

Abstract
Nickel/photoredox catalysis is used to synthesize indolines in one step from iodoacetanilides and alkenes. Very high regioselectivity for 3-substituted indoline products is obtained for both aliphatic and styrenyl olefins. Mechanistic investigations indicate that oxidation to Ni(III) is necessary to perform the difficult C–N bond-forming reductive elimination, producing a Ni(I) complex which in turn is reduced to Ni(0). This process serves to further demonstrate the utility of photoredox catalysts as controlled single electron transfer agents in multi-oxidation state nickel catalysis.
Among the many advantages of using nickel in the development of new synthetic methods1 is the availability of various oxidation states for catalysis, including Ni(0/I/II/III) and even a recently reported well-defined Ni(IV) species.2 Nickel-catalyzed methods have generally fallen into either those proposed to proceed via a Ni(0/II) pathway (e.g., aryl cross-coupling,3 reductive coupling4) or those proposed to proceed via a Ni(I/III) pathway (e.g., Csp3 cross-coupling5). However, controlled access to additional oxidation states in a single catalytic cycle can lead to novel and interesting chemistry (Scheme 1). For example, Weix and coworkers recently reported a cross-electrophile coupling with a concurrent polar Ni(0/II) oxidative addition of an aryl halide and radical Ni(III/I/II) oxidative addition/reductive elimination of an alkyl electrophile.6 The simultaneous recent reports from the research groups of Molander7 and MacMillan and Doyle8 also describe aryl–alkyl cross-coupling by using visible light photoredox catalysis9 to generate alkyl carbon radicals which can be trapped by Ni(II) intermediates.10 Then, after reductive elimination, the subsequent Ni(I) species is reduced by the same photoredox catalyst to Ni(0) to complete the catalytic cycle. This field continues to grow rapidly, giving access to novel reaction manifolds.11 Herein, we report a nickel-catalyzed synthesis of indolines, likely enabled by a mechanistically distinct direct oxidation and reduction of nickel intermediates.
Scheme 1.

Recent examples of catalytic reactions involving four different oxidation states of nickel.
Our development of nickel-catalyzed Mizoroki–Heck reactions of aliphatic olefins12 prompted us to consider whether an appropriately positioned nitrogen-based functional group could intercept the Ni–alkyl migratory insertion intermediate and participate in a direct Csp3–N reductive elimination, thus providing indoline products (Scheme 2a). Such reductive eliminations are known to be accelerated by oxidation to Ni(III) in stoichiometric reactions,13 so we reasoned that this reaction might be appropriate for multi-oxidation state nickel catalysis. This approach would also be analogous to the Larock indole synthesis (Scheme 2a).14
Scheme 2.

Synthesis of indolines and indoles via an intermolecular alkene or alkyne annulation strategy.
However, in contrast to indoles, where convergent annulation strategies are common, most methods to synthesize indolines, a common motif in natural products and pharmaceutically active compounds, involve multiple steps.15 Generally, either the Csp3–N bond or the Csp2–Csp3 bond is formed first, followed by a cyclization to form the five-membered ring. Perhaps surprisingly, given the ubiquity of the Larock indole synthesis, a general indoline synthesis of 2-haloaniline derivatives with alkenes has not been reported, although a few alkene annulation methods have been developed (Scheme 2b). This approach presents two main challenges: C–N bond reductive elimination must outcompete β-hydride elimination (forming Heck-type products), and Csp3–N reductive elimination is inherently challenging.16 These two issues were first addressed by Larock and coworkers, who reported indoline formation using 1,3-dienes, which cannot undergo β-H elimination.17 C–N bond formation also presumably proceeds via external nucleophilic attack on a π-allyl intermediate. Other intermolecular annulation approaches involve the use of alkenes without available β-H atoms, first reported by Catellani and coworker,18 or forego use of alkenes altogether.19 Perhaps the most general approach thus far was disclosed by Glorius and coworkers earlier this year.20 They reported using an internal diazinecarboxylate oxidant and Rh(III)-catalyzed directed C–H functionalization to couple arenes with electron-poor alkenes yielding 2-substituted aminoindolines. The 1-amino group can then be removed in a subsequent step to reveal the NH-indoline.
We began our development of a complementary and general approach to indoline synthesis by testing conditions similar to those of Heck reactions (Table 1, entry 1), and found that for a variety of electrophiles, aniline protecting groups, bases, and phosphine and N-type ligands, only Heck products were observed. Finally, upon using the N-heterocyclic carbene (NHC) ligand IPr, we were able to detect a small amount of indoline product (entry 2). We were gratified to see that the indoline product was produced in high selectivity (>19:1) compared to the sum of all other products. This suggests that, not only was β-H elimination suppressed, but also that alkene migratory insertion had been highly selective for arene insertion at the internal position, a difficult step given the electronically unbiased nature of aliphatic terminal alkenes. Although similar results had been seen in nickel-catalyzed Heck reactions previously, only sterically demanding phosphine ligands had been used successfully. 12 After this initial result, we attempted optimization of various reaction parameters,21 but quickly discovered that catalytic turnover from Ni(II) was likely not feasible since formation of indoline products was only observed upon exposure of the reaction mixture to air during workup. Csp3–N bond reductive elimination from low-valent Group 10 metals is known in rare cases,18,22 and Hillhouse and coworker had previously demonstrated that it could occur stoichiometrically after extended reaction times at room temperature when β-H elimination was not possible.13a However, consistent with our results, they demonstrated that reductive elimination was made far more facile by oxidation to Ni(III) by a number of stoichiometric oxidants, including oxygen.
Table 1.
Reaction optimization.
| ||||
|---|---|---|---|---|
| X; R | ligand | base | yield 6a | |
| 1 | OTf; Piv (1) | phosphine, PyBOX, phenanthroline-type | various organic/inorganic | 0% |
| 2 | 1 | IPr | LiOt-Bu | 7% |
| 3 | Cl (2), OTf (3), Br (4), I (5); Ac | IPr | LiOt-Bu | 10–14%b |
| 4c | Br; Ac (4) | IPr | 2,6-lutidine | 5% |
| 5c,d | 4 | IPr, Ru(bpy)3 | 2,6-lutidine | 15% |
| 6c,d | 4 | IPr, Ru(bpy)3, 66 h | Et3N | 33% |
| 7c,d | I; Ac (5) | IPr, Ru(bpy)3, 48 h | Et3N | 90% |
| 8c,e | 5 | IPr, Ru(bpy)3, 26 h | Et3N | 88% |
| 9c,f | 5 | IPr, Ru(bpy)3, 26 h | Et3N | 9%g |
yield 6 determined by GC using dodecane as an internal standard, selectivity 6 to sum of Heck (7), isomerized Heck (e.g., 9), and 2-alkyl indoline (8) >19:1 unless otherwise noted
≥10:1
Acetone (0.22 M)
Compact fluorescent light (CFL), (2 mol %) Ru(bpy)3(PF6)2
blue LEDs, (1 mol %) Ru(bpy)3(PF6)2
reaction run in absence of light, (1 mol %) Ru(bpy)3(PF6)2
9:1.
If oxidation to Ni(III) is necessary for reductive elimination, single electron reduction of the Ni(I) species formed therein would also be necessary to transition from a stoichiometric to a catalytic manifold, a scenario, in theory, fulfilled by a photoredox catalytic cycle. Spurred by the recent reports of Molander7 and Doyle and MacMillan,8 we were excited to find that the addition of the visible light photoredox catalyst Ru(bpy)3(PF6)2 under conditions similar to those reported by Molander (acetone, 2,6-lutidine) produced significantly more indoline product than the same reaction without photoredox catalyst (Table 1, entries 4, 5). Further optimization using triethylamine furnished, for the first time, yields in excess of the catalyst loading (entry 6). Use of aryl iodides rather than bromides and blue LEDs to shorten reaction times provided the optimal conditions for excellent yields of indoline product (entry 8). Again, we were gratified to see that under these conditions, indoline products were produced in high selectivities (>19:1) over Heck products.
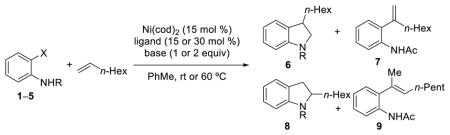
With these optimized conditions in hand, we explored the scope of this transformation (Scheme 3). Substitution at the 4- or 5-positions of the 2-iodoacetanilide electrophile was well-tolerated with both electron donating (e.g., 6b) and electron withdrawing (e.g., 6c) substituents. However, substitution at the 3- or 6- positions of the arene resulted in substantially lower yields (6f and 6g). In several cases, reaction yields were improved by slightly elevated temperatures (35 °C) and longer reaction times, although generally greater amounts of products corresponding to β-H elimination were observed at higher temperatures.
Scheme 3. Scope of aryl electrophilea.

aAll yields are isolated yields. Selectivity 6 to sum of 7–9 >19:1 unless otherwise noted. Reaction conditions: Ni(cod)2 (15 mol %), IPr (16 mol %), Ru(bpy)3(PF6)2 (1 mol %), Et3N (2 equiv), Acetone (0.22 M), blue LEDs, rt, 26 b Ru(bpy)3(PF6)2 (2 mol %), CFL, 35 °C, 48 h. c 7:1 d GC yield.
The substrate scope of alkene coupling partners is significantly broader (Scheme 4). A variety of functional groups are well tolerated in the indoline formation, including trimethylsilyl groups (6i), protected alcohols and amines (6j, 6n), ketones (6m), and alkyl chlorides (6o). Styrene was also a competent coupling partner and gave the same 3-substituted indoline product 6p as aliphatic alkenes despite the overwhelming preference for arene insertion to the terminal position of such alkenes. For styrenyl substrates, increased amounts of Heck products were observed, but 2-substituted indoline products were not observed by 1H NMR analysis. Styrenes substituted with electron donating and electron withdrawing groups were also competent reaction partners (6q, 6r). Only terminal monosubstituted alkenes were reactive under these conditions; internal and 1,1-disubstituted alkenes gave no desired product.
Scheme 4. Scope of alkene coupling partnera.

aAll yields are isolated yields. Selectivity 6 to sum of 7–9 >19:1 unless otherwise noted. Reaction conditions: Ni(cod)2 (15 mol %), IPr (16 mol %), Ru(bpy)3(PF6)2 (1 mol %), Et3N (2 equiv), Acetone (0.22 M), blue LEDs, rt, 26 h b Ru(bpy)3(PF6)2 (2 mol %), CFL, 35 °C, 48 h. c 5:1 d 12:1 e 9:1 f 8:1 g 11:1.
We next set out to probe the mechanism of this reaction. The intermediacy of a discrete Heck reaction followed by hydroamination was ruled out by reacting 5d with 1-octene in the presence of independently synthesized Heck product 7a (Scheme 5a).23 No formation of product 6a arising from 7a was observed, suggesting that it is not a competent intermediate. Next, we sought to investigate whether oxidation to Ni(III) was indeed responsible for product formation (Scheme 5b). 2-Iodoacetanilide (5a) was submitted to stoichiometric reaction conditions without photoredox catalyst for 12 hours. This mixture was then split into portions and exposed to various oxidants, before filtration through alumina under inert atmosphere and GC analysis. When no oxidant was added, very little indoline was observed, and a number of other products were formed in trace amounts. However, when stirred open to air or PhI(OAc)2 was added, significant amounts of product 6a (75 and 60% respectively) were observed with good selectivity for the desired indoline product. The addition of catalytic Ru(bpy)3(PF6)2 surprisingly did not produce significant amounts of 6a, possibly because of the larger than normal excess of Ni species compared to photoredox catalyst leading to unselective or unproductive oxidation/reduction and/or to photoredox catalyst death. Given that stoichiometric oxidants do successfully increase the amount of C–N bond formation as expected, a necessary oxidation to Ni(III) prior to product formation is still likely.
Scheme 5. Mechanistic studies.

aGC yield using dodecane as an internal standard.
We provisionally propose the catalytic cycle shown in Scheme 6, and several lines of evidence support the steps depicted. Crystal structures of oxidative addition and migratory insertion complexes were obtained,24 and the formation of complexes 10 and 12 were studied by 1H NMR.21 The feasibility of the proposed redox steps were further examined by performing cyclic voltammetry (CV) studies on Ni(0) complex 10 and Ni(II) complex 12. The Ni(0/I) oxidation of 10 was irreversible, so an exact E1/2 value could not be calculated.25 The potential wave of the Ni(I/0) transition of 10 peaks at −1.12V vs. Fc/Fc+ (or ~−0.74V vs. SCE)26 suggesting that it should be easily reduced by Ru(bpy)3+ [E1/2II/I = −1.33V vs. SCE in MeCN].9 The Ni(III/II) transition of 12 is reversible, and E1/2III/II = −0.20V vs. Fc/Fc+ (or ~+0.18V vs. SCE), suggesting that it should also be easily oxidized by *Ru(bpy)32+ [E1/2II/I = +0.77V vs. SCE in MeCN].9 The selectivity for desired reaction formation in the presence of so many redox-active species rather than off-cycle redox events is impressive.27 Also, for the first time, the photoredox cycle appears to oxidize and reduce nickel directly, rather than generating free carbon-based radicals which are then trapped by nickel. Several mechanistic puzzles still remain, however, including why both iodide and N-acetyl protecting group are necessary for good reaction yields21 and the exact role of triethylamine, so further mechanistic studies are needed to further elucidate the mechanism of this complex transformation.
Scheme 6.

Proposed mechanism.
In summary, the first general annulation reaction of 2-iodoaniline derivatives and terminal alkenes to form indolines has been developed. Using a nickel/NHC catalytic system, high selectivities for insertion to the terminal position of both aliphatic and styrenyl alkenes is achieved. Moreover, Csp3–N bond formation is achieved by leveraging the various oxidation states available to nickel within the same reaction system using photoredox catalysis as a powerful tool. We envision that using similar dual catalytic systems will enable other reactions for which reductive elimination would otherwise be particularly challenging.
Supplementary Material
Acknowledgments
Support for this work was provided by the NIGMS (GM63755) and NSF (Graduate Research Fellowship to S.Z.T.). We are grateful to Megan Jackson for help in obtaining cyclic voltammograms, Dr. Peter Müller for X-ray crystallography, Li Li for HRMS data, and Dr. Eric Standley, Dr. Kim Jensen, and Dr. Matthew Katcher for helpful discussions.
Footnotes
Note
The authors declare no competing financial interests.
Experimental procedures and spectral data for all unknown compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ananikov VP. ACS Catal. 2015;5:1964. [Google Scholar]
- 2.Camasso NM, Sanford MS. Science. 2015;347:1218. doi: 10.1126/science.aaa4526. [DOI] [PubMed] [Google Scholar]
- 3.Yamaguchi J, Muto K, Itami K. Eur J Org Chem. 2013:19. [Google Scholar]
- 4.(a) Montgomery J. Angew Chem, Int Ed. 2004;43:3890. doi: 10.1002/anie.200300634. [DOI] [PubMed] [Google Scholar]; (b) Standley EA, Tasker SZ, Jensen KL, Jamison TF. Acc Chem Res. 2015;48:1503. doi: 10.1021/acs.accounts.5b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The radical nature of oxidative addition of an alkyl halide often involves the brief intermediacy of a Ni(II) species as well: Breitenfeld J, Ruiz J, Wodrich MD, Hu X. J Am Chem Soc. 2013;135:12004. doi: 10.1021/ja4051923.Schley ND, Fu GC. J Am Chem Soc. 2014;136:16588. doi: 10.1021/ja508718m.
- 6.(a) Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920. doi: 10.1021/ja9093956. [DOI] [PubMed] [Google Scholar]; (b) Biswas S, Weix DJ. J Am Chem Soc. 2013;135:16192. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prier CK, Rankic DA, MacMillan DWC. Chem Rev. 2013;113:5322. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Recent studies have also suggested trapping of a radical by Ni(0), rather than Ni(II): Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. J Am Chem Soc. 2015;137:4896. doi: 10.1021/ja513079r.
- 11.(a) Noble A, McCarver SJ, MacMillan DWC. J Am Chem Soc. 2015;137:624. doi: 10.1021/ja511913h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Primer DN, Karakaya I, Tellis JC, Molander GA. J Am Chem Soc. 2015;137:2195. doi: 10.1021/ja512946e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xuan J, Zeng TT, Chen JR, Lu LQ, Xiao WJ. Chem Eur J. 2015;21:4962. doi: 10.1002/chem.201500227. [DOI] [PubMed] [Google Scholar]
- 12.(a) Tasker SZ, Gutierrez AC, Jamison TF. Angew Chem, Int Ed. 2014;53:1858. doi: 10.1002/anie.201308391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Matsubara R, Gutierrez AC, Jamison TF. J Am Chem Soc. 2011;133:19020. doi: 10.1021/ja209235d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Koo K, Hillhouse GL. Organometallics. 1995;14:4421. [Google Scholar]; (b) Lin BL, Clough CR, Hillhouse GL. J Am Chem Soc. 2002;124:2890. doi: 10.1021/ja017652n. [DOI] [PubMed] [Google Scholar]
- 14.Larock RC, Yum EK. J Am Chem Soc. 1991;113:6689. [Google Scholar]
- 15.Liu D, Zhao G, Xiang L. Eur J Org Chem. 2010:3975. [Google Scholar]
- 16.Hartwig JF. Nature. 2008;455:314. doi: 10.1038/nature07369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Larock RC, Berrios-Peña N, Narayanan K. J Org Chem. 1990;55:3447. [Google Scholar]
- 18.Catellani M, Del Rio A. Russ Chem Bull. 1998;47:928. [Google Scholar]
- 19.(a) Thansandote P, Raemy M, Rudolph A, Lautens M. Org Lett. 2007;9:5255. doi: 10.1021/ol702472u. [DOI] [PubMed] [Google Scholar]; (b) Gilmore CD, Allan KM, Stoltz BM. J Am Chem Soc. 2008;130:1558. doi: 10.1021/ja0780582. [DOI] [PubMed] [Google Scholar]
- 20.Zhao D, Vásquez-Céspedes S, Glorius F. Angew Chem, Int Ed. 2015;54:1657. doi: 10.1002/anie.201410342. [DOI] [PubMed] [Google Scholar]
- 21.See Supporting Information for more details.
- 22.Ni: Miura T, Morimoto M, Yamauchi M, Murakami M. J Org Chem. 2010;75:5359. doi: 10.1021/jo1008756.Pd: Marquard SL, Rosenfeld DC, Hartwig JF. Angew Chem, Int Ed. 2010;49:793. doi: 10.1002/anie.200904032.Hanley PS, Marquard SL, Cundari TR, Hartwig JF. J Am Chem Soc. 2012;134:15281. doi: 10.1021/ja307558x.
- 23.Qin L, Ren X, Lu Y, Li Y, Zhou J. Angew Chem, Int Ed. 2012;51:5915. doi: 10.1002/anie.201201806. [DOI] [PubMed] [Google Scholar]
- 24.CCDC 1403188 and 1403189 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 25.Pletcher D, Greef R, Peat R, Peter LM, Robinson J. Instrumental Methods in Electrochemistry. Horwood; Chichester: 2001. [Google Scholar]
- 26.Pavlishchuk VV, Addison AW. Inorg Chim Acta. 2000;298:97. [Google Scholar]
- 27.The need for Et3N or i-Pr2EtN for good yields is curious since the large excess of these amines relative to Ni should result in their oxidation by *Ru(bpy)32+ as well. Whether amine oxidation modulates the oxidizing strength of the photoredox catalyst acting as a SET relay for Ni oxidation, acts to build up the reducing Ru(bpy)3+, or is irrelevant (with Et3N acting solely as a base) is unclear.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.