

Abstract
Astrocytes sense changes in neural activity and extracellular space composition. In response, they exert homeostatic mechanisms critical for maintaining neural circuit function, such as buffering neurotransmitters, modulating extracellular osmolarity and calibrating neurovascular coupling. In addition to upholding normal brain activities, astrocytes respond to diverse forms of brain injury with heterogeneous and progressive changes of gene expression, morphology, proliferative capacity and function that are collectively referred to as reactive astrogliosis. Traumatic brain injury (TBI) sets in motion complex events in which noxious mechanical forces cause tissue damage and disrupt central nervous system (CNS) homeostasis, which in turn trigger diverse multi-cellular responses that evolve over time and can lead either to neural repair or secondary cellular injury. In response to TBI, astrocytes in different cellular microenvironments tune their reactivity to varying degrees of axonal injury, vascular disruption, ischemia and inflammation. Here we review different forms of TBI-induced astrocyte reactivity and the functional consequences of these responses for TBI pathobiology. Evidence regarding astrocyte contribution to post-traumatic tissue repair and synaptic remodeling is examined, and the potential for targeting specific aspects of astrogliosis to ameliorate TBI sequelae is considered.
Introduction
Responses to injury and disease in the central nervous system (CNS) involve multiple neural and non-neural cell types that interact over time in an effort to maintain homeostasis, protect viable cells, clear debris and preserve function (Burda and Sofroniew, 2014). Astrocytes are pivotal responders to all forms of CNS insults through diverse potential changes commonly referred to as reactive astrogliosis. In the healthy CNS, astrocytes play critical roles in maintaining the homeostasis of ions, transmitters, water and blood flow that are critical for neural circuit function. Many aspects of astrocyte responses to CNS damage and disease have been reviewed (Sofroniew and Vinters, 2010; Pekny and Pekna, 2014; Sofroniew, 2014b). In this article we will focus on astrocyte responses to, and roles in, traumatic brain injury (TBI).
Multiple forms and severities of TBI and tissue damage
Traumatic brain injury (TBI) can be caused by a wide variety of stimuli and encompasses a large range of severities (Graham et al., 2000). There is increasing recognition that TBI can not only have direct and immediately recognizable consequences, but also has the potential for long-term and gradually evolving sequelae, such as increased susceptibility for behavioral disturbances, seizure disorders or neurodegenerative disease (Kovacs et al., 2014; Sharp et al., 2014). It is also becoming clear that clinically mild forms of TBI that initially do not cause overt symptoms or easily detectable tissue damage can have long-term consequences, particularly if repetitive. To understand and address the consequences of TBI, there is a need for a better understanding of the cell biological consequences of different forms and severities of TBI and how they evolve over time and give rise to different forms and severities of tissue damage. The potentially beneficial or sometimes harmful effects of cellular responses to TBI such as reactive astrogliosis, are determined by a multitude of potential specific signaling events that can vary considerably with different forms and severities of CNS insults (Sofroniew, 2009; Burda and Sofroniew, 2014; Sofroniew, 2014b). Thus it is important to understand how different triggering events can lead to different cellular responses and different forms of pathology.
Tissue pathology and its functional consequences resulting from TBI are heterogeneous and determined largely by (i) the mechanical properties of the injury, (ii) degree of injury severity and (iii) the anatomical location of the injury. For analytical purposes it is useful to differentiate the features and origins of focal and diffuse tissue damage, which are associated with different types of astrocyte responses (Fig. 1). It deserves emphasis that clinically mild, moderate or severe TBI have the potential exhibit all of these forms of tissue damage to varying degrees and across varying expanses of tissue.
Figure 1. Reactive astrogliosis following TBI is a graded and heterogeneous response that reflects the severity of CNS tissue damage.

A, In response to mild or moderate tissue damage, astrocytes undergo hypertrophic reactive astrogliosis that includes molecular, structural and functional changes. Different forms of tissue pathology, such as local axonal injury and degeneration, blood brain barrier (BBB) disruption with inflammatory cell extravasation, deafferentation and synapse degeneration due to distal axon injury, or exposure to PAMPs associated with peripheral bacterial or viral infection, can all uniquely influence astrocyte function and in different combinations can drive specific forms of astrogliosis. These hypertrophic reactive astrocytes are intermingled among viable neural cells in areas of injured, but surviving and functioning neural tissue. B, Severe tissue damage elicits neural and glial cell degeneration, vascular breakdown and a robust innate and adaptive immune response, leading to the formation of tissue compartments with distinct forms of reactive astrogliosis. Immediately adjacent to the injury, astrocytes proliferate and intertwine to form an astroglial scar (AS) that surrounds and restricts the spread of the intense inflammatory response in the lesion core. These scar forming astrocytes are present in areas that contain few if any surviving neural cells, and their main interactions are with non-neural cells in tissue lesions. Adjacent to the astrocyte scar, features characteristic of mild or moderate brain trauma are present and taper with distance from the lesion core (LC). In these areas reactive astrocytes undergo changes in morphology and function characteristic of hypertrophic reactive astrogliosis as described in A, and these reactive astrocytes interact with injured but surviving cells in the perilesion perimeter (PLP). Astrocyte reactivity in the PLP may also influence neurons and glia in the healthy tissue (HT) distal to the injury.
Focal tissue damage
Focal tissue damage after TBI can arise in several ways, for example from direct impact that produces brain contusion with intra- and/or extra-axial hemorrhage, or from penetrating injuries that cause direct parenchymal laceration and hemorrhage. As a result, focal TBI lesions form due to abrupt and indiscriminate cell death of a majority of neural cells in circumscribed regions (Fig. 1B). Such focal lesions can vary considerably in size, and can encompass large areas or can be restricted to small clusters of cells (Myer et al., 2006; Villapol et al., 2014).
Diffuse tissue damage
The initial insult leading to diffuse tissue damage after TBI results from tissue strain due to inertial forces, such as occurs with rapid head acceleration/deceleration in an automotive crash or in blast injuries (Gennarelli et al., 1982). Unlike the easily detectable gross tissue disruption produced by focal TBI lesions, primary diffuse TBI pathology is difficult to detect by current neuroimaging methods, particularly in its acute stages. Rather, tissue deformation produces sub-lethal damage to neurons, glia and vascular cells, which leads to chronic progressive cellular injury due to oxidative damage, osmotic imbalance, ischemia and inflammation (Fig. 1A). As such, diffuse TBI is often only diagnosed conclusively at the microscopic level during postmortem evaluation, by vascular breakdown and hallmark diffuse axonal injury that manifests as axonal swellings, plasmalemmal disruption, disconnection and Wallerian degeneration (Johnson et al., 2013). The extent of diffuse axonal injury correlates with injury severity and the plane of mechanical loading (Smith et al., 2000), with regions of white-grey matter interface or enriched white matter (e.g cortical gyri, corpus callosum, brain stem) being particularly susceptible to strain injury (Gennarelli et al., 1982; Meythaler et al., 2001).
In this regard, it is important to realize that tissue damage after TBI is seldom purely focal or diffuse, with a single case often involving a multiplicity of focal and diffuse lesions (Graham et al., 2000; Skandsen et al., 2010). Areas of focal tissue damage are invariably surrounded by tapering perimeters of diffuse tissue damage and its associated cellular changes and responses (Fig. 1B). For example, an acute contusive TBI may produce a gross focal lesion at the site of impact, while generating rapid head acceleration that evokes more diffuse damage by way of compressive countercoup injury and rotational tissue shearing (Ommaya and Gennarelli, 1974). Indeed, animal models of focal contusive and percussive TBI demonstrate diffuse cellular perturbations in regions distal to the epicenter of focal damage (Singleton and Povlishock, 2004; Vlodavsky et al., 2005). Conversely, after a diffuse TBI caused by severe acceleration-deceleration, the coalescence of multiple small vascular deficits created by diffuse multiple small shear injuries may lead to tissue lesions similar in nature to focal lesions (Shih et al., 2013). Accordingly, in many if not most cases the traumatized brain may be comprised of lesions of disparate pathogenesis, involving a range of cellular microenvironments, including synapses, axons and the vascular/parenchymal interface of the blood brain barrier.
A number of animal models of TBI have been designed to investigate the complex detrimental biomechanical and molecular mechanism of underlying brain trauma, and which exhibit the features focal and diffuse tissue damage in varying degrees. In-depth analysis of the most common models and their utility for investigating specific features of TBI pathology has been the focus of excellent review articles (Xiong et al., 2013).
Astrocytes
Astrocytes tile the brain and spinal cord. They localize to all cellular environments and thereby exist as common denominators among injury compartments containing differing degrees of cell death, neuronal and axonal injury, inflammation and vascular injury (Fig. 1). Astrocytes are key players in the multicellular response to CNS trauma and disease (Burda and Sofroniew, 2014). Their critical roles in healthy CNS and their general responses to CNS insults have been reviewed extensively elsewhere (Sofroniew and Vinters, 2010; Burda and Sofroniew, 2014; Pekny and Pekna, 2014). Noteworthy is the increasing recognition that astrocytes exhibit structural, molecular and functional diversity in healthy CNS and in responses to CNS insults (Zhang and Barres, 2010; Anderson et al., 2014). Here, we will focus on astrocyte responses to, and roles in, different forms and severities of CNS tissue damage after TBI (Fig. 1). In particular we consider the role of TBI induced mechanical forces in the initiation of astrocyte reactivity and astrocyte-microglial interactions. We consider functional consequences of such responses with respect to severity of tissue damage and regulation of inflammation, cerebrovascular integrity and post-traumatic circuit plasticity and remodeling. We will discuss how certain mechanisms of astrocyte reactivity have the potential to dysfunction and become maladaptive, with serious repercussions for surrounding neurons and glia. Understanding mechanisms that trigger and regulate astrocyte reactivity in TBI, and the beneficial or detrimental consequences of reactivity-associated changes in astrocyte functions for disease progression, has the potential to identify novel targets for ameliorating post-traumatic brain injury and promoting tissue repair.
TBI mechanopathogenesis as a trigger of astrocyte reactivity
Dissecting the means by which mechanical forces translate into cellular dysfunction and damage is fundamental to understanding and developing therapeutic strategies for TBI, in particular for milder forms of TBI that may cause widespread diffuse tissue damage but little or no severe focal tissue damage. Supra-threshold dynamic strain accompanying the spectrum of TBI can profoundly influence cell behavior and function without causing gross tissue destruction. For example, although axons are capable of sustaining minor membrane deformations, they are rigid structures within an elastic extracellular surround (Javid et al., 2014) and deleterious mechanical stress experienced at the onset of TBI-induced damage produces immediate plasmalemmal instability and cytoskeletal disassembly (Povlishock, 1993; Pettus and Povlishock, 1996; Singleton and Povlishock, 2004). Astrocytes appear equally susceptible to membrane distortions and poration (Cullen et al., 2011). How astrocytes transduce physical strain associated with more diffuse forms of tissue damage after TBI into subsequent changes in cell function is incompletely understood, but evidence is emerging. One mechanism could involve activation of astrocyte mechanosensitive ion channels elicited by traumatic membrane deformation (Fig. 2). Indeed, astrocytes express a number of mechanotransducing ion channels (Bowman et al., 1992; Islas et al., 1993), as well as non-traditional, stretch-sensitive cation channels such as N-methyl-D-aspartic acid receptors and “BK” potassium channels, which may all contribute to the rapid influx of extracellular calcium and sodium observed in physically stressed astrocytes (Rzigalinski et al., 1997; Floyd et al., 2005). Other in vitro studies demonstrate diverse astroglial responses to physical strain in the form of plasma membrane stretching, including mitogen-activated protein kinase and protein kinase B (AKT) signaling, elevations in intracellular calcium and adenosine triphosphate (ATP) release (Ahmed et al., 2000; Verderio and Matteoli, 2001; Neary et al., 2003; Neary et al., 2005). ATP is released via connexin hemi-channels, which are abundantly expressed by astrocytes (Rouach et al., 2002; Stout et al., 2002). Other studies show that astrocyte stretch injury can lead to secretion of vasoactive molecules such as endothelin-1 and isoprostanes (Hoffman et al., 2000; Ostrow et al., 2011), as well as inositol triphosphate signaling induction and altered sensitivity to extracellular glutamate and inflammatory cytokines (B. A. Rzigalinski, 1998; Ralay Ranaivo et al., 2011). Astrocytes may also release matrix metalloproteinase-9 in response to mechanical strain (Pan et al., 2012). These diverse effects could have a wide variety of consequences for astrocyte functions (Fig. 2) that in turn impact on neural circuit function and circuit plasticity during circuit reorganization and attempts to maintain or restore neurological functions. Much work is needed to gain a better understanding of these events.
Figure 2. Astrocytes sense and respond to mechanical strain after TBI.

Physical strain deforms flexible networks of intermediate filaments within astrocytes and activates ion influx through mechanosensitvie cation channels. Rises in intracellular calcium cause astrocyte ATP release that signals in an autocrine or paracrine manner, driving multiple intra- and inter-cellular signaling pathways and inducing the release of endothelin-1, MMP9 and glutamate. Depending on the severity of the mechanical insult, astrocyte reactivity may involve complex changes in phenotype and function that respond to and influence neuroinflammatory responses to injury as well as mechanisms of secondary TBI pathogenesis. Trauma also causes astrocytes to release GFAP and calcium-binding S100B that may serve as biomarkers of TBI severity.
It is also noteworthy that astrocytes contain dense networks of intermediate filaments such as glial fibrillary acidic protein (GFAP). TBI-associated mechanical strain transduced by these flexible intermediate filament networks could serve to encode severity of astrocyte deformation over a broad dynamic range, in part instructing an appropriate course of reactive astrogliosis, which is heterogeneous depending on the degree and location of the injury (Fig. 2). GFAP and other intermediate filaments, vimentin and nestin, are markedly upregulated following brain trauma and stroke (Li and Chopp, 1999; Liu et al., 2014). Elevated levels of astrocyte intermediate filaments appears to be related to the severity of cellular perturbation, as GFAP expression is greatest in reactive astrocytes proximal to CNS traumatic lesions, tapering in perilesion zones (Wanner et al., 2013). Studies in mice lacking both GFAP and vimentin demonstrate markedly impaired astrocyte reactivity, attenuated debris clearance and chronic blood-brain barrier dysfunction after focal traumatic injury or stroke (Pekny et al., 1999; Liu et al., 2014), indicating that these cytoskeletal proteins are required for appropriate initiation and maintenance of reactive astrogliosis. The upregulation of intermediate filaments by trauma-reactive astrocytes, as well as their direct association with known early stress-response proteins substantiates their role in the early astroglial response to TBI (reviewed in (Pekny and Lane, 2007; Toivola et al., 2010)). TBI also induces the release of GFAP and S100 calcium binding protein B (S100B) from astrocytes, which may serve as a serum and/or cerebrospinal fluid biomarkers of TBI severity (Fig. 2)(Zetterberg et al., 2013; Plog et al., 2015).
Further investigation into how astrocytes sense and respond to varying degrees of mechanical stress will allow for a clearer understanding of the initializing events and functional consequences of astrocyte reactivity in TBI. Importantly, primary injury in TBI yields immediate release of molecular stress signals by astrocytes and other surrounding injured cells, often coupled with the extravasation of volatile serum proteins across a disrupted blood brain barrier. The additive effects of initial physical and molecular disturbances strongly influences astrocyte gene expression, morphology, secretory and proliferative capacities, which impact on the CNS tissue response to TBI (Fig. 2).
Rapid astrocyte responses to TBI
Studies employing intravital time-lapse imaging have revealed astrocytes as critical early responders to TBI and suggest an essential role for astrocyte-derived ATP in stimulating other cellular responses (Kim and Dustin, 2006). Such findings are consistent with the ability of TBI mechanopathogenic forces to initiate various astrocyte signaling events, including ATP release via connexin hemi-channels as discussed above. It is noteworthy that ATP is also released by dying and injured cells. Regardless of source, TBI-induced extracellular ATP induces a rapid rise in cytoplasmic calcium in reactive astrocyte networks surrounding acute traumatic brain lesions. This calcium signaling precedes the (i) polarization of astrocyte processes towards the site of injury and (ii) recruitment of dynamically motile microglia and neutrophils, which occur within minutes following trauma and which are all ATP and connexin hemi-channel-dependent (Davalos et al., 2005; Kim and Dustin, 2006; Roth et al., 2014). In vitro studies also demonstrate ATP-dependent astrocyte calcium signaling with subsequent ATP release, just prior to microglial activation (Guthrie et al., 1999; Verderio and Matteoli, 2001). Astrocyte connexin-mediated ATP signaling has also been implicated in driving astroglial and microglial reactivity in traumatically injured spinal cord (Huang et al., 2012). These observations support a model in which primary mechanical tissue damage induced by TBI rapidly triggers ATP release from astrocytes and other cells, which in turn triggers a wave of inter-astrocyte calcium signaling and astrocyte-derived ATP release that rapidly recruits microglia to injury sites and leads to both microglial and astrocyte reactivity responses (Fig. 3). Notably, initially released astrocyte-derived ATP can be recognized as indirectly neuroprotective, as recruitment of innate immune cells to traumatic brain lesions is ATP/connexin-dependent and essential for the survival of local parenchymal and meningeal cells (Roth et al., 2014). In addition, these initial events can also set in motion longer term cellular interactions that, depending on the severity of the inducing trauma and degree of tissue damage, can in a context dependent manner lead to different forms and functions of reactive astrogliosis.
Figure 3. ATP gradients from astrocytes direct the initial innate immune response to TBI.

(1) Non-reactive astrocytes have the capacity to sense and release ATP. (2) Local trauma triggers ATP release from injured cells. (3) ATP signaling to other astrocytes causes a rapid and persistent rise in intracellular calcium in the surrounding astroglial network. (4) Calcium-induced release of ATP via astrocyte connexin hemi-channels generates an ATP gradient that signals to innate immune cells (5) activating and recruiting them to the site of injury.
Different forms and functions of reactive astrogliosis
As discussed above, TBI varies greatly in severity of cellular and tissue damage. There is now substantial evidence that astrocytes tune their responses to the nature and severity of CNS insults (Fig. 1A,B), resulting in different forms of reactive astrogliosis that have been discussed in detail elsewhere (Sofroniew and Vinters, 2010; Burda and Sofroniew, 2014; Sofroniew, 2014b). In broad terms, reactive astrogliosis can now be defined as a finely graded continuum of multiple potential changes that range from reversible alterations in gene expression and cell hypertrophy within preserved individual astrocyte domains (Fig. 1A), to scar formation that involves substantial cell proliferation and permanent rearrangement of tissue structure (Fig. 1B) (Sofroniew and Vinters, 2010; Sofroniew, 2014b). Thus, astrogliosis is both complex and heterogeneous. Of particular interest here is that CNS trauma induces clear gradients in astrogliosis that vary with distance from lesions and with injury intensity (Wanner et al., 2013). From a functional perspective, emerging evidences points towards critical roles for different forms of reactive astrocytes in a variety of post-injury mechanisms including (i) regulation of inflammation, (ii) isolation of lesions and protection of adjacent neural tissue, (iii) regulation of the blood brain barrier and (iv) synaptic plasticity and neural circuit reorganization, as discussed in the following sections.
Reactive astrocytes regulate TBI-associated inflammation
TBI with severe focal tissue damage triggers inflammatory mechanisms essential for clearance of debris (Burda and Sofroniew, 2014). Astrocytes play key roles in this process. Astrocytes can both respond to and produce many immunomodulatory molecules, including cytokines, chemokines and inflammatory mediators such as danger-associate molecular patterns (DAMPs) and alarmins released by stressed, injured or dying cells. Prototypical DAMPs, including high-mobility group box 1 (HMGB1), heat shock proteins and S100 proteins, signal through pattern recognition receptors on phagocytic immune cells to promote clearance of cytotoxic cellular debris and decrease inflammation (reviewed in (Neal, 2012; Burda and Sofroniew, 2014)). Though pattern recognition receptors, such as toll-like receptors (TLRs) and receptor for advanced glycation end products (RAGE), are traditionally expressed by microglia and macrophages, they are also expressed by astrocytes (Ponath et al., 2007; Gorina et al., 2011). Stimulation of astrocyte pattern recognition receptors by DAMPs results in nuclear-factor-κB (NFκB) signaling, the production of proinflammtory cytokines like tumor necrosis factor α (TNFα), α-chemokines, as well as inflammatory mediators cyclooxygenase-2 and matrix metalloproteinase 9 (MMP-9) (Pedrazzi et al., 2007; Ponath et al., 2007; Gorina et al., 2011). Intriguingly, amyloid-β mediated activation of astrocyte RAGE stimulates astrocytes themselves to become phagocytic and engulf extracellular amyloid-β (Jones et al., 2013), possibly implicating reactive astrocytes in the clearance of neuro- and gliotoxic amyloid protein after injury. Additionally, pathogen-associated molecular patterns (PAMPs) from bacterial pathogens, such as lipopolysaccharide also bind astrocyte pattern recognition receptors and elicit expression of immunomodulatory and pro-inflammatory molecules (Hoarau et al., 2011; Hamby et al., 2012). Therefore peripheral infections, which can occur after TBI due to peripheral immune suppression (Lenz et al., 2007), could result in the extravasation of PAMPs into the CNS across damaged blood brain barrier. PAMP signaling to reactive astrocytes and innate immune cells could potentiate pro-inflammatory signaling that would recruit additional monocytes, neutrophils and lymphocytes into the injured brain. Clinical epidemiological evidence is also emerging that peripheral infections have a negative impact on neurological outcome after spinal cord injury (Failli et al., 2012).
Pattern recognition receptor-mediated NFκB signaling in astrocytes also results in cell swelling implicated in cytotoxic edema, a major pathophysiologic mechanism underlying the harmful increase in intracranial pressure following TBI (Jayakumar et al., 2014). Correspondingly, HMGB1 release following TBI-induced tissue damage can signal to microglia, inducing secretion of IL-6 that signals reactive astrocytes to upregulate the AQP4 water channel involved in astroglial water uptake (Laird et al., 2014). Therefore, while the DAMP signaling to astrocytes elicits their communication to phagocytic immune cells necessary to promote clearance of potentially toxic debris, it may also be maladaptive by directly contributing to cytotoxic edema and the deleterious production of inflammatory mediators. In this regard, inhibition of NFκB signaling specifically in astrocytes reduces inflammation following CNS trauma (Brambilla et al., 2005; Brambilla et al., 2009). Additionally, increasing concentrations of the DAMPs HMGB1 and mitochondrial DNA in cerebrospinal fluid correlates with greater disability in TBI patients (Walko et al., 2014). However, NFκB signaling in astrocytes may also produce beneficial effects after brain injury, as it can result in their production and secretion of the neuro- and glioprotective growth factors, brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) (Zaheer et al., 2001). Likewise, reactive astrocytes can release DAMPs like HMGB1 that signals to endothelial cells and their progenitors to promote neurovascular remodeling and BBB repair after brain injury (Hayakawa et al., 2012b; Hayakawa et al., 2012a).
It is important to recognize that the astrocytic response to pro- and anti-inflammatory cytokines and diverse inflammatory mediators is context-dependent. Different combinations of inflammation-associated molecules at different times will elicit disparate astroglial responses. This includes, but is by no means limited to the production of immunomodulatory molecules, cytokines, chemokines, growth factors and extracellular proteases (Sofroniew, 2014a). Though daunting from an investigational standpoint, this phenomenon confers reactive astrocytes with the ability to respond to and communicate with innate and adaptive immune cells, neurons, glia and vascular cells, and positions them as hub cells in the tissue response to TBI (Fig. 1,2,3).
Scar-forming reactive astrocytes form functional barriers around tissue lesions
Experimental evidence indicates that reactive astrocytes are instrumental in preserving injured but salvageable tissue. In response to focal tissue damage or inflammation, reactive astrocytes form scar borders that segregate damaged and inflamed tissue from adjacent potentially viable neural tissue (Sofroniew and Vinters, 2010; Burda and Sofroniew, 2014; Sofroniew, 2014b). These scar-borders are comprised almost entirely of newly proliferated astrocytes that do not observe discrete individual cellular domains and have elongated processes that intertwine extensively (Wanner et al., 2013). Scar-forming proliferative reactive astrocytes that surround areas of severe brain injury exhibit barrier functions that are instrumental in regulating the expanse of tissue injury, inflammation and instructing wound repair (Fig. 1B). TBI studies using transgenic models of astrocyte ablation have been particularly informative as to the specific roles being played by reactive astrocytes proximal to sites of brain trauma. For example, genetic ablation of the proliferative scar forming reactive astrocytes responding to focal penetrating brain damage results in a greatly intensified and prolonged neuroinflammatory response by both innate and adaptive immune cells, with pronounced neurodegeneration (Bush et al., 1999). Following CNS trauma, reactive astrocytes proliferate and dynamically intertwine to form a barrier that isolates inflammatory cells into numerous clusters, effectively containing an otherwise harmful inflammatory response to the site of injury (Wanner et al., 2013). Reactive astrocytes may play a similar role in quelling the inflammatory response to diffuse brain trauma that results in microvascular disruption and axonal injury that precipitate only a moderate innate inflammatory footprint (Csuka et al., 2000; Lin and Wen, 2013).
It is interesting that astrocytes act to isolate and protect CNS tissue not only under injury-reactive conditions, but also under non-reactive conditions in healthy tissue. For instance, the glial limitans, a barrier of astrocytic endfeet continuous with nearly all neurovascular basal lamina of the CNS is central to a selectively permeable blood brain barrier (Abbott et al., 2006), as well as the efficient exchange of cerebrospinal and interstitial fluid by the more recently described glymphatic system (Iliff et al., 2012; Nedergaard, 2013).
Still unclear are the mechanisms underlying how reactive astrocytes initially determine the locations of scar borders that separate potentially viable neural tissue from tissue that is irreversibly damaged and ceded to inflammatory degradation. Astrocytes express a number of neuroimmune-regulators (NI-Regs) that could spatially restrict inflammation by binding respective NI-Reg receptors on macrophages to directly inhibit phagocytic activity or obstruct members of the complement cascade by way of complement regulatory proteins (Burda and Sofroniew, 2014). Reactive astrocytes are also capable of inducing T-cell apoptosis and may thereby control the spread of peripheral lymphocyte-mediated inflammation following severe TBI. For example, following TBI, reactive astrocytes upregulate CD95 and CD95R to induce lymphocyte apoptosis through a CD95/CD95R-dependent mechanism, though astrocytes themselves remain protected (Bechmann et al., 2000; Bechmann et al., 2002; Griffiths et al., 2010).
Reactive astrocytes regulate the blood brain barrier
There is a long history of studying astrocyte roles in blood brain barrier function in health and disease (Abbott et al., 2006). Advances were greatly accelerated by transgenic mouse models that allowed dissection of cellular and molecular mechanisms of blood brain barrier function in vivo. For example, an early observation using transgenic mice demonstrated that blood brain barrier repair after penetrating TBI is critically dependent on the presence of newly proliferated scar-forming astrocytes (Bush et al., 1999). Recent findings using transgenic mice are beginning to define in vivo molecular mechanism through which astrocytes influence blood brain barrier integrity in different ways. There is now evidence that astrocyte secrete molecules that either open or close the blood brain barrier. For example, in response to stimulation by the pro-inflammatory cytokine IL-β, astrocytes generate and release vasoactive endothelial growth factor (VEGF) that increases blood-brain barrier permeability and promotes leukocyte extravasation (Argaw et al., 2009; Argaw et al., 2012). In addition, apolipoprotein E (APOE) secretion by astrocytes suppresses a cyclophilin A-NFκB-matrix metaloproteinase-9 pathway in pericytes that increases blood brain barrier permeability and is pro-inflammatory (Bell et al., 2012). In this context it is noteworthy that polymorphisms of APOE are associated with various disease mechanisms such that APOE4 allele in humans worsens prognosis in Alzheimer’s disease and after TBI and stroke (Mahley and Huang, 2012; Yamagata et al., 2013). In transgenic mice, modulation of astrocytes towards APOE4 production leads to blood brain barrier disruption, predisposing to inflammation (Bell et al., 2012). Under different circumstances, astrocytes release molecules that reduce blood brain barrier permeability and promote its repair. In this regard, astrocyte released molecules that act on endothelia to reduce blood brain barrier permeability after CNS injury include Sonic hedge hog (Shh) (Alvarez et al., 2011; Alvarez et al., 2013) and retinoic acid (Mizee et al., 2014). In addition, an astrocyte/microglial axis also likely to involve astrocyte-derived ATP gradients seems play a role in the maintenance of the blood brain barrier early after TBI (Roth et al., 2014). Thus, astrocytes are emerging as pivotal regulators of endothelial blood brain barrier properties that can, via specific molecular mechanisms, act to open, maintain or restore barrier functions, and do so in a context dependent manner as regulated by specific signaling events.
In this context is deserves mention that many reactive astrocytes alter their expression and cellular distribution of AQP4 so as to lose their heavy polarization of AQP4 along endothelial surfaces, with consequent impact on tissue fluid homeostasis and edema formation after TBI as reviewed elsewhere (Papadopoulos and Verkman, 2013). The potential impact of astrogliosis on the recently described glymphatic system and its proposed mechanisms for clearance from extracellular space into CSF of molecules and molecular fragments, including potential biomarkers of TBI, is intriguing (Iliff et al., 2012; Thrane et al., 2014). Recent evidence suggests a critical role for the glymphatic system in the clearance of CNS-endogenous biomarkers, including GFAP and S100B, into the blood following TBI (Plog et al., 2015).
Reactive astrocytes and neural circuit functions
It deserves emphasis that after mild or diffuse TBI, as well as in perimeter areas around focal CNS lesions, large areas of functioning neural tissue contain astrocytes exhibiting mild to moderate reactive astrogliosis (Burda and Sofroniew, 2014). These hypertrophic reactive astrocytes exhibit varying degrees of molecular, structural and functional changes that generally stop short of proliferation and scar formation (Fig. 1A), and these reactive astrocytes continue to interact with synapses and neurons as prior to the insult (Fig. 4). In healthy CNS tissue, astrocytes perform diverse functions critical for the function of mature neural circuits (Zhang et al., 2003; Schummers et al., 2008; Allen et al., 2012). The degree to which changes associated with reactive astrogliosis impacts on neural circuit function and neural circuit reorganization after CNS injuries is an area of intense interest and investigation, as discussed in the next sections.
Figure 4. Contribution of astrocyte-mediated synaptic scaling to post-traumatic epileptogenesis. A.
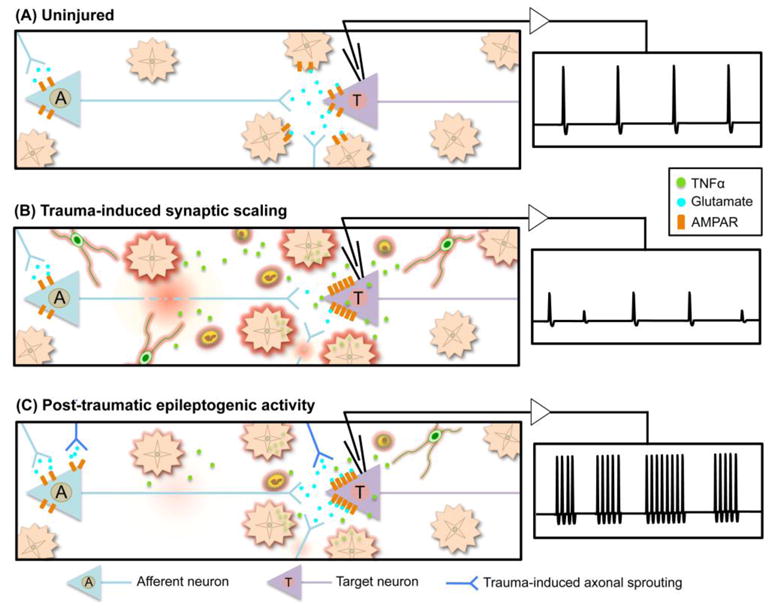
, In the uninjured brain, astrocytes buffer ions and neurotransmitters from the synaptic space and are actively involved in maintenance of neural activity and firing rate. B, TBI induced neuronal and axonal injury reduces afferent input to target neurons. The resulting reduction in synaptic activity elicits excitatory synaptic scaling, in part, by reactive astrocyte-derived TNFα, which stimulates AMPAR insertion into the postsynaptic membrane of target neurons, thereby increasing excitability. Infiltrating inflammatory cells also produce local gradients of TNFα, further amplifying post-synaptic excitability. C, Partial or complete recovery of traumatized afferent axonal input, and/or newly formed connections formed through trauma-induced axonal sprouting can drive epileptogenic burst firing in the post-traumatic hyperexcitable target neuron. Lingering inflammation and associated TNFα can continue to potentiate the synaptic scaling and hyperexcitability.
Reactive astrocytes regulate injury-induced synapse remodeling
A recent series of ground breaking studies from the Barres lab shows that in the developing CNS, astrocytes serve multiple essential roles in the formation of functional synapses (Clarke and Barres, 2013). Astrocyte-secreted thrombospondins (TSPs) and hevin work in tandem to induce structural formation of excitatory synapses (Christopherson et al., 2005; Kucukdereli et al., 2011). Astrocyte-derived Gypican-4 and -6, recruit AMPA glutamate receptors to synapses to induce their functionality (Allen et al., 2012). In addition, astrocytes play critical roles in the synapse pruning and phagocytosis that are critical for the development of functional neural circuits (Stevens et al., 2007; Chung et al., 2013). These seminal findings are now also providing a molecular basis with which to start dissecting how reactive astrocytes may impact on synapse formation and pruning after injury and disease, including TBI. For example, a recent study shows that following extracranial facial nerve transection, reactive astrocytes extend hypertrophic processes around neighboring traumatized motor neurons, promote their survival and mediate the recovery of functional synapses with excitatory afferents in a thrombospondin- and signal transducer and activator of transcription 3 (STAT3)-dependent mechanism (Tyzack et al., 2014). Expression of TSP-1 and TSP-2 by astrocytes is also increased after stroke and TSP-1/2 knockout mice demonstrate significantly diminished axonal sprouting that contributed to impaired functional recovery (Liauw et al., 2008). Much additional work is needed, but these early findings point towards important roles for reactive astrocytes in post-traumatic synaptic and neural circuit reorganization.
Reactive astrocytes mediate post-TBI changes in the perineuronal net
Astrocytes may also influence synaptic reorganization through production of extracellular matrix molecules that contribute to formation and modification of the perineuronal net, which is a rich network of extracellular matrix and cell adhesion proteins that encapsulates and is thought to stabilize synapses (Wang and Fawcett, 2012). These specialized structures contain a mixture of proteoglycans, glycoproteins and glycosaminoglycans, including chondroitin sulfate proteoglycans (CSPGs), tenascins (R and C), hyaluronan and link proteins, produced by both neurons and glia (Carulli et al., 2006; Wang and Fawcett, 2012). Formation of perineuronal nets during development is neural activity-dependent and associated with the closure of critical periods of plasticity and thereby act to inhibit formation of aberrant synaptic connections (McRae et al., 2007; Wang and Fawcett, 2012). In the normal brain, perineuronal nets are the predominant site of CSPG deposition (Carulli et al., 2006), however these sulfated proteoglycans and other perineuronal net proteins are upregulated with CNS trauma, where they may regulate post-traumatic collateral sprouting and synaptic reorganization. The upregulation of CSPGs in response to CNS trauma has been studied extensively, in part due to their nature as plasticity-restricting proteins and their accumulation within and around traumatic CNS lesions (Yi et al., 2012) reviewed in (Busch and Silver, 2007).
In response to TBI, astrocytes upregulate the expression of select CSPGs and other perineuronal net-related proteins, most of which have been demonstrated to have both permissive and restrictive effects on injury-induced axonal sprouting. For example, the CSPG neurocan, which is expressed normally only by developing astrocytes and neurons, is robustly expressed by reactive astrocytes in the hippocampus following traumatic deafferentation from the entorhinal cortex (Haas et al., 1999). In vitro axon outgrowth assays demonstrate an inhibitory effect of neurocan on developing axons (Friedlander et al., 1994), although in vivo, expression of neurocan is dense within zones of extensive axon sprouting, both following injury and in development (Fukuda et al., 1997; Haas et al., 1999). Brain trauma also causes astrocytes to increase expression of tenascin-C, which although capable of directly inhibiting axon outgrowth in vitro, localizes to zones of post-traumatic sprouting in the denervated hippocampus (Deller et al., 1997; Treloar et al., 2009). Such conflicting data may, in part, be explained by differential expression of adhesion molecules by developing or reactive sprouting axons (Andrews et al., 2009).
TBI-reactive changes in CPSG production are not uniform throughout the lesion area. For example, while the lesion core of a cortical contusion contains and is immediately surrounded by deposits of sulfated proteoglycans, there is a significant decrease in CSPGs in the perilesion perimeter (Harris et al., 2009; Harris et al., 2010; Yi et al., 2012). Indeed, astrocytes surrounding traumatic brain lesions are highly immuno-reactive for the CSPGs versican, neurocan and aggrecan (Harris et al., 2009), but appear to downregulate their expression further from the injury site. In fact, reduction in CSPG deposition within the perilesion area is maximal at 7 days post injury and correlates with a reduction in perineuronal net density and a remarkable increase in the number of growth associated protein-43-postiive neurons (Harris et al., 2010). In contrast to these findings, astrocytes surrounding traumatically deafferented hippocampal neurons appear to upregulate expression of neurocan specifically within regions of active sprouting (Haas et al., 1999). Therefore, like developmental axon guidance cues, reactive astrocytes may alter their spatiotemporal expression of plasticity-restricting perineuronal net proteins to push, pull and hem sprouting axons towards new functionally beneficial synaptic targets while preventing aberrant and potentially deleterious connections. Notably, ischemic brain injury in vimentin and GFAP double knockout mice leads to a compromised reactive gliosis, yielding greater amounts of CSPG in the perilesion area and significantly attenuated motor axon remodeling (Liu et al., 2014). This suggests that reactive astrocytes are involved in the alteration of perilesion CSPGs and play a substantial role in mediating post-traumatic circuit remodeling after brain injury. The precise signaling mechanisms that regulate astrocyte CSPG expression or drive perineuronal net breakdown in the perilesion area are unclear, but reactive astrocytes release of MMP-9 which can breakdown perineuronal net proteins (Pan et al., 2012). Thus reactive astrocytes have the potential to influence synaptic plasticity or stability via the breakdown or production of perineuronal nets.
Reactive astrocytes contribute to post-traumatic homeostatic synaptic plasticity
Regulation of synaptic drive and firing rate is central to maintaining stable and precise information transfer through vastly cross-connected neural circuits. Under normal physiologic circumstances, alterations in activity brought about by deviations in the strength of particular synapses due to long-term potentiation (LTP) or depression (LDP) can potentially destabilize associated networks (Malenka and Bear, 2004; Turrigiano and Nelson, 2004). Stabilizing phenomena, collectively referred to as homeostatic synaptic plasticity, respond to perturbations in activity by balancing excitatory and inhibitory drive through modulation of global synaptic strength (Turrigiano and Nelson, 2004). Homeostatic synaptic plasticity appears also to be an important mechanism for maintaining circuit function after TBI, when damage to neurons and axons results in decreased input to and from affected brain regions (Fig. 4A,B). Recent evidence points towards roles for reactive astrocytes in this process. Excitatory synaptic scaling, a form of homeostatic synaptic plasticity, tunes global excitatory synaptic strength to fluctuations in firing rate by increasing postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) density across all of a neurons’ synapses (Turrigiano et al., 1998; Beattie et al., 2002). This process has been demonstrated to rely, in part, on the production of the cytokine TNFα, which is secreted by astrocytes and possibly other glia in response to decreased excitatory input (Beattie et al., 2002; Stellwagen and Malenka, 2006; Turrigiano, 2006)(Fig. 4B). Reactive astrocyte-derived TNFα may also contribute to homeostatic synaptic plasticity after TBI such that TNFα released by reactive astrocytes following traumatic hippocampal denervation appears responsible for increased excitatory synaptic strength of local dentate granule cells (Becker et al., 2013). This suggests that reactive astrocytes play a role in maintaining neuronal excitability, and thereby neurologic function, after TBI (Fig. 4B).
Reactive astrocytes and post-traumatic epilepsy
Epilepsy can be a secondary pathology following TBI as a result of injury-induced circuit reorganization (Hunt et al., 2013). Computational models suggest that a post-traumatic inflammatory response could elicit epileptogenic activity by hijacking the astrocyte-mediated excitatory synaptic scaling system (Savin et al., 2009). Similar models also implicate synaptic scaling in the creation of epileptic burst firing within the deafferented cortex (Houweling et al., 2005). Interestingly, the overexpression of TNFα by astrocytes in the uninjured brain has been shown to elicit spontaneous seizure activity (Akassoglou et al., 1997). Thus, several lines of evidence suggest that although reactive-astrocyte derived TNFα may help to maintain neural excitability and neurological function after TBI by potentiating excitatory transmission (Fig. 4B), hyperphysiologic levels of TNFα after TBI have the potential to drive dysregulated synaptic scaling that results in harmful hyperexcitability of post-injury neural circuits (Fig. 4C). In such a model, an intermittent reduction or loss of excitatory input to a neuron following TBI could induce the production TNFα by reactive astrocytes to drive increased AMPAR insertion to the post-synaptic membrane to sustain neuronal excitability in response to reduced levels presynaptic glutamate release. As part of the inflammatory response to brain trauma, activated microglia and neutrophils recruited to the injury by reactive astrocytes (Fig. 3) also contribute to the production of local TNFα gradients that potentiate the excitatory synaptic scaling mechanism (Fig. 4B) (Turtzo et al., 2014). Activated microglia also release the chemokine stromal cell-derived factor 1, which binds its receptor, CXCR4 on astrocyte to elicit even further TNFα secretion into the (peri)synaptic space (Bezzi et al., 2001). Trauma-induced collateral sprouting of local axons, stimulated in part by astrocyte-derived molecules as discussed above, could result in simultaneous synaptic remodeling and the formation of compensatory excitatory inputs upon the deafferented neuron (Fig. 4C). These events, alone or in combination with the recovery of injured excitatory synapses on the now hyperexcitable neuron, have the potential to drive irregular and potentially epileptogenic excitatory transmission in the post-traumatic brain (Fig. 4C). It is also noteworthy that the absence of β1-integrin expression by astrocytes elicits spontaneous seizure activity in uninjured mouse brain (Robel et al., 2015), strengthening the argument that astrocyte dysfunction may be a causal factor in certain seizure disorders. The experimental examples of TNFα and β1-integrin also raise the interesting possibly that vulnerability to seizure disorders may be influenced by genetic polymorphisms in astrocytes. Nevertheless, it deserve emphasis that the majority of experimental and clinical circumstances that elicit astrogliosis in seizurigenic brain regions are not associated with seizures, suggesting that normally functioning astrogliosis per se is not an absolute seizure trigger and that specific molecular circumstances are involved when seizurigenesis occurs.
Concluding remarks
Recent progress demonstrates that astrocyte responses and roles in TBI are complex and determined by specific signaling mechanisms in a context-dependent manner that is related to the nature and severity of tissue damage. There is now substantive evidence that reactive astrocytes perform essential functions in the regulation and restriction of inflammation and the preservation of tissue and function. In addition, there is increasing evidence that reactive astrocytes play critical roles in post-TBI synaptic plasticity and the reorganization of neural circuits. It is also becoming clear that dysfunctions of reactive astrogliosis can occur, either through gain of abnormal effects or loss of normal functions, and contribute to post-TBI disorders, such as susceptibility to seizures. As a result, reactive astrocytes are increasingly recognized as potential targets for novel therapeutic strategies in TBI. For example, the anticonvulsant and anti-inflammatory drug levetiracetam may be useful in maintaining astrocyte connexin expression and healthy membrane potential (Haghikia et al., 2008; Stienen et al., 2011). Such a therapy could assist in the prevention of secondary injury due to enduring inflammation and closely related post-traumatic epilepsy discussed above. Opioid antagonists and agonists may also be potent regulators of astrocyte reactivity (Block et al., 2013). As we learn more about the specific astrocyte roles and underlying mechanisms regulating TBI pathophysiology additional potential therapeutic targets will emerge. Much work is needed in this area.
Highlights.
Astrocytes respond to diverse forms of brain injury with heterogeneous and progressive changes of gene expression, morphology, proliferative capacity and function that are collectively referred to as reactive astrogliosis.
In response to TBI, astrocytes in different cellular microenvironments tune their reactivity to varying degrees of axonal injury, vascular disruption, ischemia and inflammation.
TBI-reactive astrocytes significantly contribute to post-traumatic tissue repair and synaptic remodeling following brain trauma.
Acknowledgments
Work in the authors’ laboratory is supported by grants from the National Institutes of Health (NS057624, NS084030), The Dr. Miriam and Sheldon G. Adelson Medical Foundation and Wings for Life.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature Cited
- Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53. doi: 10.1038/nrn1824. [DOI] [PubMed] [Google Scholar]
- Ahmed SM, Rzigalinski BA, Willoughby KA, Sitterding HA, Ellis EF. Stretch-induced injury alters mitochondrial membrane potential and cellular ATP in cultured astrocytes and neurons. J Neurochem. 2000;74:1951–1960. [PubMed] [Google Scholar]
- Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol. 1997;158:438–445. [PubMed] [Google Scholar]
- Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486:410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez JI, Katayama T, Prat A. Glial influence on the blood brain barrier. Glia. 2013;61:1939–1958. doi: 10.1002/glia.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonniere L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science. 2011;334:1727–1731. doi: 10.1126/science.1206936. [DOI] [PubMed] [Google Scholar]
- Anderson MA, Ao Y, Sofroniew MV. Heterogeneity of reactive astrocytes. Neurosci Lett. 2014;565:23–29. doi: 10.1016/j.neulet.2013.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews MR, Czvitkovich S, Dassie E, Vogelaar CF, Faissner A, Blits B, Gage FH, ffrench-Constant C, Fawcett JW. Alpha9 integrin promotes neurite outgrowth on tenascin-C and enhances sensory axon regeneration. J Neurosci. 2009;29:5546–5557. doi: 10.1523/JNEUROSCI.0759-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106:1977–1982. doi: 10.1073/pnas.0808698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, Mahase S, Dutta DJ, Seto J, Kramer EG, Ferrara N, Sofroniew MV, John GR. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–2468. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzigalinski BA, JTW, Willoughby KA, Ellis EF. Intracellular Free Calcium Dynamics in Stretch-Injured Astrocytes. Journal of Neurochemistry. 1998;70:2377–2385. doi: 10.1046/j.1471-4159.1998.70062377.x. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Lossau S, Steiner B, Mor G, Gimsa U, Nitsch R. Reactive astrocytes upregulate Fas (CD95) and Fas ligand (CD95L) expression but do not undergo programmed cell death during the course of anterograde degeneration. Glia. 2000;32:25–41. doi: 10.1002/1098-1136(200010)32:1<25::aid-glia30>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Steiner B, Gimsa U, Mor G, Wolf S, Beyer M, Nitsch R, Zipp F. Astrocyte-induced T cell elimination is CD95 ligand dependent. J Neuroimmunol. 2002;132:60–65. doi: 10.1016/s0165-5728(02)00311-9. [DOI] [PubMed] [Google Scholar]
- Becker D, Zahn N, Deller T, Vlachos A. Tumor necrosis factor alpha maintains denervation-induced homeostatic synaptic plasticity of mouse dentate granule cells. Frontiers in cellular neuroscience. 2013;7:257. doi: 10.3389/fncel.2013.00257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485:512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A. CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci. 2001;4:702–710. doi: 10.1038/89490. [DOI] [PubMed] [Google Scholar]
- Block L, Bjorklund U, Westerlund A, Jorneberg P, Biber B, Hansson E. A new concept affecting restoration of inflammation-reactive astrocytes. Neuroscience. 2013;250:536–545. doi: 10.1016/j.neuroscience.2013.07.033. [DOI] [PubMed] [Google Scholar]
- Bowman CL, Ding JP, Sachs F, Sokabe M. Mechanotransducing ion channels in astrocytes. Brain Res. 1992;584:272–286. doi: 10.1016/0006-8993(92)90906-p. [DOI] [PubMed] [Google Scholar]
- Brambilla R, Hurtado A, Persaud T, Esham K, Pearse DD, Oudega M, Bethea JR. Transgenic inhibition of astroglial NF-kappa B leads to increased axonal sparing and sprouting following spinal cord injury. J Neurochem. 2009;110:765–778. doi: 10.1111/j.1471-4159.2009.06190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Bracchi-Ricard V, Hu WH, Frydel B, Bramwell A, Karmally S, Green EJ, Bethea JR. Inhibition of astroglial nuclear factor kappaB reduces inflammation and improves functional recovery after spinal cord injury. J Exp Med. 2005;202:145–156. doi: 10.1084/jem.20041918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burda JE, Sofroniew MV. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron. 2014;81:229–248. doi: 10.1016/j.neuron.2013.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch SA, Silver J. The role of extracellular matrix in CNS regeneration. Curr Opin Neurobiol. 2007;17:120–127. doi: 10.1016/j.conb.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- Carulli D, Rhodes KE, Brown DJ, Bonnert TP, Pollack SJ, Oliver K, Strata P, Fawcett JW. Composition of perineuronal nets in the adult rat cerebellum and the cellular origin of their components. J Comp Neurol. 2006;494:559–577. doi: 10.1002/cne.20822. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311–321. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csuka E, Hans VH, Ammann E, Trentz O, Kossmann T, Morganti-Kossmann MC. Cell activation and inflammatory response following traumatic axonal injury in the rat. Neuroreport. 2000;11:2587–2590. doi: 10.1097/00001756-200008030-00047. [DOI] [PubMed] [Google Scholar]
- Cullen DK, Vernekar VN, LaPlaca MC. Trauma-induced plasmalemma disruptions in three-dimensional neural cultures are dependent on strain modality and rate. J Neurotrauma. 2011;28:2219–2233. doi: 10.1089/neu.2011.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Deller T, Haas CA, Naumann T, Joester A, Faissner A, Frotscher M. Up-regulation of astrocyte-derived tenascin-C correlates with neurite outgrowth in the rat dentate gyrus after unilateral entorhinal cortex lesion. Neuroscience. 1997;81:829–846. doi: 10.1016/s0306-4522(97)00194-2. [DOI] [PubMed] [Google Scholar]
- Failli V, Kopp MA, Gericke C, Martus P, Klingbeil S, Brommer B, Laginha I, Chen Y, DeVivo MJ, Dirnagl U, Schwab JM. Functional neurological recovery after spinal cord injury is impaired in patients with infections. Brain. 2012;135:3238–3250. doi: 10.1093/brain/aws267. [DOI] [PubMed] [Google Scholar]
- Floyd CL, Gorin FA, Lyeth BG. Mechanical strain injury increases intracellular sodium and reverses Na+/Ca2+ exchange in cortical astrocytes. Glia. 2005;51:35–46. doi: 10.1002/glia.20183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander DR, Milev P, Karthikeyan L, Margolis RK, Margolis RU, Grumet M. The neuronal chondroitin sulfate proteoglycan neurocan binds to the neural cell adhesion molecules Ng-CAM/L1/NILE and N-CAM, and inhibits neuronal adhesion and neurite outgrowth. J Cell Biol. 1994;125:669–680. doi: 10.1083/jcb.125.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Kawano H, Ohyama K, Li HP, Takeda Y, Oohira A, Kawamura K. Immunohistochemical localization of neurocan and L1 in the formation of thalamocortical pathway of developing rats. J Comp Neurol. 1997;382:141–152. [PubMed] [Google Scholar]
- Gennarelli TA, Thibault LE, Adams JH, Graham DI, Thompson CJ, Marcincin RP. Diffuse axonal injury and traumatic coma in the primate. Ann Neurol. 1982;12:564–574. doi: 10.1002/ana.410120611. [DOI] [PubMed] [Google Scholar]
- Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- Graham DI, McIntosh TK, Maxwell WL, Nicoll JA. Recent advances in neurotrauma. J Neuropathol Exp Neurol. 2000;59:641–651. doi: 10.1093/jnen/59.8.641. [DOI] [PubMed] [Google Scholar]
- Griffiths MR, Gasque P, Neal JW. The regulation of the CNS innate immune response is vital for the restoration of tissue homeostasis (repair) after acute brain injury: a brief review. International journal of inflammation. 2010;2010:151097. doi: 10.4061/2010/151097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie PB, Knappenberger J, Segal M, Bennett MV, Charles AC, Kater SB. ATP released from astrocytes mediates glial calcium waves. J Neurosci. 1999;19:520–528. doi: 10.1523/JNEUROSCI.19-02-00520.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas CA, Rauch U, Thon N, Merten T, Deller T. Entorhinal cortex lesion in adult rats induces the expression of the neuronal chondroitin sulfate proteoglycan neurocan in reactive astrocytes. J Neurosci. 1999;19:9953–9963. doi: 10.1523/JNEUROSCI.19-22-09953.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghikia A, Ladage K, Hinkerohe D, Vollmar P, Heupel K, Dermietzel R, Faustmann PM. Implications of antiinflammatory properties of the anticonvulsant drug levetiracetam in astrocytes. J Neurosci Res. 2008;86:1781–1788. doi: 10.1002/jnr.21639. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J Neurosci. 2012;32:14489–14510. doi: 10.1523/JNEUROSCI.1256-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NG, Carmichael ST, Hovda DA, Sutton RL. Traumatic brain injury results in disparate regions of chondroitin sulfate proteoglycan expression that are temporally limited. J Neurosci Res. 2009;87:2937–2950. doi: 10.1002/jnr.22115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris NG, Mironova YA, Hovda DA, Sutton RL. Pericontusion axon sprouting is spatially and temporally consistent with a growth-permissive environment after traumatic brain injury. J Neuropathol Exp Neurol. 2010;69:139–154. doi: 10.1097/NEN.0b013e3181cb5bee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Pham LD, Katusic ZS, Arai K, Lo EH. Astrocytic high-mobility group box 1 promotes endothelial progenitor cell-mediated neurovascular remodeling during stroke recovery. Proc Natl Acad Sci U S A. 2012a;109:7505–7510. doi: 10.1073/pnas.1121146109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Miyamoto N, Seo JH, Pham LD, Kim KW, Lo EH, Arai K. High-mobility group box 1 from reactive astrocytes enhances the accumulation of endothelial progenitor cells in damaged white matter. J Neurochem. 2012b doi: 10.1111/jnc.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoarau JJ, Krejbich-Trotot P, Jaffar-Bandjee MC, Das T, Thon-Hon GV, Kumar S, Neal JW, Gasque P. Activation and control of CNS innate immune responses in health and diseases: a balancing act finely tuned by neuroimmune regulators (NIReg) CNS Neurol Disord Drug Targets. 2011;10:25–43. doi: 10.2174/187152711794488601. [DOI] [PubMed] [Google Scholar]
- Hoffman SW, Rzigalinski BA, Willoughby KA, Ellis EF. strocytes Generate Isoprostanes in Response to Trauma or Oxygen Radicals. Journal of Neurotrauma. 2000:17. doi: 10.1089/neu.2000.17.415. [DOI] [PubMed] [Google Scholar]
- Houweling AR, Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Homeostatic synaptic plasticity can explain post-traumatic epileptogenesis in chronically isolated neocortex. Cerebral cortex. 2005;15:834–845. doi: 10.1093/cercor/bhh184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Han X, Li X, Lam E, Peng W, Lou N, Torres A, Yang M, Garre JM, Tian GF, Bennett MV, Nedergaard M, Takano T. Critical role of connexin 43 in secondary expansion of traumatic spinal cord injury. J Neurosci. 2012;32:3333–3338. doi: 10.1523/JNEUROSCI.1216-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt RF, Boychuk JA, Smith BN. Neural circuit mechanisms of post-traumatic epilepsy. Frontiers in cellular neuroscience. 2013;7:89. doi: 10.3389/fncel.2013.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Science translational medicine. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islas L, Pasantes-Morales H, Sanchez JA. Characterization of stretch-activated ion channels in cultured astrocytes. Glia. 1993;8:87–96. doi: 10.1002/glia.440080204. [DOI] [PubMed] [Google Scholar]
- Javid S, Rezaei A, Karami G. A micromechanical procedure for viscoelastic characterization of the axons and ECM of the brainstem. Journal of the mechanical behavior of biomedical materials. 2014;30:290–299. doi: 10.1016/j.jmbbm.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Jayakumar AR, Tong XY, Ruiz-Cordero R, Bregy A, Bethea JR, Bramlett HM, Norenberg MD. Activation of NF-kappaB mediates astrocyte swelling and brain edema in traumatic brain injury. J Neurotrauma. 2014;31:1249–1257. doi: 10.1089/neu.2013.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. 2013;246:35–43. doi: 10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RS, Minogue AM, Connor TJ, Lynch MA. Amyloid-beta-induced astrocytic phagocytosis is mediated by CD36, CD47 and RAGE. Journal of neuroimmune pharmacology : the official journal of the Society on NeuroImmune Pharmacology. 2013;8:301–311. doi: 10.1007/s11481-012-9427-3. [DOI] [PubMed] [Google Scholar]
- Kim JV, Dustin ML. Innate response to focal necrotic injury inside the blood-brain barrier. J Immunol. 2006;177:5269–5277. doi: 10.4049/jimmunol.177.8.5269. [DOI] [PubMed] [Google Scholar]
- Kovacs SK, Leonessa F, Ling GS. Blast TBI Models, Neuropathology, and Implications for Seizure Risk. Frontiers in neurology. 2014;5:47. doi: 10.3389/fneur.2014.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukdereli H, Allen NJ, Lee AT, Feng A, Ozlu MI, Conatser LM, Chakraborty C, Workman G, Weaver M, Sage EH, Barres BA, Eroglu C. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc Natl Acad Sci U S A. 2011;108:E440–449. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird MD, Shields JS, Sukumari-Ramesh S, Kimbler DE, Fessler RD, Shakir B, Youssef P, Yanasak N, Vender JR, Dhandapani KM. High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of toll-like receptor 4. Glia. 2014;62:26–38. doi: 10.1002/glia.22581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38:1336–1345. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Li Y, Chopp M. Temporal profile of nestin expression after focal cerebral ischemia in adult rat. Brain Res. 1999;838:1–10. doi: 10.1016/s0006-8993(99)01502-4. [DOI] [PubMed] [Google Scholar]
- Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- Lin Y, Wen L. Inflammatory response following diffuse axonal injury. International journal of medical sciences. 2013;10:515–521. doi: 10.7150/ijms.5423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Li Y, Cui Y, Roberts C, Lu M, Wilhelmsson U, Pekny M, Chopp M. Beneficial effects of gfap/vimentin reactive astrocytes for axonal remodeling and motor behavioral recovery in mice after stroke. Glia. 2014;62:2022–2033. doi: 10.1002/glia.22723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Apolipoprotein e sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–885. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- McRae PA, Rocco MM, Kelly G, Brumberg JC, Matthews RT. Sensory deprivation alters aggrecan and perineuronal net expression in the mouse barrel cortex. J Neurosci. 2007;27:5405–5413. doi: 10.1523/JNEUROSCI.5425-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meythaler JM, Peduzzi JD, Eleftheriou E, Novack TA. Current concepts: diffuse axonal injury-associated traumatic brain injury. Archives of physical medicine and rehabilitation. 2001;82:1461–1471. doi: 10.1053/apmr.2001.25137. [DOI] [PubMed] [Google Scholar]
- Mizee MR, Nijland PG, van der Pol SM, Drexhage JA, van Het Hof B, Mebius R, van der Valk P, van Horssen J, Reijerkerk A, de Vries HE. Astrocyte-derived retinoic acid: a novel regulator of blood-brain barrier function in multiple sclerosis. Acta neuropathologica. 2014;128:691–703. doi: 10.1007/s00401-014-1335-6. [DOI] [PubMed] [Google Scholar]
- Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- Neal JWDM, Hoarau JJ, Gasque P. The CNS Innate Immune System and the Emerging Roles of the Neuroimmune Regulators (NIRegs) in Response to Infection, Neoplasia and Neurodegeneration. In: DJK, editor. Recent Advances in Immunology to Target Cancer, Inflammation and Infections. intechopen; 2012. pp. 201–240. [Google Scholar]
- Neary JT, Kang Y, Willoughby KA, Ellis EF. Activation of extracellular signal-regulated kinase by stretch-induced injury in astrocytes involves extracellular ATP and P2 purinergic receptors. J Neurosci. 2003;23:2348–2356. doi: 10.1523/JNEUROSCI.23-06-02348.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neary JT, Kang Y, Tran M, Feld J. Traumatic injury activates protein kinase B/Akt in cultured astrocytes: role of extracellular ATP and P2 purinergic receptors. J Neurotrauma. 2005;22:491–500. doi: 10.1089/neu.2005.22.491. [DOI] [PubMed] [Google Scholar]
- Nedergaard M. Neuroscience. Garbage truck of the brain. Science. 2013;340:1529–1530. doi: 10.1126/science.1240514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ommaya AK, Gennarelli TA. Cerebral concussion and traumatic unconsciousness. Correlation of experimental and clinical observations of blunt head injuries. Brain. 1974;97:633–654. doi: 10.1093/brain/97.1.633. [DOI] [PubMed] [Google Scholar]
- Ostrow LW, Suchyna TM, Sachs F. Stretch induced endothelin-1 secretion by adult rat astrocytes involves calcium influx via stretch-activated ion channels (SACs) Biochem Biophys Res Commun. 2011;410:81–86. doi: 10.1016/j.bbrc.2011.05.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Wang H, Wang X, Zhu L, Mao L. The absence of Nrf2 enhances NF-kappaB-dependent inflammation following scratch injury in mouse primary cultured astrocytes. Mediators of inflammation. 2012;2012:217580. doi: 10.1155/2012/217580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14:265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrazzi M, Patrone M, Passalacqua M, Ranzato E, Colamassaro D, Sparatore B, Pontremoli S, Melloni E. Selective proinflammatory activation of astrocytes by high-mobility group box 1 protein signaling. J Immunol. 2007;179:8525–8532. doi: 10.4049/jimmunol.179.12.8525. [DOI] [PubMed] [Google Scholar]
- Pekny M, Lane EB. Intermediate filaments and stress. Exp Cell Res. 2007;313:2244–2254. doi: 10.1016/j.yexcr.2007.04.023. [DOI] [PubMed] [Google Scholar]
- Pekny M, Pekna M. Astrocyte reactivity and reactive astrogliosis: costs and benefits. Physiol Rev. 2014;94:1077–1098. doi: 10.1152/physrev.00041.2013. [DOI] [PubMed] [Google Scholar]
- Pekny M, Johansson CB, Eliasson C, Stakeberg J, Wallen A, Perlmann T, Lendahl U, Betsholtz C, Berthold CH, Frisen J. Abnormal reaction to central nervous system injury in mice lacking glial fibrillary acidic protein and vimentin. J Cell Biol. 1999;145:503–514. doi: 10.1083/jcb.145.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- Plog BA, Dashnaw ML, Hitomi E, Peng W, Liao Y, Lou N, Deane R, Nedergaard M. Biomarkers of Traumatic Injury Are Transported from Brain to Blood via the Glymphatic System. J Neurosci. 2015;35:518–526. doi: 10.1523/JNEUROSCI.3742-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponath G, Schettler C, Kaestner F, Voigt B, Wentker D, Arolt V, Rothermundt M. Autocrine S100B effects on astrocytes are mediated via RAGE. J Neuroimmunol. 2007;184:214–222. doi: 10.1016/j.jneuroim.2006.12.011. [DOI] [PubMed] [Google Scholar]
- Povlishock JT. Pathobiology of traumatically induced axonal injury in animals and man. Annals of emergency medicine. 1993;22:980–986. doi: 10.1016/s0196-0644(05)82738-6. [DOI] [PubMed] [Google Scholar]
- Ralay Ranaivo H, Zunich SM, Choi N, Hodge JN, Wainwright MS. Mild stretch-induced injury increases susceptibility to interleukin-1beta-induced release of matrix metalloproteinase-9 from astrocytes. J Neurotrauma. 2011;28:1757–1766. doi: 10.1089/neu.2011.1799. [DOI] [PubMed] [Google Scholar]
- Robel S, Buckingham SC, Boni JL, Campbell SL, Danbolt NC, Riedemann T, Sutor B, Sontheimer H. Reactive astrogliosis causes the development of spontaneous seizures. J Neurosci. 2015;35:3330–3345. doi: 10.1523/JNEUROSCI.1574-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N, Avignone E, Meme W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biology of the cell/under the auspices of the European Cell Biology Organization. 2002;94:457–475. doi: 10.1016/s0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Rzigalinski BA, Liang S, McKinney JS, Willoughby KA, Ellis EF. Effect of Ca2+ on in vitro astrocyte injury. J Neurochem. 1997;68:289–296. doi: 10.1046/j.1471-4159.1997.68010289.x. [DOI] [PubMed] [Google Scholar]
- Savin C, Triesch J, Meyer-Hermann M. Epileptogenesis due to glia-mediated synaptic scaling. Journal of the Royal Society, Interface/the Royal Society. 2009;6:655–668. doi: 10.1098/rsif.2008.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schummers J, Yu H, Sur M. Tuned responses of astrocytes and their influence on hemodynamic signals in the visual cortex. Science. 2008;320:1638–1643. doi: 10.1126/science.1156120. [DOI] [PubMed] [Google Scholar]
- Sharp DJ, Scott G, Leech R. Network dysfunction after traumatic brain injury. Nature reviews Neurology. 2014;10:156–166. doi: 10.1038/nrneurol.2014.15. [DOI] [PubMed] [Google Scholar]
- Shih AY, Blinder P, Tsai PS, Friedman B, Stanley G, Lyden PD, Kleinfeld D. The smallest stroke: occlusion of one penetrating vessel leads to infarction and a cognitive deficit. Nat Neurosci. 2013;16:55–63. doi: 10.1038/nn.3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton RH, Povlishock JT. Identification and characterization of heterogeneous neuronal injury and death in regions of diffuse brain injury: evidence for multiple independent injury phenotypes. J Neurosci. 2004;24:3543–3553. doi: 10.1523/JNEUROSCI.5048-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skandsen T, Kvistad KA, Solheim O, Strand IH, Folvik M, Vik A. Prevalence and impact of diffuse axonal injury in patients with moderate and severe head injury: a cohort study of early magnetic resonance imaging findings and 1-year outcome. Journal of neurosurgery. 2010;113:556–563. doi: 10.3171/2009.9.JNS09626. [DOI] [PubMed] [Google Scholar]
- Smith DH, Nonaka M, Miller R, Leoni M, Chen XH, Alsop D, Meaney DF. Immediate coma following inertial brain injury dependent on axonal damage in the brainstem. Journal of neurosurgery. 2000;93:315–322. doi: 10.3171/jns.2000.93.2.0315. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2014a;20:160–172. doi: 10.1177/1073858413504466. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Astrogliosis. Cold Spring Harbor perspectives in biology. 2014b doi: 10.1101/cshperspect.a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta neuropathologica. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131:1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- Stienen MN, Haghikia A, Dambach H, Thone J, Wiemann M, Gold R, Chan A, Dermietzel R, Faustmann PM, Hinkerohe D, Prochnow N. Anti-inflammatory effects of the anticonvulsant drug levetiracetam on electrophysiological properties of astroglia are mediated via TGFbeta1 regulation. Br J Pharmacol. 2011;162:491–507. doi: 10.1111/j.1476-5381.2010.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Thrane AS, Rangroo Thrane V, Nedergaard M. Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci. 2014 doi: 10.1016/j.tins.2014.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivola DM, Strnad P, Habtezion A, Omary MB. Intermediate filaments take the heat as stress proteins. Trends in cell biology. 2010;20:79–91. doi: 10.1016/j.tcb.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treloar HB, Ray A, Dinglasan LA, Schachner M, Greer CA. Tenascin-C is an inhibitory boundary molecule in the developing olfactory bulb. J Neurosci. 2009;29:9405–9416. doi: 10.1523/JNEUROSCI.2356-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. More than a sidekick: glia and homeostatic synaptic plasticity. Trends in Molecular Medicine. 2006;12:458–460. doi: 10.1016/j.molmed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Turtzo LC, Lescher J, Janes L, Dean DD, Budde MD, Frank JA. Macrophagic and microglial responses after focal traumatic brain injury in the female rat. J Neuroinflammation. 2014;11:82. doi: 10.1186/1742-2094-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyzack GE, Sitnikov S, Barson D, Adams-Carr KL, Lau NK, Kwok JC, Zhao C, Franklin RJ, Karadottir RT, Fawcett JW, Lakatos A. Astrocyte response to motor neuron injury promotes structural synaptic plasticity via STAT3-regulated TSP-1 expression. Nature communications. 2014;5:4294. doi: 10.1038/ncomms5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Matteoli M. ATP mediates calcium signaling between astrocytes and microglial cells: modulation by IFN-gamma. J Immunol. 2001;166:6383–6391. doi: 10.4049/jimmunol.166.10.6383. [DOI] [PubMed] [Google Scholar]
- Villapol S, Byrnes KR, Symes AJ. Temporal dynamics of cerebral blood flow, cortical damage, apoptosis, astrocyte-vasculature interaction and astrogliosis in the pericontusional region after traumatic brain injury. Frontiers in neurology. 2014;5:82. doi: 10.3389/fneur.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky E, Palzur E, Feinsod M, Soustiel JF. Evaluation of the apoptosis-related proteins of the BCL-2 family in the traumatic penumbra area of the rat model of cerebral contusion, treated by hyperbaric oxygen therapy: a quantitative immunohistochemical study. Acta neuropathologica. 2005;110:120–126. doi: 10.1007/s00401-004-0946-8. [DOI] [PubMed] [Google Scholar]
- Walko TD, 3rd, Bola RA, Hong JD, Au AK, Bell MJ, Kochanek PM, Clark RS, Aneja RK. Cerebrospinal fluid mitochondrial DNA: a novel DAMP in pediatric traumatic brain injury. Shock. 2014;41:499–503. doi: 10.1097/SHK.0000000000000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Fawcett J. The perineuronal net and the control of CNS plasticity. Cell and tissue research. 2012;349:147–160. doi: 10.1007/s00441-012-1375-y. [DOI] [PubMed] [Google Scholar]
- Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33:12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M. Animal models of traumatic brain injury. Nat Rev Neurosci. 2013;14:128–142. doi: 10.1038/nrn3407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Nakayama C, Suzuki K. Dietary beta-carotene regulates interleukin-1beta-induced expression of apolipoprotein E in astrocytes isolated from stroke-prone spontaneously hypertensive rats. Neurochem Int. 2013;62:43–49. doi: 10.1016/j.neuint.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Yi JH, Katagiri Y, Susarla B, Figge D, Symes AJ, Geller HM. Alterations in sulfated chondroitin glycosaminoglycans following controlled cortical impact injury in mice. J Comp Neurol. 2012;520:3295–3313. doi: 10.1002/cne.23156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaheer A, Yorek MA, Lim R. Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion. Neurochemical research. 2001;26:1293–1299. doi: 10.1023/a:1014241300179. [DOI] [PubMed] [Google Scholar]
- Zetterberg H, Smith DH, Blennow K. Biomarkers of mild traumatic brain injury in cerebrospinal fluid and blood. Nature reviews Neurology. 2013;9:201–210. doi: 10.1038/nrneurol.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol. 2010;20:588–594. doi: 10.1016/j.conb.2010.06.005. [DOI] [PubMed] [Google Scholar]