coupling.
Control of Regioselectivity in Alkyne Reductive Coupling
Questions of regioselectivity are inherent in reactions of alkynes. Although good-to-excellent selectivity could be achieved by varying the electronic nature of the substituents (such as aryl–alkyl alkynes), we wondered if the use of more general directing groups could secure similarly high levels of selectivity. First, we observed that 1,3-enynes also provided high levels of selectivity, and the resulting diene 15 could be selectively reduced to give alkyl-substituted allylic alcohols (16) that would be otherwise inaccessible (Scheme 4a).9 Moreover, the conjugated alkene preferentially dictates the regioselectivity of addition, even directly opposite an aryl group. DFT calculations suggested that this intriguing preference may be due to a slightly different oxidative cyclization transition state involving a formal 1,4-attack and a transient η3-allylmetal complex (17).10 These 1,3-enyne substrates were also successfully coupled with ketones.11
SCHEME 4.
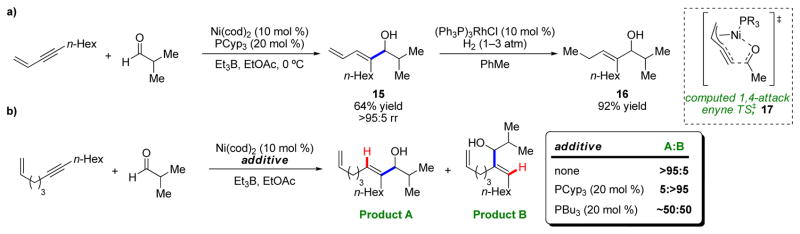
Alkene directing effects in alkyne–aldehyde reductive coupling reactions for a) 1,3-enynes and b) tethered enynes.
Next, we decided to investigate whether a remote, tethered alkene could be a suitable directing group. To our surprise, we found that not only could the reaction with the aldehyde be directed to the distal carbon of the alkyne (Product A; >95:5 rr), but by the addition of tricyclopentylphosphine (PCyp3) the selectivity was completely reversed to the proximal carbon (Product B; 5:>95 rr) (Scheme 4b).12 Interestingly, addition of a less sterically demanding ligand, tributylphosphine (PBu3), provided no selectivity (~1:1 rr). To rationalize these results, we considered square planar intermediate 18, in which nickel coordinates both the alkyne and tethered alkene, leaving room for one more weakly coordinating ligand (L) (Scheme 5). If no strongly coordinating ligands are present, the aldehyde replaces L (19), and the reaction proceeds to form the C–C bond at the carbon atom distal to the tethered alkene (Product A). However, if a strongly coordinating phosphine ligand is present, it will replace L (20a or 21). The sterically demanding PCyp3 disfavors simultaneous coordination of two phosphine ligands, meaning the aldehyde will replace the coordinating alkene while preserving the geometry of the complex (20b), presumably by an associative mechanism, to eventually produce Product B. Less bulky phosphine ligands, such as PBu3, preferentially replace both L and the alkene (21), forming Products A and B unselectively. These mechanistic proposals were corroborated by examining product diastereoselectivity using chiral 1,6-enyne substrates.13
SCHEME 5.

Mechanistic rationale of tethered alkene reductive coupling regioselectivity.
Finally, it is important to note that other solutions to issues of alkyne regioselectivity in reductive couplings have been reported. Montgomery and coworkers has demonstrated that carbene ligands of various steric bulk can be employed to achieve high levels of regioselectivity for both aldehyde reductive coupling product regioisomers.14 More recently, they disclosed an impressive solution for regiocontrol, altering whether the stereo-determining oxidative cyclization is rate determining or reversible by changing the ligand and silane reducing agent.15
Reductive Coupling of Alkynes and Imines
Intrigued by the success of reductive coupling to provide ready access to allylic alcohols, we were keen to investigate whether alkynes could be coupled with imines to form allylic amines. The reduced electrophilicity of imines provided a challenge and necessitated a complete change in reaction conditions. After extensive investigation we found that alkylative (rather than reductive) coupling products 22 could be formed in good yields and regioselectivities using protic solvents in combination with electron-rich phosphines and triethylborane (Scheme 6a).16 Although reductive coupling products were not formed, the scope of the three-component coupling reaction was significantly improved by the use of aryl and vinyl boronic acids. (23, Scheme 6b). Several years later, Zhou and coworkers determined that reductive coupling products could be formed selectively by using electron-poor N-tosylimines, PBu3, and aprotic solvents.17
SCHEME 6.

Selected examples of reductive coupling of alkynes and imines using a) borane and b) boronic acid alkylating agents. c) Enantioselective reductive coupling of imines.
Use of chiral phosphine (+)-NMDPP, which worked well for the coupling of aldehydes, provided products with only moderate enantioselectivity. Fortunately, by employing P-chiral ferrocenyl phosphine 25 in combination with a removable TBSOCH2CH2-protecting group, the resulting alkylative coupling products containing an allylic amine and tetrasubstituted alkene (26) were formed in good yields with good-to-excellent regioselectivity and good enantioselectivity (Scheme 6c).18
Reductive Coupling of Alkynes and Epoxides
After having explored the reductive coupling of various π-components, we next sought to study whether one of these π-components could be exchanged for an activated σ-component. In 2003, we reported the reductive coupling of alkynes with terminal epoxides to afford homoallylic alcohols (29, Scheme 7a).19 By conducting the reaction without added solvent, the products were obtained in moderate-to-good yields. The transformation proceeds with stereospecific syn-addition to the alkyne and with excellent regioselectivity for both the alkyne and epoxide. Experiments with enantiomerically enriched epoxides demonstrated, in accordance with the proposed mechanism, that the stereochemistry of the internal C–O bond is preserved in the product. The reaction was also conducted in an intramolecular fashion, which proceeds with excellent endo-selectivity (>95:5) in the ring-opening, regardless of the linking group and ring size formed (Scheme 7b).
SCHEME 7.

a) Inter- and b) intramolecular reductive couplings of alkynes and epoxides.
In a later study, we demonstrated that the intramolecular reaction could be performed by employing an inexpensive, air-stable nickel(II) salt as a precatalyst and 2-propanol as the reducing reagent (Scheme 8).20 Deuterium-labeling studies demonstrated that the terminal carbon of the epoxide undergoes inversion of configuration in the reaction (33). This result, along with the preserved stereochemistry of the epoxide, is indicative of a mechanism involving a nickelaoxetane intermediate (35). Oxidative addition of the terminal epoxide C–O bond with Ni(0) occurs with inversion, by way of nucleophilic ring-opening, to afford 35. Next, migratory insertion (with retention) of the alkyne forms 36, which after transmetallation (Et3B) or protonolysis (i-PrOH) to form 37, undergoes β-hydride elimination followed by reductive elimination to release the product and the Ni(0)-catalyst.
SCHEME 8.

Mechanism of the reductive coupling reaction of alkynes and epoxides.
APPLICATION OF REDUCTIVE COUPLING TO TOTAL SYNTHESIS
Although the discussion thus far has focused on new reaction development, our group has always sought to create methodologies addressing specific unmet needs in complex molecule synthesis. To this end, we have tested our reductive coupling methods in the total synthesis of a number of natural products, particularly in pushing the boundaries of reductive macrocyclizations.
As mentioned in the introduction, our initial inspiration for pursuing the development of reductive coupling methodology came from the allylic alcohol moiety found in the sesterterpene terpestacin. Indeed, the alkyne–aldehyde reductive coupling reaction allowed us to readily access this motif and rapidly assemble (−)-terpestacin (1, Scheme 9).21 Although dialkyl alkynes such as 39 can present regioselectivity challenges, we were gratified that our P-chiral ferrocenyl phosphine ligands favored the desired diastereo- and regioisomers to produce 41. Moreover, by simply changing the antipode of the ligand, we were afforded complete stereocontrol over the isolated allylic alcohol stereocenter. This allowed us to also access 11-epi-terpestacin via the same synthetic pathway, which was originally reported as being the structure of the natural product siccanol. In fact, we were able to reassign the structure of siccanol as being identical to (−)-terpestacin.
SCHEME 9.
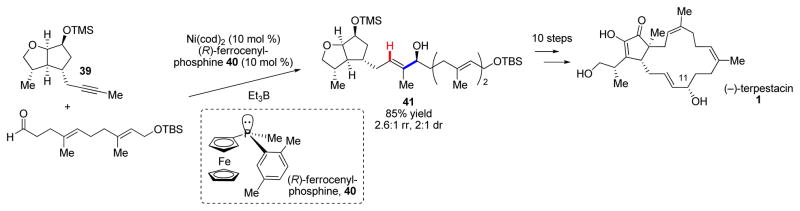
Total synthesis of (−)-terpestacin and 11-epi-terpestacin using an enantioselective alkyne–aldehyde reductive coupling reaction to couple key fragments.
Another molecule whose synthesis was amenable to our developed methods is amphidinolide T1 (2, Scheme 10).22 In this case, both the homoallylic alcohol and the α-hydroxyketone motifs were synthesized via reductive coupling of alkynes with an epoxide and an aldehyde, respectively. The transformations proceeded with good yields and diastereoselectivity, including the macrocyclization event which forms the 18-membered ring product 42, with only a single C13-epimer observed, solely under substrate control. A similar approach allowed us to access the related amphidinolide T4.23
SCHEME 10.
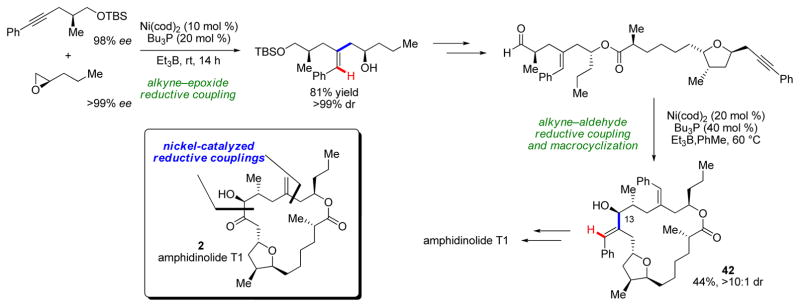
Total synthesis of amphidinolide T1 leveraging both alkyne–epoxide and alkyne–aldehyde reductive couplings.
Additionally, reductive couplings and cyclizations were explored for the synthesis of (+)-acutiphycin (43)24 and have allowed us to achieve the total syntheses of (−)-gloeosporone (44)25 and pumiliotoxins 209F (45) and 251D (46)26 (Figure 2).
FIGURE 2.
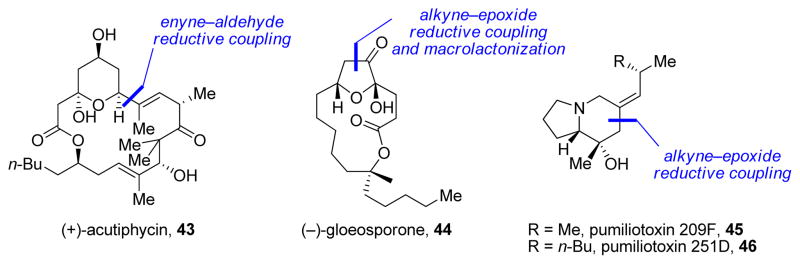
Applications of nickel-catalyzed methods to natural product synthesis.
REDUCTIVE COUPLING OF ALLENES
During the development of multicomponent reductive couplings of alkynes, we questioned whether other types of C–C π-bonds could undergo similar transformations. We first decided to study allenes, and in 2005 we reported a highly stereoselective and regiospecific reductive coupling of allenes, aldehydes, and silanes to furnish allylic alcohol products 50 in moderate-to-good yields (Scheme 11a).27
SCHEME 11.

a) Highly selective coupling of allenes, aldehydes, and silanes. b) Proposed mechanism.
Several noteworthy aspects of this transformation deserve further comment. The reaction displays a remarkable site selectivity in which coupling of the least reactive (central sp-hybridized) carbon atom of allene is observed. Furthermore, the use of NHC ligands provided complete transfer of enantiomeric purity from the allene to the product. This transfer of axial chirality turned out to be highly ligand dependent, with aliphatic phosphines, such as PCyp3, causing significant erosion of enantiomeric purity. Lastly, the alkene moiety is formed with excellent Z-selectivity. This makes the protocol complementary to the previously developed coupling of alkynes and aldehydes which leads to allylic alcohol products with excellent E-selectivity (Scheme 1). The formed Z-geometry is noteworthy since it corresponds to a syn-addition of the aldehyde and the hydride to the more hindered face of the allene (syn to the R2 substituent). In addition, there appears to be an inherent selectivity for addition across the sterically more hindered double bond of the allene as shown for products 50b–d.
A possible explanation for these observed selectivities was provided by a deuterium labeling study (Scheme 11b). When DSiEt3 was used, deuterium incorporation occurred with excellent diastereoselectivity (>19:1) at a single site (55). Based on this result, the reaction is proposed to proceed via complex 52, where Ni(0) coordinates to the less hindered allene face and least sterically congested alkene. The aldehyde would then coordinate opposite to the methyl substituent. Insertion of the aldehyde forms metallacycle 53, which after π-bond metathesis with the silane is believed to afford η3-allylnickel complex 54. Next, reductive elimination occurs to form the least congested alkene, accounting for the location of the deuterium incorporation.
CROSS–COUPLING OF AZIRIDINES
During our investigation of reductive coupling reactions, we became interested in the application of aziridines as a σ-bond electrophile. Despite numerous attempts, we have not been able to develop a reductive coupling of aziridines and alkynes to form homoallylic amines. However, we and others have recently developed highly selective nickel-catalyzed coupling reactions of aziridines with organozinc reagents (Scheme 12a).
SCHEME 12.

a) Highly regioselective cross–coupling of N-tosyl aziridines and alkylzinc reagents. b) Proposed mechanism for azanickelacyclobutane formation.
Seminal work by Hillhouse demonstrated that (bpy)Ni(0) complexes undergo oxidative insertion into the terminal position of aliphatic monosubstituted aziridines to form stable, isolable azanickelacyclobutane complexes.28 Despite this intriguing discovery, no nickel-catalyzed cross coupling reactions of aziridines were developed until Doyle and coworkers reported a Negishi-type reaction in 2012.29 In this study, they showed that styrene-derived N-tosylaziridines undergo regioselective coupling with aliphatic organozinc reagents at the more reactive benzylic position. Subsequently, less reactive aliphatic N-tosylaziridines were successfully coupled by using an N-sulfonyl group containing an unsaturated ester in the 2-position of the arylsulfonyl group.30 Although the catalytic system displayed excellent reactivity, the method provided only moderate regioselectivity and furnished the products as a mixture of inseparable isomers.
Inspired by the pioneering studies of Hillhouse and the successful development of a catalytic coupling reaction based on this activation by Doyle, we were able to develop a highly selective (>20:1 rr) and efficient coupling of aliphatic N-tosylaziridines with aliphatic organozinc reagents affording sulfonamide products 57 in good to excellent yields (Scheme 12a).31 Furthermore, we developed an air-stable nickel(II)chloride/ligand precatalyst that can be handled and stored outside a glovebox, thus avoiding the use of air-sensitive Ni(cod)2. Finally, through examination of catalytic intermediates for competency and deuterium-labeling studies, we were able to devise a likely mechanism for this transformation. First the nickel(0) catalyst, generated from the precatalyst by two consecutive transmetallation and reductive elimination reactions, undergoes insertion into the least hindered, terminal C–N bond of the aziridine to form azanickelacyclobutane 59. The oxidative insertion proceeds via an SN2-type mechanism (inversion) followed by C–C bond rotation (58) and ring-closure to form 59 (Scheme 12b), similar to the mechanism of epoxide oxidative addition shown in Scheme 8. Subsequent transmetallation with the organozinc reagent and reductive elimination releases product 57 (as a ZnBr adduct).
ALKENES AS NUCLEOPHILES
Coupling Reactions of Alkenes and Aldehydes
Throughout the course of our work with alkynes and allenes, we aimed to extend these methods to other types of π-nucleophiles, particularly alkenes. These investigations bore their first fruit in 2005, when we disclosed our first example of a nickel-catalyzed alkene–aldehyde coupling (Scheme 13). Closely related to both the carbonyl–ene and Prins reactions, this multi-component reaction couples terminal olefins (including ethylene), aldehydes, and silyl triflates to form silyl-protected allylic32 or homoallylic alcohols.33 In contrast to prototypical carbonyl–ene chemistry, less substituted alkenes, such as ethylene and terminal alkenes, are more reactive coupling partners than di-, tri-, and tetrasubstituted alkenes. The key component in this reaction is the highly electrophilic silyl triflate promoter. A number of groups, for example that of Tamaru,34 have demonstrated the important role that Lewis acid additives, such as Et2Zn and Et3B, play in the activation of weakly electrophilic π-components. Subsequently, Ogoshi demonstrated that silyl triflate additives can also fulfill this role and can allow even poor external nucleophiles, such as alkenes, to react with the electrophilic nickel species more easily.35
SCHEME 13.

The nickel-catalyzed carbonyl–ene reaction.
Ultimately, three distinct reaction protocols were devised to enable the selective synthesis of both the allylic (A, or branched) and homoallylic (H, or linear) isomers.36 Initially, P(o-anis)3 was found to be an outstanding ligand for ethylene (Scheme 13, Conditions A, 63a). However, monosubstituted alkenes required a different ligand for successful reaction, favoring the allylic product (Conditions B, 63b). Unfortunately, yields were only moderate, and the selectivity for the allylic product was generally not better than ~2:1. Subsequently, conditions were developed that afforded the homoallylic products in good yield and with excellent selectivity (Conditions C, 63c). However, despite having devised methods to provide both the allylic and homoallylic products, the selectivity for the allylic product remained inadequate, so we returned to this reaction to attempt to improve the selectivity. Extensive optimization and mechanistic investigation were reported after the two initial communications,37 and the results of this investigation suggested that nickel complexes containing NHC ligands, such as IPr, were competent to support the early steps of the coupling reaction with excellent allylic selectivity, but that catalytic turnover did not take place. Thus, a campaign was initiated to turn NHC-ligated nickel complexes into viable catalysts.38
Ultimately, we determined that the catalytic cycle was likely stalling subsequent to β-hydride elimination. Work of Yamamoto and coworkers39 had demonstrated that difficult reductive eliminations could be induced by the coordination of an electron-deficient alkene to the metal center. The addition of 3-trifluoromethylstyrene slightly improved the yield of the desired product, prompting us to examine other π-accepting ligands that could fulfill the same role; indeed, P(OPh)3 was identified as an extremely effective additive. The combination of Ni(cod)2, IPr, and P(OPh)3 (Scheme 13, Conditions D) provided the desired silyl-protected allylic alcohol products in excellent yields and with essentially perfect A:H selectivity. Intriguingly, both P(OPh)3 and IPr were incapable of catalyzing the reaction, yet the combination of these ligands forms the ideal catalyst system. We believe this results from the fact that IPr coordinates much more strongly to nickel, and that it is not until the IPr-nickel hydride has formed that P(OPh)3 can coordinate to nickel, eventually resulting in reductive elimination and catalyst turnover.
Coupling Reactions of Alkenes and Enones
An extension of this work was subsequently developed in which the alkene could act as an alkenylmetal equivalent, as before, but instead of 1,2-addition to the carbonyl, 1,4-addition across an enone or enal (65) could be effected (Scheme 14).40 The position of reactivity on the alkene is determined by the identity of R1—an alkyl group causes reaction at the internal position (66b), whereas an aryl group reverses the reactivity to form a linear product (66c).
SCHEME 14.
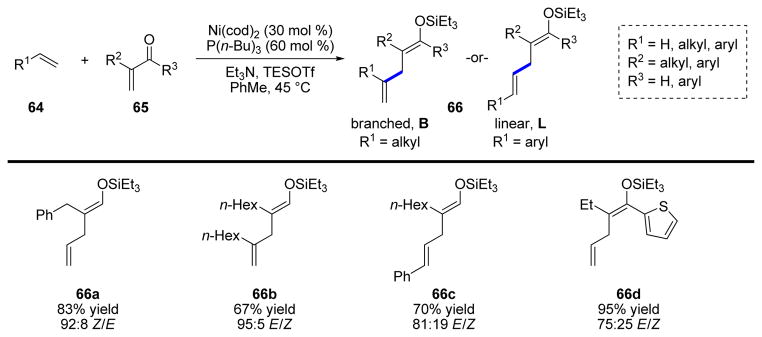
Nickel-catalyzed conjugate addition of alkenes to enals and enones.
Allylic Substitution Reactions
After demonstrating the use of alkenes as nucleophiles in conjugate additions, we sought to examine a reaction that could instead provide skipped dienes (1,4-dienes) more generally. We envisioned an allylic substitution reaction with simple alkenes as a way to obtain these valuable products, and our initial efforts found that allylic carbonates (67), as well as several other allylic substrates, reacted smoothly with ethylene (Scheme 15a) and terminal alkenes (Scheme 15b). The reaction produces 1,4-dienes in good yield and with outstanding selectivity for linear (as opposed to branched) products, for 1,4-dienes instead of isomerized 1,3-dienes, and for production of E-alkene isomers rather than Z (68a–68c).41 Additionally, when terminal alkenes were used, excellent selectivity for reaction at the internal position of the alkene was obtained (69a–69c).
SCHEME 15.

Allylic substitution reactions of a) ethylene and b) terminal aliphatic alkenes.
The Mizoroki–Heck Reaction
Having demonstrated the use of terminal alkenes as alkenylmetal equivalents, we became interested in one of the classic examples of such a reaction, the Mizoroki–Heck reaction (MHR). Often grouped more closely with cross-coupling, the MHR is mechanistically quite similar to allylic substitution reactions and is an indispensable means to synthesize alkenes. Under conditions closely related to those for allylic carbonates, the desired allylbenzenes were obtained in outstanding yields with a wide range of substituted benzyl chlorides and ethylene (71, Scheme 16a).42 Fortuitously, only a slight change to the conditions was necessary to allow successful reaction with terminal alkenes, yielding substituted allylbenzenes with outstanding regioselectivity for reaction at the internal position of the alkene (>95:5 branched/linear in almost all cases) (73, Scheme 16b).
SCHEME 16.

Mizoroki–Heck benzylation of a) ethylene and b) terminal alkenes and c) the development of an air-stable Ni(II) precatalyst.
Subsequently, we decided to investigate monosubstituted alkenes as substrates in more detail and improve the method with respect to practicality and cost (Scheme 16c).43 A critical enabling factor was the development of an air-stable Ni(II) precatalyst for this reaction, which was crucial because it alleviated the need to use expensive and highly air-sensitive Ni(cod)2. This change allows all reactions to be set up entirely without the use of a glovebox or even any air-free techniques, which greatly simplifies the use of this chemistry. This investigation significantly expanded the substrate scope and systematically examined functional group compatibility of the reaction, as well as substituting TMSOTf for the far more expensive TESOTf. These air-stable Ni σ-aryl precatalysts were further explored in subsequent studies and can be prepared from numerous ligands.44
Following the success of the benzylic MHR, we sought to examine related reactions in which this catalyst system could be applicable. One logical extension was, rather than benzyl electrophiles, to examine aryl electrophiles. Obtaining the branched product of a MHR with electronically unbiased alkenes was, at the time, an open problem in this field,45 and therefore piqued our interest.
Unfortunately, the developed conditions, as well as many related conditions, generally performed poorly with aryl chlorides and triflates instead of benzyl chlorides. Moving from benzyl to aryl electrophiles, although a subtle change, removes the possibility for η3-allylnickel intermediates and thus a distinct ligand sphere is required. Indeed, bidentate phosphine ligands proved essential, and in combination with the appropriate base, provided the desired branched styrene products in good yield and excellent regioselectivity of >95:5 (branched product to all other isomers produced) (Scheme 17).46 Importantly, aryl electrophiles other than triflates could also be employed in the reaction when TESOTf was also added to perform a counterion exchange and intercept a common cationic Ni–OTf species.
SCHEME 17.

Branch-selective Mizoroki–Heck arylation of terminal aliphatic alkenes.
CONCLUSIONS
Though our interest in nickel-catalysis initially arose from the utility of reductive coupling reactions to reach structural motifs such as allylic alcohols found in many natural products, the breadth of methodology developed in our group over the past 15 years far outstripped our initial plans. We were successfully able to couple readily available alkynes with π-donors, such as aldehydes and imines, as well as σ-donors in the form of epoxides to furnish ubiquitous allylic alcohols and amines and homoallylic alcohols, respectively. One clear omission, and place for future developments, lies in the reductive coupling reactions of aziridines to afford homoallylic amines. However, we were able to develop a nickel-catalyzed cross-coupling reaction of aziridines, demonstrating that oxidative addition, at least, is feasible.
In addition, we have expanded the range of π-components for nickel-catalyzed coupling reactions beyond alkynes to allenes and alkenes. Coupling of alkenes in particular proved extremely fruitful, not only with aldehydes as coupling partners, but with allylic, benzylic, and aryl electrophiles as well. We foresee that many of the modes of reactivity and mechanistic insights gained over the course of our study will add to the current exciting developments in the field of nickel catalysis. For instance, leveraging many of the different oxidation states that are readily available to nickel47 could provide access to distinct modes of activity in addition to those displayed by Ni(0)/Ni(II) catalytic cycles discussed in this Account. For these reasons, we eagerly look forward to contributing to new developments within the field of nickel catalysis.
Figure 1.
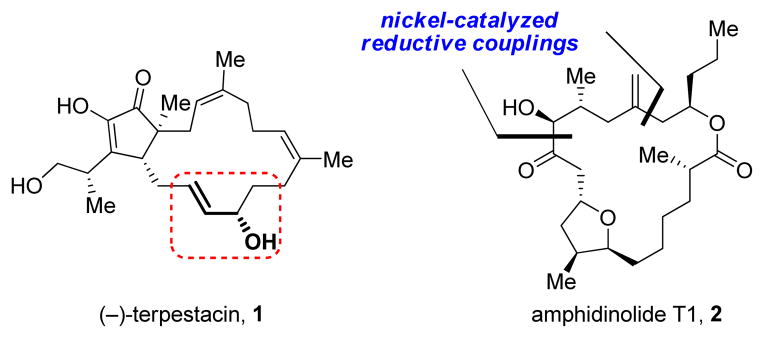
Natural product inspirations for nickel-catalyzed methodology.
Acknowledgments
We would like to thank the dozens of current and former group members who have contributed to this research program over the past 15 years. Their creativity, insight, and dedication have made this venture not only fruitful, but also very enjoyable. Generous financial support has been provided by many organizations, in particular the National Institute of General Medical Sciences, the National Science Foundation, numerous pharmaceutical companies, and several graduate and postdoctoral fellowship agencies.
Biographies
Eric A. Standley received a B.S. in Chemistry and a B.A. in German from Boise State University in 2010, and completed his Ph.D. with Prof. Timothy F. Jamison at the Massachusetts Institute of Technology in 2015. He will begin a postdoctoral fellowship with Prof. Frank Glorius at the University of Münster, Germany in fall of 2015.
Sarah Z. Tasker received a B.S. in Chemistry from Calvin College in 2010, and is currently pursuing her doctoral studies in the laboratory of Prof. Timothy F. Jamison at the Massachusetts Institute of Technology.
Kim L. Jensen received his B.S. in Chemistry from Aarhus University, Denmark, in 2007, where he continued on to receive his Ph.D. under the supervision of Prof. Karl Anker Jørgensen. In 2013, he joined the laboratory of Prof. Timothy F. Jamison as a postdoctoral fellow, and has recently departed for a position at LEO Pharma in Denmark.
Timothy F. Jamison received his undergraduate education at the University of California, Berkeley. He undertook his PhD studies at Harvard University with Prof. Stuart L. Schreiber and then moved to the laboratory of Prof. Eric N. Jacobsen at Harvard University. In 1999, he began his independent career at the Massachusetts Institute of Technology, where his research program focuses on the development of new methods of organic synthesis and their implementation in the total synthesis of natural products.
References
- 1.Tamaru Y, editor. Modern Organonickel Chemistry. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- 2.Montgomery J. Nickel-Catalyzed Reductive Cyclizations and Couplings. Angew Chem, Int Ed. 2004;43:3890–3908. doi: 10.1002/anie.200300634. [DOI] [PubMed] [Google Scholar]
- 3.Oblinger E, Montgomery J. A New Stereoselective Method for the Preparation of Allylic Alcohols. J Am Chem Soc. 1997;119:9065–9066. [Google Scholar]
- 4.Huang W-S, Chan J, Jamison TF. Highly Selective Catalytic Intermolecular Reductive Coupling of Alkynes and Aldehydes. Org Lett. 2000;2:4221–4223. doi: 10.1021/ol006781q. [DOI] [PubMed] [Google Scholar]
- 5.McCarren PR, Liu P, Cheong PHY, Jamison TF, Houk KN. Mechanism and Transition-State Structures for Nickel-Catalyzed Reductive Alkyne–Aldehyde Coupling Reactions. J Am Chem Soc. 2009;131:6654–6655. doi: 10.1021/ja900701g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahandru GM, Liu G, Montgomery J. Ligand-Dependent Scope and Divergent Mechanistic Behavior in Nickel-Catalyzed Reductive Couplings of Aldehydes and Alkynes. J Am Chem Soc. 2004;126:3698. doi: 10.1021/ja049644n. [DOI] [PubMed] [Google Scholar]
- 7.Miller KM, Huang WS, Jamison TF. Catalytic Asymmetric Reductive Coupling of Alkynes and Aldehydes: Enantioselective Synthesis of Allylic Alcohols and α-Hydroxy Ketones. J Am Chem Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y. [DOI] [PubMed] [Google Scholar]
- 8.Colby EA, Jamison TF. P-Chiral, Monodentate Ferrocenyl Phosphines, Novel Ligands for Asymmetric Catalysis. J Org Chem. 2003;68:156–166. doi: 10.1021/jo0264123. [DOI] [PubMed] [Google Scholar]
- 9.Miller KM, Luanphaisarnnont T, Molinaro C, Jamison TF. Alkene-Directed, Nickel-Catalyzed Alkyne Coupling Reactions. J Am Chem Soc. 2004;126:4130–4131. doi: 10.1021/ja0491735. [DOI] [PubMed] [Google Scholar]
- 10.Liu P, McCarren P, Cheong PH-Y, Jamison TF, Houk KN. Origins of Regioselectivity and Alkene-Directing Effects in Nickel-Catalyzed Reductive Couplings of Alkynes and Aldehydes. J Am Chem Soc. 2010;132:2050–2057. doi: 10.1021/ja909562y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller KM, Jamison TF. Highly Regioselective, Catalytic Asymmetric Reductive Coupling of 1,3-Enynes and Ketones. Org Lett. 2005;7:3077–3080. doi: 10.1021/ol051075g. [DOI] [PubMed] [Google Scholar]
- 12.Miller KM, Jamison TF. Ligand-Switchable Directing Effects of Tethered Alkenes in Nickel-Catalyzed Additions to Alkynes. J Am Chem Soc. 2004;126:15342–15343. doi: 10.1021/ja0446799. [DOI] [PubMed] [Google Scholar]
- 13.Moslin RM, Jamison TF. Mechanistic Implications of Nickel-Catalyzed Reductive Coupling of Aldehydes and Chiral 1,6-Enynes. Org Lett. 2006;8:455–458. doi: 10.1021/ol052719n. [DOI] [PubMed] [Google Scholar]
- 14.Malik HA, Sormunen GJ, Montgomery J. A General Strategy for Regiocontrol in Nickel-Catalyzed Reductive Couplings of Aldehydes and Alkynes. J Am Chem Soc. 2010;132:6304–6305. doi: 10.1021/ja102262v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson EP, Montgomery J. Regiocontrol in Catalytic Reductive Couplings through Alterations of Silane Rate Dependence. J Am Chem Soc. 2015;137:958–963. doi: 10.1021/ja511778a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel SJ, Jamison TF. Catalytic Three-Component Coupling of Alkynes, Imines, and Organoboron Reagents. Angew Chem, Int Ed. 2003;42:1364–1367. doi: 10.1002/anie.200390349. [DOI] [PubMed] [Google Scholar]
- 17.Zhou C-Y, Zhu S-F, Wang L-X, Zhou Q-L. Enantioselective Nickel-Catalyzed Reductive Coupling of Alkynes and Imines. J Am Chem Soc. 2010;132:10955–10957. doi: 10.1021/ja104505t. [DOI] [PubMed] [Google Scholar]
- 18.Patel SJ, Jamison TF. Asymmetric Catalytic Coupling of Organoboranes, Alkynes, and Imines with a Removable (Trialkylsilyloxy)ethyl Group—Direct Access to Enantiomerically Pure Primary Allylic Amines. Angew Chem, Int Ed. 2004;43:3941–3944. doi: 10.1002/anie.200460044. [DOI] [PubMed] [Google Scholar]
- 19.Molinaro C, Jamison TF. Nickel-Catalyzed Reductive Coupling of Alkynes and Epoxides. J Am Chem Soc. 2003;125:8076–8077. doi: 10.1021/ja0361401. [DOI] [PubMed] [Google Scholar]
- 20.Beaver MG, Jamison TF. Ni(II) Salts and 2-Propanol Effect Catalytic Reductive Coupling of Epoxides and Alkynes. Org Lett. 2011;13:4140–4143. doi: 10.1021/ol201702a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan J, Jamison TF. Synthesis of (−)-Terpestacin via Catalytic, Stereoselective Fragment Coupling: Siccanol Is Terpestacin, Not 11-epi-Terpestacin. J Am Chem Soc. 2003;125:11514–11515. doi: 10.1021/ja0373925. [DOI] [PubMed] [Google Scholar]
- 22.Colby EA, O’Brien KC, Jamison TF. Synthesis of Amphidinolide T1 via Catalytic, Stereoselective Macrocyclization. J Am Chem Soc. 2004;126:998–999. doi: 10.1021/ja039716v. [DOI] [PubMed] [Google Scholar]
- 23.Colby EA, O’Brien KC, Jamison TF. Total Syntheses of Amphidinolides T1 and T4 via Catalytic, Stereoselective, Reductive Macrocyclizations. J Am Chem Soc. 2005;127:4297–4307. doi: 10.1021/ja042733f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moslin RM, Jamison TF. Total Synthesis of (+)-Acutiphycin. J Org Chem. 2007;72:9736–9745. doi: 10.1021/jo701821h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trenkle JD, Jamison TF. Macrocyclization by Nickel-Catalyzed, Ester-Promoted, Epoxide–Alkyne Reductive Coupling: Total Synthesis of (−)-Gloeosporone. Angew Chem, Int Ed. 2009;48:5366–5368. doi: 10.1002/anie.200902079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Woodin KS, Jamison TF. Total Synthesis of Pumiliotoxins 209F and 251D via Late-Stage, Nickel-Catalyzed Epoxide–Alkyne Reductive Cyclization. J Org Chem. 2007;72:7451–7454. doi: 10.1021/jo071132e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.(a) Ng S-S, Jamison TF. Highly Enantioselective and Regioselective Nickel-Catalyzed Coupling of Allenes, Aldehydes, and Silanes. J Am Chem Soc. 2005;127:7320–7321. doi: 10.1021/ja0521831. [DOI] [PubMed] [Google Scholar]; (b) Ng S-S, Jamison TF. Enantioselective and Regioselective Nickel-Catalyzed Multicomponent Coupling of Chiral Allenes, Aromatic Aldehydes, and Silanes. Tetrahedron. 2005;61:11405–11417. doi: 10.1021/ja0521831. [DOI] [PubMed] [Google Scholar]; (c) Ng SS, Jamison TF. Nickel-Catalyzed Coupling of Terminal Allenes, Aldehydes, and Silanes. Tetrahedron. 2006;62:11350–11359. [Google Scholar]
- 28.Lin BL, Clough CR, Hillhouse GL. Interactions of Aziridines with Nickel Complexes: Oxidative-Addition and Reductive-Elimination Reactions that Break and Make C–N Bonds. J Am Chem Soc. 2002;124:2890–2891. doi: 10.1021/ja017652n. [DOI] [PubMed] [Google Scholar]
- 29.Huang C-Y, Doyle AG. Nickel-Catalyzed Negishi Alkylations of Styrenyl Aziridines. J Am Chem Soc. 2012;134:9541–9544. doi: 10.1021/ja3013825. [DOI] [PubMed] [Google Scholar]
- 30.Nielsen DK, Huang C-Y, Doyle AG. Directed Nickel-Catalyzed Negishi Cross Coupling of Alkyl Aziridines. J Am Chem Soc. 2013;135:13605–13609. doi: 10.1021/ja4076716. [DOI] [PubMed] [Google Scholar]
- 31.Jensen KL, Standley EA, Jamison TF. Highly Regioselective Nickel-Catalyzed Cross-Coupling of N-Tosylaziridines and Alkylzinc Reagents. J Am Chem Soc. 2014;136:11145–11152. doi: 10.1021/ja505823s. [DOI] [PubMed] [Google Scholar]
- 32.Ng S-S, Jamison TF. Simple Alkenes as Substitutes for Organometallic Reagents: Nickel-Catalyzed, Intermolecular Coupling of Aldehydes, Silyl Triflates, and Alpha Olefins. J Am Chem Soc. 2005;127:14194–14195. doi: 10.1021/ja055363j. [DOI] [PubMed] [Google Scholar]
- 33.Ho C-Y, Ng S-S, Jamison TF. Nickel-Catalyzed, Carbonyl–Ene-Type Reactions: Selective for α-Olefins and More Efficient with Electron-Rich Aldehydes. J Am Chem Soc. 2006;128:5362–5363. doi: 10.1021/ja061471+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura M, Fujimatsu H, Ezoe A, Shibata K, Shimizu M, Matsumoto S, Tamaru Y. Nickel-Catalyzed Homoallylation of Aldehydes and Ketones with 1,3-Dienes and Complementary Promotion by Diethylzinc or Triethylborane. Angew Chem, Int Ed. 1999;38:397–400. doi: 10.1002/(SICI)1521-3773(19990201)38:3<397::AID-ANIE397>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 35.Ogoshi S, Oka M-a, Kurosawa H. Direct Observation of Oxidative Cyclization of η2-Alkene and η2-Aldehyde on Ni(0) Center. Significant Acceleration by Addition of Me3SiOTf. J Am Chem Soc. 2004;126:11802–11803. doi: 10.1021/ja0460716. [DOI] [PubMed] [Google Scholar]
- 36.Ho C-Y, Schleicher KD, Chan C-W, Jamison TF. Catalytic Addition of Simple Alkenes to Carbonyl Compounds by Use of Group 10 Metals. Synlett. 2009;16:2565–2582. doi: 10.1055/s-0029-1217747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng S-S, Ho C-Y, Jamison TF. Nickel-Catalyzed Coupling of Alkenes, Aldehydes, and Silyl Triflates. J Am Chem Soc. 2006;128:11513–11528. doi: 10.1021/ja062866w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ho C-Y, Jamison TF. Highly Selective Coupling of Alkenes and Aldehydes Catalyzed by [Ni(NHC){P(OPh)3}]: Synergy Between a Strong σ-Donor and a Strong π-Acceptor. Angew Chem, Int Ed. 2007;46:782–785. doi: 10.1002/anie.200603907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto T, Yamamoto A, Ikeda S. Study of Organo(dipyridyl)nickel Complexes. I. Stability and Activation of the Alkyl–Nickel Bonds of Dialkyl(dipyridyl)nickel by Coordination with Various Substituted Olefins. J Am Chem Soc. 1971;93:3350–3359. [Google Scholar]
- 40.Ho C-Y, Ohmiya H, Jamison TF. α-Olefins as Alkenylmetal Equivalents in Catalytic Conjugate Addition Reactions. Angew Chem, Int Ed. 2008;47:1893–1895. doi: 10.1002/anie.200705163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsubara R, Jamison TF. Nickel-Catalyzed Allylic Substitution of Simple Alkenes. J Am Chem Soc. 2010;132:6880–6881. doi: 10.1021/ja101186p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsubara R, Gutierrez AC, Jamison TF. Nickel-Catalyzed Heck-Type Reactions of Benzyl Chlorides and Simple Olefins. J Am Chem Soc. 2011;133:19020–19023. doi: 10.1021/ja209235d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Standley EA, Jamison TF. Simplifying Nickel(0) Catalysis: An Air-Stable Nickel Precatalyst for the Internally Selective Benzylation of Terminal Alkenes. J Am Chem Soc. 2013;135:1585–1592. doi: 10.1021/ja3116718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Standley EA, Smith SJ, Müller P, Jamison TF. A Broadly Applicable Strategy for Entry into Homogeneous Nickel(0) Catalysts from Air-Stable Nickel(II) Complexes. Organometallics. 2014;33:2012–2018. doi: 10.1021/om500156q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.While this work was underway, Zhou and coworkers published three related, palladium-catalyzed methods for the branch-selective MHR. See Qin L, Ren X, Lu Y, Li Y, Zhou J. Intermolecular Mizoroki–Heck Reaction of Aliphatic Olefins with High Selectivity for Substitution at the Internal Position. Angew Chem, Int Ed. 2012;51:5915–5919. doi: 10.1002/anie.201201806.Qin L, Hirao H, Zhou J. Regioselective Heck Reaction of Aliphatic Olefins and Aryl Halides. Chem Commun. 2013;49:10236–10238. doi: 10.1039/c3cc45911j.Zou Y, Qin L, Ren X, Lu Y, Li Y, Zhou J. Selective Arylation and Vinylation at the α Position of Vinylarenes. Chem Eur J. 2013;19:3504–3511. doi: 10.1002/chem.201203646.
- 46.Tasker SZ, Gutierrez AC, Jamison TF. Nickel-Catalyzed Mizoroki–Heck Reaction of Aryl Sulfonates and Chlorides with Electronically Unbiased Terminal Olefins: High Selectivity for Branched Products. Angew Chem, Int Ed. 2014;53:1858–1861. doi: 10.1002/anie.201308391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tasker SZ, Standley EA, Jamison TF. Recent Advances in Homogeneous Nickel Catalysis. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]