

Abstract
The proteolytic activity of cathepsin B in complex breast cell lysates have been measured with alternating current voltammetry (ACV) using ferrocene (Fc)-labeled-tetrapeptides immobilized on nanoelectrode arrays (NEAs) fabricated with vertically aligned carbon nanofibers (VACNFs). Four types of breast cells have been tested, including normal breast cells (HMEC), transformed breast cells (MCF-10A), breast cancer cells (T47D), and metastatic breast cancer cells (MDA-MB-231). The detected protease activity was found increased in cancer cells, with the MDA-MB-231 metastatic cancer cell lysate showing the highest cathepsin B activity. The equivalent cathepsin B concentration in MDA-MB-231 cancer cell lysate was quantitatively determined by spiking recombinant cathepsin B into the immunoprecipitated MDA-MB-231 lysate and the HMEC whole cell lysate. The results illustrated the potential of this technique as a portable multiplex electronic device for cancer diagnosis and treatment monitoring through rapid profiling the activity of specific cancer-relevant proteases.
Keywords: Electrochemical protease profiling, nanoelectrode array, vertically aligned carbon nanofibers, proteolytic activity, cancer formation
Graphical Abstract
The proteolytic activity of cathepsin B in breast cell lysates have been measured with alternating current voltammetry using ferrocene-labeled-tetrapeptides immobilized on nanoelectrode arrays fabricated with vertically aligned carbon nanofibers. Four types of breast cells have been tested, including normal breast cells (HMEC), transformed breast cells (MCF-10A), breast cancer cells (T47D), and metastatic breast cancer cells (MDA-MB-231). The detected protease activity was found increased in cancer cells, with the highest value in MDA-MB-231 cell lysate. This technique is promising as a portable multiplex electronic device for cancer diagnosis and treatment monitoring through rapid profiling the activity of specific cancer-relevant proteases.
Introduction
Breast cancer is the most common type of cancers, with about 235,000 new cases expected in the United States in 2014, followed by prostate and lung cancer.1 The most useful prognostic parameters for cancer recurrence and progression are the tumor grade, the depth of tumor penetration (stage) and the presence of carcinoma in situ.2 Quantitative detection of molecular biomarkers that are involved in breast tumor progression is another important avenue for cancer diagnosis and treatment. It is known that cancer metastasis involves proteases, a group of enzymes that cause proteolysis to degrade the extracellular matrix (ECM) components and intercellular cohesive structures of neighboring cells leading to the activation of growth and angiogenic factors,3, 4 which could serve as biomarkers for diagnosis.5 Among them, cathepsin B is a representative prognostic marker for human lung cancer6 and breast cancer.7
Cathepsin B is a papain-family cysteine protease synthesized as glycosylated proenzyme of ∼37 kDa and subsequently converted to the active form of ∼25 kDa [by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)] after the proteolysis of the signal sequence.8, 9 Of the cysteine proteases, cathepsin B has been most often implicated in invasive and malignant phenotype of tumors including brain, breast, lung, pancreas, bladder, prostate and stomach.10 Enzyme-linked immunosorbent assay (ELISA) has been commonly used to measure the cathepsin B concentrations in urine, serum, and tissue lysates. Kotaska et al. found that cathepsin B concentrations have been significantly elevated in the serum and urine samples from bladder cancer patients and correlated well with grading and invasivity of tumors.2 Chen's group claimed that the serum level of cathepsin B was significantly higher in the lung cancer patients compared to the healthy controls.11 Using immunochemical staining of tissue section and immunoblotting of cell lysates, Nouh et al. found that there was a positive correlation between the expression of cathepsin B and the number of positive metastatic lymph nodes in inflammatory breast cancer.7 By studying the microdissected frozen tissue sections of human tumors from colon and breast carcinoma, a group of researchers found a significant increase in the level of cathepsin B in regions of invasive tumor as compared to normal epithelium from the same patients.12, 13
Although ELISA and immunohistochemical methods can sensitively detect the concentration of specific proteases, they are typically time-consuming and can only measure one protease each time. In contrast, electrochemical technology has superior advantages such as its simple electronic readout and potential for simultaneous detection of multiple proteases using an electrode array, making it more suitable for rapid point-of-care diagnosis, treatment monitoring, and drug (protease inhibitor) screening. In addition, it detects the activity instead of concentration of proteases, which is more relevant to biological processes. However, there is very limited study of the activities of cancer-related proteases in complex human tissues or cancer cell lysates using electrochemical methods. In previous studies, we have demonstrated an electrochemical technique in detecting the proteolytic kinetics of two cancer-related proteases legumain and cathepsin B in purified solutions and complex whole tissue lysates, respectively, using nanoelectrode arrays (NEAs) fabricated with vertically aligned carbon nanofibers (VACNFs), a special type of carbon nanotubes.14, 15 Specific tetrapeptides with a ferrocene (Fc) group at the distal end were attached to the exposed VACNF tip and selectively cleaved by the proteases. The unique structure of VACNFs allowed using a high-frequency (∼1 kHz) alternating current voltammetry (ACV) to monitor the small electrochemical signal from Fc.14-16 In this report, we further apply this technique on detection of cathepsin B activities in four types of cell lysates including normal and breast cancer cells. The cathepsin B activities were found increased from normal to cancer cells and reached highest value in metastatic cancer cell lysate. A specific inhibitor clearly suppressed the activity of cathepsin B in the cell lysate. Our results showed that this NEA-based electrochemical method has great potential as a portable multiplex system for simple and rapid protease profiling of lysate, serum or urine samples toward early cancer diagnosis, treatment monitoring, and drug screening.
Methods
Materials and Reagents
All reported measurements in this study used purified recombinant human cathepsin B containing proenzyme (∼60%) and mature (∼40%) cathepsin B, purchased from R&D Systems Inc. (Minneapolis, MN, USA). The molecular weights are 37 and 25 kDa, respectively, predicted from the sequence. Thus the average molecular weight of ∼33.8 kDa was used in all concentration calculations. Only a few control experiments in immunoprecipitation study used cathepsin B purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). In normal activity measurements, cathepsin B and the breast cell lysates were activated for 15 min in 5 mM dithiothreitol (DTT) and 25 mM 2-(4-morpholino)ethanesulfonic acid (MES) buffer (pH 5.0) to convert the proenzyme into the active form. The details of other reagents are listed in the Supplementary Information (SI). The synthesis of cathepsin B specific tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc and other peptides including tripeptide Arg-Phe-Gly-NH-CH2-Fc, dipeptide Phe-Gly-NH-CH2-Fc and modified single amino acid Gly-NH-CH2-Fc for studying cleaving sites is also described in the SI (Scheme S1). The cathepsin B inhibitor GC-373 was prepared17 in house as described in the SI.
Cell lines and correlation with cancer stages
The selected cell lines were used in representing different stages of cancer formation. Human mammary epithelial cells (HMEC), obtained from Lonza (Annandale, NJ, USA), are primary cells, derived from adult female breast tissue. HMECs are positive for cytokertins 14 and 18 and negative for cytokeratin 19. MCF-10A cell line, obtained from American Type Cell Culture (ATCC) (Manassas, MA, USA), is a non-tumorigenic epithelial cell line derived from a 36 year old female. These cells have no signs of terminal differentiation or senescence. They respond to insulin, epidermal growth factor (EGF), and glucocorticoids. T47D cell line, obtained from ATCC, was isolated by I. Keydar from a pleural effusion obtained from a 54 year old female patient with an infiltrating ductal carcinoma of the breast. MDA-MB-231 cell line, obtained from ATCC, was isolated from a metastatic site of a 51 year old female patient with breast adenocarcinoma.
Fabrication of VACNF NEAs
The fabrication procedures of VACNF NEAs followed previous reports.14, 15, 18-21 Briefly, randomly distributed VACNFs with average length of ∼5 μm and an areal density of ∼1×109 CNFs/cm2 were grown on Cr coated Si(100) wafer using a plasma enhanced chemical vapor deposition (PECVD) system (Aixtron, CA, USA). A nickel film with ∼22 nm thickness was coated as the catalyst for CNF growth. The diameter of the CNFs distributed from ∼100 to 200 nm. Dielectric SiO2 was deposited on the as-grown CNFs using chemical vapor deposition (CVD) of ∼400-500 mTorr tetraethyl-orthosilicate precursor. Reactive ion etching (RIE) system (Nano-Master Inc., Austin, TX, USA) was performed to selectively etch away desired amount of SiO2, leaving only a tip length of ∼20 to 200 nm of the longest VACNFs to expose above the SiO2 matrix, forming a randomly distributed NEA with a neighboring distance of ∼0.5-2 μm (Supplementary Figure S1).
As detailed in the SI, the SiO2 surface of VACNF NEAs was first passivated with ethylene glycol to reduce nonspecific adsorption of biomolecules before the measurements. The VACNF tips were then cleaned and activated by electrochemical etching. The Fc-appended tetrapeptides were covalently linked to the VACNF NEA by forming an amide bond between the –(CH2)4NH2 linker and the –COOH group at the exposed CNF tips. In order to stabilize electrochemical signals, the unreacted –COOH sites at the VACNF NEA was further reacted with 6-aminohexanol as diluent agents.
Electrochemical measurements of enzymatic activity
The electrochemical measurements were carried out with an electrochemical analyzer (Model 440A, CH Instruments, Austin, TX, USA) in a TEFLON cell with a total volume of 250 μL w sealed against the VACNF NEA chip with a 3-mm i.d. O-ring. Three-electrode configuration was used in all measurements, including a VACNF NEA working electrode, an Ag/AgCl (3M KCl) reference electrode, and a coiled Pt wire counter electrode. The electrolyte was 25 mM MES (pH = 5.0). The ACV was measured by applying a 800 Hz and 150 mV amplitude AC voltage on the DC staircase waveform sweeping from -0.1 V to +0.75 V at 10 mV·s-1 scan rate.
Enzyme activity measurements with fluorescence assays
Fluorescence assay was performed on a GloMax-Multi+ Microplate Multimode Reader (Promega, Madison, WI, USA). Cathepsin B specific fluorogenic peptide substrate benzyloxycarbonyl-leucine-arginine-7-amino-4-methylcoumarin (Z-Leu-Arg-AMC) (Bachem, Torrance, CA, USA) was used in the fluorescence assay to measure enzyme activity. The cleavage at the site between arginine and AMC dye by cathepsin B resulted in the increase of fluorescence intensity. The half maximal inhibitory concentration (IC50) of the cathepsin B inhibitor GC-373 was measured with this method as detailed in the SI.
Western blot and immunoprecipitation of cancer cell lysates
Cells were harvested and lysed in lysis buffer (20 mM Tris pH 7.5, 0.5 mM EDTA, 0.5 mM EGTA, 0.5% Triton X-100) at 1:1,000 dilution of protease inhibitors (Sigma-Aldrich, Saint Louis, MO, USA). Whole-cell extract (25 μg) was resolved by 10% SDS-PAGE and transferred to nitrocellulose membrane (Midwest Scientific, Saint Louis, MO, USA) that was blocked in 5% milk for an hour at room temperature and then incubated with monoclonal antibodies (1:1,000). Western blots were detected by enhanced chemiluminescence detection reagents (Pierce, Rockford, IL, USA) and visualized by Fluorchem E imaging system (ProteinSimple, Santa Clara, CA, USA).
The procedure for obtaining whole-cell extract (WCE) for immunoprecipitation (IP) was the same as that in Western blotting. First, 500 μg WCE was pre-cleared with 20 μL of protein A/G-agarose beads from Santa Cruz Biotechnology (Santa Cruz, CA, USA) for 30 minutes. Then, the mixture was centrifuged at 2,000 rpm for 5 minutes. The supernatant was collected and incubated with the primary antibody of cathepsin B (1:1000, giving the total amount of 1.4 μg) from Santa Cruz Biotechnologies (Santa Cruz, CA, USA) overnight at 4°C. Twenty μl of protein A/G-agarose beads was added and incubated for additional 2 h at 4°C on a rocker. Samples were centrifuged at 2,000 rpm for 5 minutes and pellet (A/G-agarose complex) was washed three times with lysis buffer (20 mM Tris pH 7.5, 0.5 mM EDTA, 0.5 mM EGTA, 0.5% Triton X-100). Samples were run on 10% SDS-PAGE and immunoblotted with cathepsin B as described in Western blot analysis. In case of multiple steps of IP, samples were incubated with cathepsin B antibodies for 2 or 3 times prior to Western blot analysis. This repeated step allows the complete removal of cathepsin B in whole cell lysate. Intensities of the bands were digitized using Un-Scan-It software (Silk Scientific Inc., Orem, Utah, USA). The supernatant after the IP procedure was used in some proteolysis experiments as a blank control.
Results
Principles of ACV detection of protease activities using NEAs
Figure 1 shows the schematic illustration of cleavage of tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc attached on the CNF tips by cathepsin B. In ACV measurements, the Fc attached to the CNFs provides a reliable oxidation peak at ∼0.25 V versus Ag/AgCl (3M KCl), as illustrated in Figure S2A. The AC peak current ip,acv can be obtained by subtracting the linear background. The value of ip,acv remained relatively stable before adding the enzyme. After adding cathepsin B, the tetrapeptide was cleaved at the site between arginine and phenylalanine, which has been proved by HPLC and MS analyses in our previous study.14, 15 The fragment containing Fc was thus released into the bulk solution causing the peak current ip,acv to decrease (Figure S2B). The kinetics of the proteolysis can be monitored by continuously repeated ACV measurements (Figure S2C). The kinetic curve can be fit with an exponential decay that corresponds to cathepsin B proteolysis on top of a slowly varying linear background drift,
Figure 1.
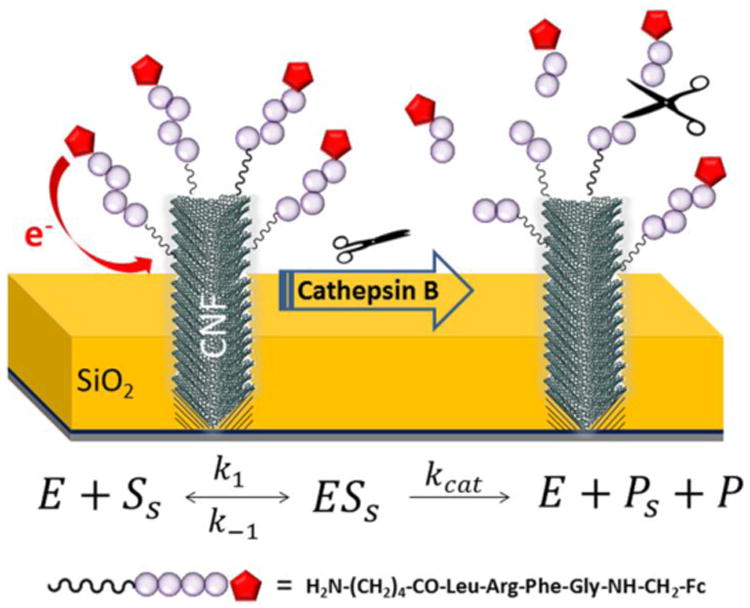
Schematic diagram of the cleavage of tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc at the VACNF NEA tip by cathepsin B. The cleavage of the peptide at the site between arginine (Arg) and phenylalanine (Phe) releases Fc into bulk solution and caused an exponential decay of the corresponding electrochemical signal.
| (1) |
The value of i0 corresponds to the total amount of surface-attached Fc-peptide at the beginning, which varies with the surface area of VACNFs in the NEAs.15, 16, 22 According to our previous study, the variation can be normalized by using the relative change represented by the “extracted proteolytic signal” S, as defined below:15
| (2) |
Obviously, the extracted function S only corresponds to the exponential component of the kinetic curve, making the analysis much simpler.
The proteolysis kinetics on the VACNF NEAs can be explained with a modified Michaelis-Menten model for heterogeneous enzymatic reactions14, 15, 23:
| (3) |
where E, Ss, ESs, Ps and P represent the enzyme (cathepsin B in this case), the surface-attached peptide substrate, the enzyme-substrate complex, the surface-attached product and the product released to the solution (containing Fc), respectively. The reaction rate ν can be defined as
| (4) |
where ΓSs represents the density of surface-attached peptide substrate, kcat is the dissociation rate constant and KM = (kcat+k-1)/k1 is Michaelis-Menten constant. Since “extracted proteolytic signal” S is proportional to the density of surface-attached peptide substrate (i.e. ΓSs ∝ S), Equation 4 can also be written as
| (5) |
Combining Equations 2 and 5, it gives
| (6) |
Hence the decay time constant τ of S over time is directly related to the protease activity (kcat/KM)·[E0].
Cell characterization: Western blot analysis and IP assay
The expression of cathepsin B in normal breast cells HMEC, non-tumorigenic breast cells MCF-10A, breast cancer cells T47D and metastatic cancer cells MDA-MB-231 was first subjected to Western blot analysis. It is known that cathepsin B presents in two forms, i.e. proenzyme and active form, with the molecular weight of ∼37 kDa and ∼25 kDa, respectively, by SDS-PAGE.8 Incubated in activation buffer consisting of 5 mM DTT and 25 mM MES converts a portion of pro cathepsin B into the active enzyme.8 The Western blot results in Figure 2A, clearly show the presence of both pro and active forms of cathepsin B in WCE of all breast cells, with the content of the active form generally higher than that of the pro form. It needs to be noted that the band intensity among the four cell lysates in Figure 2A varied due to different sample loading and immunoblotting conditions, as indicated by the control Western blot results of actin in these samples (see Figure S3). To obtain reliable quantitative analysis, Cathepsin B was then precipitated out from the same amount of WCEs using cathepsin B antibody. The pellets (the immunoprecipitated fraction) containing the concentrated and purified cathepsin B were then subjected to Western blot analyses all in one run. Figure 2B and Table S1 illustrate that, with the same initial loading, the total integrated pixel intensity of cathepsin B in the Western blot bands increased in cancer cell lysates. The linear relationship of the Western blot band intensity with the amount of enzyme was verified with experiments using purified cathepsin B (Figure S4 in SI). After one round of IP, small but detectable amount of cathepsin B still present in the supernatant of the lysate (shown in 1X column of Figure 2A). However, no obvious amount of cathepsin B was found in the supernatant after 2 to 3 times of IP (shown in the 3X column of Figure 2A), indicating a complete removal of cathespin B in the sample.
Figure 2.
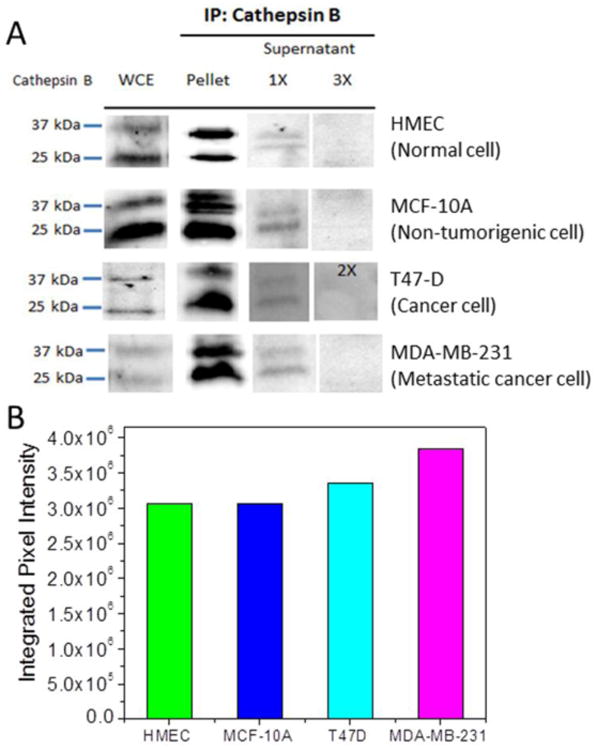
(A) Western blot analysis of cathepsin B expression in four types of breast cells: Normal breast cells (HMEC), transformed breast cells (MCF-10A), breast cancer cells (T47D), and metastatic breast cancer cells (MDA-MB-231). The first column shows that the whole cell extract (WCE) of all 4 types of cells consist both proenzyme (∼ 37 kDa) and active (∼ 25 kDa) cathepsin B. (Note that the experimental conditions were varied and thus this set of data is only for qualitative assessment). About 0.5 mg of WCE was used for immunoprecipitation (IP) assay with 1.4 μg of cathepsin B antibody. The immunoprecipitated fraction was subjected to Western blot analysis using cathepsin B antibody. Pellet includes pulldown complex with cathepsin B antibody. Supernatant with one round of immunoprecipitation denotes as 1X and supernatant with two and three rounds of immunoprecipitation denotes as 2X and 3X, respectively. (B) The quantitative cathepsin B expression represented by the total integrated pixel intensity of cathepsin B bands in a set of Western blot analyses under the same conditions from the same HMEC, MCF-10A, T47D, and MDA-MB-231 cell lysate loading. The cathepsin B level increased in cancer cell lysates.
Electrochemical study of proteolysis of breast cancer cell lysates (MDA-MB-231)
The electrochemical detection of cathepsin B activity in the breast cancer cell followed the method in our previous study.15 The direct lysate of breast cancer cell MDA-MB-231 and that with cathepsin B removed by two rounds of IP (denoted as 2IP) have been studied. The cathepsin B specific tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc was immobilized on VACNF NEAs as the substrate for the enzymatic reaction. Twenty five μL of the activated lysate sample was added into the electrochemical cell containing 250 μL of 25 mM MES (pH 5.0) while ACV signal from the Fc-appended tetrapeptide was repeatedly recorded. In Figure 3A, the final total protein concentration of the lysate is 7.28 μg·mL-1. Adding both lysate samples caused kinetic exponential decay in the extracted electrochemical signal S. The fitted time constants are τ1 = 215 s for MDA-MB-231 cell lysate (pink dots) and τ2 = 533 s for the cathepsin B-removed lysate (blue triangle). Cathepsin B-removed lysate (by 2IP) showed 2.5 times slower kinetic decay rate compared to the direct lysate. In order to confirm that the difference of decay rate is indeed due to the removal of cathepsin B, 13.3 nM cathepsin B was spiked into the 7.28 μg·mL-1 cathepsin B-removed MDA-MB-231 lysate (green inverted triangle). The decay time was shortened from τ2 = 533 s to τ3 = 352 s.
Figure 3.
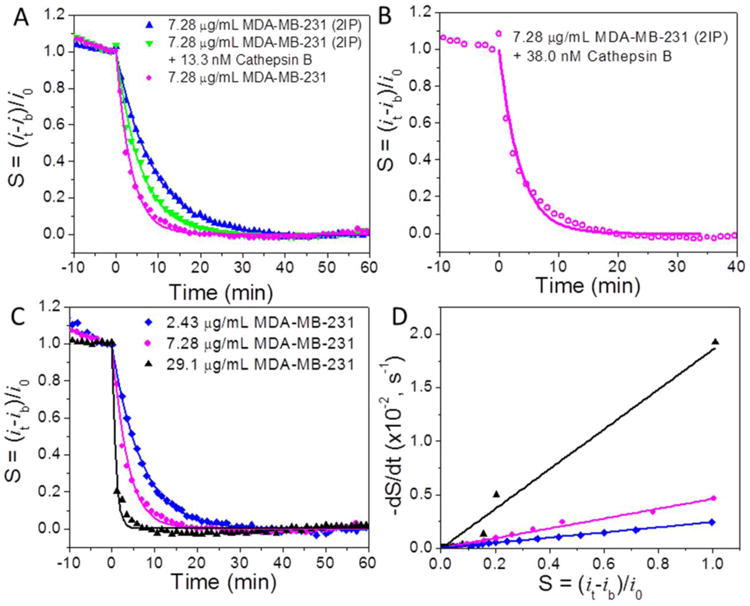
The normalized proteolysis curves of (A) 7.28 μg·mL-1 MDA-MB-231 cell extract with cathepsin B partially removed by two times immunoprecipitation (2IP) (blue triangle), this sample spiked with ∼13.3 nM (0.45 μg·mL-1) purified cathepsin B (green inverted triangle), and 7.28 μg·mL-1 MDA-MB-231 whole cell extract (pink dots); (B) 7.28 μg·mL-1 2IP MDA-MB-231 cell extract spiked with ∼38.0 nM (1.30 μg·mL-1) purified cathepsin B and (C) 2.43 μg·mL-1 (blue diamond), 7.28 μg·mL-1 (pink dots) and 29.1 μg·mL-1 (black triangle) MDA-MB-231 whole cell extract. (D) Plot of −dS/dt vs S during the proteolytic reaction by the cell extracts demonstrated in panel A. The slope is equal to the inverse of the exponential decay, i.e. 1/τ. All ACV measurements were carried out at f = 800 Hz and AC voltage amplitude V0 = 150 mV.
According to Equation 6, the value of kcat/KM can be derived by:
| (7) |
which gives kcat/KM = 7.22 × 104 M-1s-1. This value is slightly larger than our previous result, 4.90 × 104 M-1s-1 which derived from the purified cathepsin B.15 We can also obtain the difference of cathepsin B in direct and cathepsin B-removed MDA-MB-231 lysates as following:
| (8) |
Since kcat/KM equals to 7.22 × 104 M-1s-1 according to above calculation, the difference of cathepsin B in 7.28 μg·mL-1 MDA-MB-231 lysate and cathepsin B-removed lysate, i.e. ΔE0, can be calculated as ∼38 nM. To investigate whether our calculation is legitimate, we spiked 38 nM (1.30 μg·mL-1) cathepsin B into 7.28 μg·mL-1 cathepsin B-removed MDA-MB-231 lysate. A similar decay time τ4 = 207 s (versus τ1 = 215 s in 7.28 μg·mL-1 of original MDA-MB-231 cell lysate) was obtained as shown in Figure 3B, validating that the higher proteolytic rate was indeed due to high cathepsin B concentration.
In principle, the 2IP cell lysate should not cause any exponential decay in contrast to the significant decay in Figure 3A. In previous studies,14, 15 we have demonstrated that the tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc can be selectively cleaved by cathepsin B but not legumain. However, the cell lysate contain many unknown proteases which could cleave the tetrapeptide at any possible sites. To identify a highly selective peptide remains a big challenge of today's research. Our future studies will focus on screening the peptides with the optimum selectivity from a large library of peptide substrates.
The proteolysis of tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc by different concentrations of MDA-MB-231 cell lysates was also studied. As shown in Figure 3C, the decay rate increases with the increasing concentrations of MDA-MB-231 lysates. The decay time constants are 53 s, 215 s and 410 s for 29.1, 7.28 and 2.43 μg·mL-1 MDA-MB-231 cell lysates, respectively. According to Equation 6, (kcat/KM)·[E0] can be represented by 1/τ or the slope of Figure 3D (i.e., -dS/dt versus S). The values of (kcat/KM)·[E0] derived from the slope were 1.86 × 10-2, 4.60 × 10-3 and 2.42 × 10-3 s-1 with lysate concentration at 29.1, 7.28 and 2.43 μg·mL-1, respectively. The quantity of (kcat/KM)·[E0] deviated from linear relationship at the low lysate concentration [E0] = 2.43 μg·mL-1, posing the low limit of lysate concentration for quantitative analysis. However, the results showed that this technique can reliably measure the protease above ∼2.43 μg·mL-1.
Specificity study by using cathepsin B inhibitor GC-373
To assess the specificity of the electrochemical detection of cathepsin B activity, an inhibitor GC-373 which inhibits the activity of cathepsin B was applied. The IC50 value of 9.67 nM was first measured with the fluorescence assay (Figure S6). The proteolysis of tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc by purified recombinant cathepsin B with and without inhibitor was then measured as shown in Figure 4A. Three sets of measurement were done with 30.7 nM cathepsin B: (1) with normal 15 min activation in the activation buffer (5 mM DTT and 25 mM MES buffer, pH = 5.0); (2) with extra 30 min incubation following the normal activation; and (3) with 30 min incubation with added 0.6 μM GC-373 inhibitor following the normal activation. Since the inhibitor requires 30 min incubation with the activated cathepsin B for it to take effects, experiment 2 is needed as the control. The decay time for the cathepsin B being activated for 15 min is 1287 s, giving (kcat/KM)·[E0] (= 1/τ) equals to 7.77 × 10-4 s-1. With further incubation for 30 min, the enzyme is more active, giving a smaller decay time of 397 s. After adding the GC-373 inhibitor, the decay time dramatically increases to 4,812 s. Accordingly, the cathepsin B activity (kcat/KM)·[E0] dropped by ∼12 fold from 2.52 × 10-3 s-1 to 2.08 × 10-4 s-1. The same trend was also found by using fluorescence method shown in Figure S5. The above results implied that inhibitor GC-373 is an efficient inhibitor for cathepsin B. Furthermore, Figure 4B showed that inhibitor GC-373 treated cancer cell lysate MDA-MB-231 also exhibited 3.4 fold lower proteolysis activity compared to the untreated lysate, with (kcat/KM)·[E0] of 1.37 × 10-3 s-1 vs. 4.65 × 10-3 s-1 and the decay time constant of 729 s vs. 215 s. This result illustrated that cathepsin B was the major protease responsible to the cleavage of the tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc in the electrochemical measurements, but other proteases in cancer cell lysate MDA-MB-231 also made some contributions.
Figure 4.
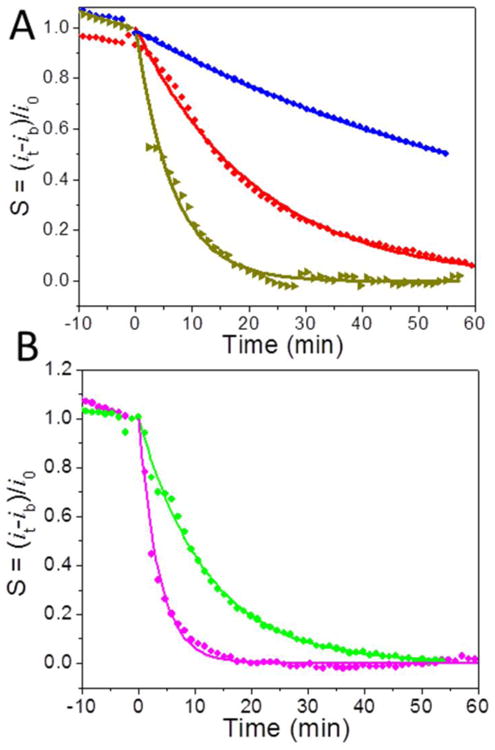
(A) The normalized proteolysis curves of 30.7 nM (0.89 μg·mL-1) purified recombinant cathepsin B after (1) normal activation procedure by incubation for 15 min in the activation buffer (5 mM DTT and 25 mM MES buffer, pH 4.0) (red dot), (2) with additional 30 min incubation after the normal activation (dark yellow triangle), and (3) adding 0.6 μM inhibitor GC-373 during the 30 min additional incubation (blue diamond). (B) The normalized proteolysis curves of 7.28 μg·mL-1 MDA-MB-231 incubated for 30 min with (green dot) and without (pink dot) adding 0.6 μM inhibitor GC-373 after the normal 15 min activation procedure.
Comparison of proteolysis kinetics of different breast cell lysates
A major goal of today's research is to develop methods that can quantitatively detect protease in complex samples and correlate the results with cancer formation. Toward this goal, one type of primary culture cells and three different breast cell lines were used to investigate whether the electrochemical detection of cathepsin B activity using VACNF NEAs could have biological significance. A solution of 25 μL of normal mammary cell HMEC lysate, non-tumorigenic breast cell MCF-10A lysate, breast cancer cell T47D, or metastatic breast cell MDA-MB-231 lysates at total protein concentration of 7.28 μg·mL-1 was added into the electrochemical cell filled with 250 μL of 25 mM MES buffer (pH = 5.0). The decay time clearly varied among these different cell lysates (Figure 5A) giving τ = 497, 311, 248 and 215 s for 7.28 μg·mL-1 of HMEC, MCF-10A, T47D, and MDA-MB-231, respectively. The values of (kcat/KM)·[E0] derived from 1/τ or the slope of −dS/dt versus S are 2.00 × 10-3, 3.20 × 10-3, 4.02 × 10-3 and 4.60 × 10-3 s-1 accordingly (Figure 5B).
Figure 5.
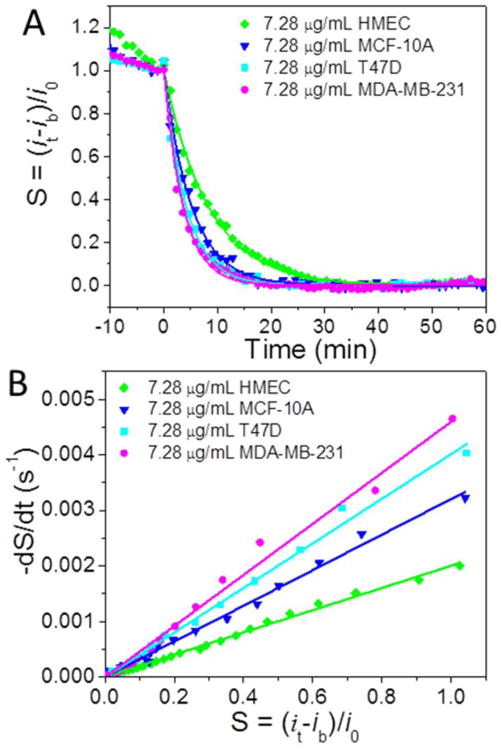
(A) The normalized proteolysis curves of 7.28 μg·mL-1 of HMEC cell extract (green diamond), MCF-10A cell extract (blue inverted triangle), T47D cell extract (cyan square) and MDA-MB-231 cell extract (pink diamond), respectively. (B) The plot of −dS/dt vs. S during the enzymatic reaction of the cell extracts in panel A.
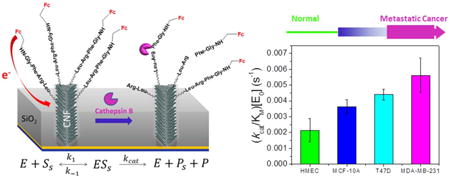
Most of the early stage mammary tumors are ER-positive and many patients could respond to anti-estrogen or endocrine therapy24-27 and later stage breast cancers become resistant to endocrine therapy and are more malignant, aggressive, invasive and metastatic28-32. HMEC is derived from normal mammary tissue, and MCF-10A is a non-tumorigenic cell line, while T47D and MDA-MB-231 cell lines are derived from breast cancer patients. MDA-MB-231 has been shown to be estrogen receptor (ER)-negative, indicating a more aggressive form of breast cancer. Excitingly, our electrochemical results showed that the values of (kcat/KM)·[E0] (= 1/τ) significantly increased in the cancer cell lysates. Comparison of the values of (kcat/KM)·[E0] of different breast cell lysates at concentration of 7.28 μg·mL-1 in bar graph is shown in Figure 6A. Triplicated experiments were carried out for each cell lysates. The statistical values of (kcat/KM)·[E0] are (2.13 ± 0.73) × 10-3, (3.62 ± 0.44) × 10-3, (4.39 ± 0.34) × 10-3 and (5.61 ± 1.09) × 10-3 s-1 for HMEC, MCF-10A, T47D and MDA-MB-231 cell lysates, respectively. This trend is correlated well with our Western blot results (Figure 2B).
Figure 6.
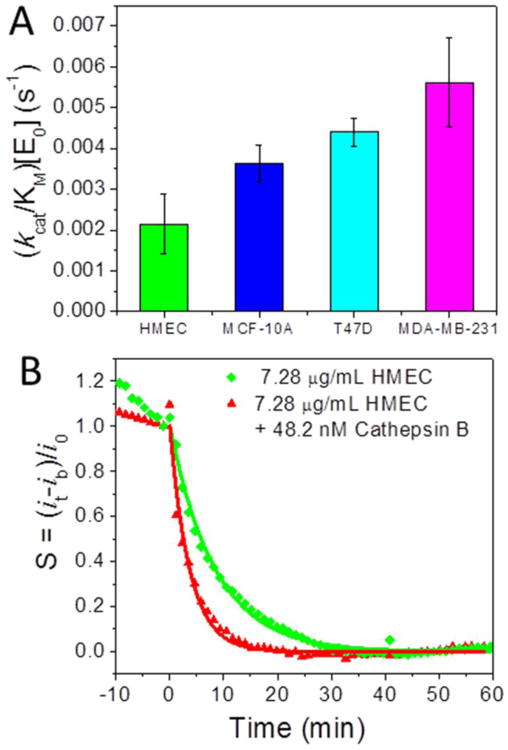
(A) Comparison of the (kcat/KM)·[E0] values derived from 7.28 μg·mL-1 HMEC, MCF-10A, T47D and MDA-MB-231 cell extracts reacting with tetrapeptide H2N-(CH2)4-CO-Leu-Arg-Phe-Gly-NH-CH2-Fc on VACNF NEAs. The error bars represent standard deviation from the mean from three measurements. (B) The normalized proteolysis curves of 7.28 μg·mL-1 HMEC cell extract spiked with 48.2 nM (1.63 μg·mL-1) purified cathepsin B. The AC measurements were carried out at f = 800 Hz and AC voltage amplitude V0 = 150 mV.
In order to estimate the overexpressed amount of cathepsin B in cancer cell lysates compared to the normal cells, an experiment was done by spiking certain amount of cathepsin B into HMEC lysate solution. According to Equation 6, the difference of cathepsin B in HMEC and MDA-MB-231 cell lysates at concentration of 7.28 μg·mL-1 can be derived as:
| (9) |
Assuming that kcat/KM equals to 7.22 × 104 M-1s-1 as obtained earlier, ΔE0 equals to 48.2 nM. This implied that 7.28 μg·mL-1 HMEC spiked with 48.2 nM purified cathepsin B should be equivalent to 7.28 μg·mL-1 MDA-MB-231. The kinetic curves after spiking 48.2 nM purified cathepsin B into 7.28 μg·mL-1 HMEC solution indeed showed a decay time constant comparable to the same concentration of MDA-MB-231 cell lysate, as shown in Figure 6B. The statistic value of the decay time from three repeating measurements was (255 ± 30) s, giving a (kcat/KM)·[E0] value of (3.97 ± 0.50) × 10-3 s-1 falling between (3.62 ± 0.44) × 10-3 s-1 for MCF-10A and (4.39 ± 0.34) × 10-3 s-1 for MDA-MB-231. The higher proteolysis rate of cancer cell lysate is indeed due to elevated cathepsin B activity.
Proteolysis by breast cell lysates has also been studied with a commercial fluorescence assay using 0.05 mg·mL-1 lysate of HMEC, MCF-10A, T47D and MDA-MB-231. As shown in Figure S7, even though this method works well in measuring proteolysis of purified recombinant cathepsin B and was used as a validation technique in our previous studies,14, 15 the results were quite diverging and unreliable in measuring cell lysates. Despite that the lysate concentration was about 10 times of that in electrochemical experiments, none of the kinetic curves except T47D lysate showed clear exponential increase in fluorescence intensity due to proteolytic cleavage of the AMC group from the peptide substrate. The fluorescence intensity was in general 10 to 100 times lower than the measurements using purified cathepsin B solutions at comparable or even lower concentrations. The measurements in MCF-10A and MDA-MB-231 cell lysates only showed noisy uncharacteristic fluctuations. None of these measurements were able to be used for quantitative analysis. It is noted that the fluorescence assay can only use a rather simple substrate Z-Leu-Arg-AMC where the signal moiety AMC has to be attached to the cleavage site Arg at the end of the peptide due to its fluorescence mechanism. The lack of specificity and strong interference likely limited this commercial fluorescence assay from measuring the proteolytic kinetics of the complex cell lysates. Further study for a better understanding of this phenomenon is still in progress.
Discussion
In this study, we have shown that cathepsin B exits as both pro and active forms in the whole cell extract of normal breast cells (HMEC), non-tumorigenic breast cells (MCF-10A) and breast cancer cells (T47D and MDA-MB-231). Our results also demonstrated that the total integrated pixel intensity from the Western blot bands, thus the amount of cathepsin B, increased for the cancer cell lysates. However, the commercial fluorescence assay is not suitable to study the proteolytic kinetics of the cathepsin B in the cell lysates. In contrast, electrochemical method applied on VACNF NEAs has shown great potential and advantage in quantitative study the complex cell lysates. We have shown that ACV measurements is a reliable tool to derive the value of (kcat/KM)·[E0] from the cancer cell lysate solutions. The spiking experiments results confirmed that the elevated proteolytic rate is due to the cathepsin B. Well-separated protruding VACNF tips immobilized with the peptide substrate likely facilitate the protease to reach and cleave the specific site of the peptides. The results from inhibitor experiment illustrated that proteases other than cathepsin B in the cancer cell lysate also contributed to the cleavage of the tetrapeptide substrate. We also found that the protease activity (i.e., the value of (kcat/KM)·[E0]) increased for the cancer cells, consistent with Western blot results. The value of (kcat/KM)·[E0] is the highest for metastatic cancer cell MDA-MB-231, then followed by cancer cell T47D and non-tumorigenic cell MCF-10A. Normal cell lysate HMEC showed the smallest value of (kcat/KM)·[E0], thus lowest protease activity. To our knowledge, this is the first report to use simple electrochemical method to quantify the protease activities in complex cell lysates and identify the cancer formation. Integrated in the multiplex system, this electrochemical biosensor can be potentially used for improving cancer diagnosis, treatment monitoring, and drug (inhibitor) screening.
Although this electrochemical technique is viable for detecting cathepsin B activity in tissue or cell lysates, it would have broader impact if it can profile protease in serum or urine samples. According to Karel Kotaska et al.'s bladder cancer study, the concentration of cathepsin B in serum is in the range of 27-126 μg·L-1 and that in urine is in the range of 0-2.54 μg·L-1.2 The lowest concentration of purified cathepsin B by using ACV on VACNF NEA in our previous study is ∼450 μg·L-1 (13.3 nM), about 4-20 times of the concentration of cathepsin B in serum and >200 times more in urine.15 In order to effectively detect the cathepsin B in serum or urine samples, the detection limit of our electrochemical method needs to be further improved. One of the approaches is to use e-beam patterned VACNF NEAs (Figure S1B) which has exhibited enhanced performance in our previous study due to more uniform SiO2 encapsulation and smaller background from nonspecifically adsorbed Fc-peptide.15 At the same time, the serum or urine samples can also be concentrated in order to effectively measure cathepsin B activity by using electrochemical method. It is noteworthy that cell culture (as used in this study) is the key to new drug discovery and screening analysis. Not only the 2D cell culture allows rapid and cost-effective approach, it also provides a simple and control environment to design a new technology. However, the 2D cell culture has its limitation due to the flat environment and lack of the complexity of cell network in biological system. In the previous study15, we have demonstrated that this electrochemical technique also works nicely in the complex tissue lysates. The next logical step is to detect the level of cathepsin B activity in clinical tumor samples.
The absolute concentration [E0] of cathepsin B also needs further study. From the difference in the proteolytic rates between the original WCE and 2IP MDA-MB-231 lysates, the concentration of removed cathepsin B was calculated to be ∼38 nM according to Equation 8. The mass percentage of cathepsin B in MDA-MB-231 cell extract can be calculated as ∼18% (1.3 μg·mL-1 cathepsin B in 7.28 μg·mL-1 total cell protein). However, according to the Western blot data (Table S1) and the calibration curve (Figure S4), the approximate percentage of cathepsin B in MDA-MB-231 is only ∼0.13 %. Besides loss of the some cathepsin B during Western blot procedures, there are two other possibilities at this stage to explain such a large discrepancy. First, the kcat/KM value of the cathepsin B in the cell lysate may be much larger than that of the purified recombinant cathepsin B. Thus more recombinant cathepsin B was needed in the spiking experiments, leading to overestimation of cathepsin B quantities. The large variation in kcat/KM value of cathepsin B is quite common in literature. For example, it varies from ∼2 × 103 M-1s-1 33 to 7 × 106 M-1s-1 34 depending on the source of cathepsin B and the nature of the peptide substrates. Second, the complex matrix of the cell lysate may have strong interference to suppress the activity of the spiked recombinant cathepsin B, making its real kcat/KM value much lower than what we measured in simple buffer solutions in previous studies, thus more cathepsin B was needed to reach the same effects. This is consistent with the known network interaction with other enzymes in complex samples.
In summary, this work demonstrated two critical features toward rapid protease profiling using an electrochemical method based on nanoelectrode arrays fabricated with vertically aligned carbon nanofibers. First, continuously repeated ACV measurement of Fc-tetrapeptide attached to VACNF NEAs has been proved to be a reliable tool to measure the proteolytic kinetics and quantify the protease activity in complex breast cell lysates. Second, the activity of cathepsin B measured by this electrochemical method was found to increase from normal to cancer cell lysates. The clear inhibitor effects indicated that this method may be used as an effective technique for inhibitor screening in the complex biological samples. This study demonstrates the feasibility for future protease profiling using a library of peptide substrates specific to interested proteases on independently addressed NEAs. It could be potentially developed into a portable multiplex electronic system for cancer diagnosis, assessment of treatment efficacy, and drug screening.
Supplementary Material
Acknowledgments
This work has been supported by Award Number R15CA159250 from the National Cancer Institute (NCI) and, in part, by Johnson Center for Basic Cancer Research at Kansas State University. The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NCI.
Footnotes
Conflict of Interest: The authors have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cancer facts and figures 2014. Atlanta: American Cancer Society; 2014. [Google Scholar]
- 2.Kotaska K, Dusek P, Prusa R, Vesely S, Babjuk M. Urine and serum cathepsin B concentrations in the transitional cell carcinoma of the bladder. J Clin Lab Anal. 2012;26:61–5. doi: 10.1002/jcla.21483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahai E. Mechanisms of cancer cell invasion. Curr Opin Genet Dev. 2005;15:87–96. doi: 10.1016/j.gde.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Lee M, Fridman R, Mobashery S. Extracellular proteases as targets for treatment of cancer metastases. Chem Soc Rev. 2004;33:401–9. doi: 10.1039/b209224g. [DOI] [PubMed] [Google Scholar]
- 5.Koblinski J, Ahram M, Sloane B. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–35. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 6.Sukoh N, Abe S, Ogura S, Isobe H, Takekawa H, Inoue K, et al. Immunohistochemical study of cathepsin B. Prognostic significance in human lung cancer. Cancer. 1994;74:46–51. doi: 10.1002/1097-0142(19940701)74:1<46::aid-cncr2820740109>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 7.Nouh MA, Mohamed MM, El-Shinawi M, Shaalan MA, Cavallo-Medved D, Khaled HM, et al. Cathepsin B: a potential prognostic marker for inflammatory breast cancer. J Tans Med. 2011;9:1–8. doi: 10.1186/1479-5876-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mort JS. Chapter 406 - Cathepsin B. In: Rawlings ND, Salvesen G, editors. Handbook of Proteolytic Enzymes. 3th. Vol. 2. San Diego: Academic Press; 2013. pp. 1784–91. [Google Scholar]
- 9.Barrett AJ, Kirschke H. Cathepsin B, cathepsin H, and cathepsin L. Methods Enzymol. 1981;80:535–61. doi: 10.1016/s0076-6879(81)80043-2. [DOI] [PubMed] [Google Scholar]
- 10.Elliott E, Sloane BF. The cysteine protease cathepsin B in cancer. Perspect Drug Discov. 1996;6:12–32. [Google Scholar]
- 11.Chen Q, Fei J, Wu L, Jiang Z, Wu Y, Zheng Y, et al. Detection of cathepsin B, cathepsin L, cystatin C, urokinase plasminogen activator and urokinase plasminogen activator receptor in the sera of lung cancer patients. Oncol lett. 2011;2:693–99. doi: 10.3892/ol.2011.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buck MR, Roth MJ, Zhuang Z, Campo E, Rozhin J, Sloane BF, et al. Increased levels of 72 kilodalton type IV collagenase and cathepsin B in microdissected human breast and colon carcinomas. Proc Am Assoc Cancer Res. 1994;35:59. [Google Scholar]
- 13.Emmert-Buck MR, Roth MJ, Zhuang Z, Campo E, Rozhin J, Sloane BF, et al. Increased gelatinanse A (MMP-2) and cathepsin B activity in invasive tumor regions of human colon cancer samples. Am J Pathol. 1994;145:1285–90. [PMC free article] [PubMed] [Google Scholar]
- 14.Swisher LZ, Syed LU, Prior AM, Madiyar FR, Carlson KR, Nguyen TA, et al. Electrochemical protease biosensor based on enhanced AC voltammetry using carbon nanofiber nanoelectrode arrays. J Phys Chem C. 2013;117:4268–77. doi: 10.1021/jp312031u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swisher LZ, Prior AM, Shishido S, Nguyen TA, Hua DH, Li J. Quantitative electrochemical detection of cathepsin B activity in complex tissue lysates using enhanced AC voltammetry at carbon nanofiber nanoelectrode arrays. Biosens Bioelectron. 2014;56:129–36. doi: 10.1016/j.bios.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Syed LU, Liu J, Prior AM, Hua DH, Li J. Enhanced electron transfer rates by AC voltammetry for ferrocenes attached to the end of enbedded carbon nanofiber nanoelectrode array. Electroanalysis. 2011;23:1709–17. doi: 10.1002/elan.201100088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandadapu SRGM, Tiew KC, Uy RAZ, Prior AM, Alliston KR, Hua DH, Kim Y, Chang KO, Groutas WC. Inhibition of norovirus 3CL protease by bisulfite adducts of transition state inhibitors. Bioorg & Med Chem Lett. 2013;23:62–5. doi: 10.1016/j.bmcl.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Ng HT, Cassell A, Fan W, Chen H, Ye Q, et al. Carbon nanotube nanoelectrode array for ultrasensitive DNA detection. Nano Lett. 2003;3:597–602. [Google Scholar]
- 19.Li J, Koehne JE, Cassell AM, Chen H, Ng HT, Ye Q, et al. Inlaid multi-walled carbon nanotube nanoelectrode arrays for electroanalysis. Electroanalysis. 2005;17:15–27. [Google Scholar]
- 20.Siddiqui S, Arumugam PU, Chen H, Li J, Meyyappan M. Characterization of carbon nanofiber electrode arrays using electrochemical impedance spectroscopy: effect of scaling down electrode size. ACS Nano. 2010;4:955–61. doi: 10.1021/nn901583u. [DOI] [PubMed] [Google Scholar]
- 21.Arumugam PU, Chen H, Siddiqui S, Weinrich JAP, Jejelowo A, Li J, et al. Wafer-scale fabrication of patterned carbon nanofiber nanoelectrode arrays: A route for development of multiplexed, ultrasensitive disposable biosensors. Biosens Bioelectron. 2009;24:2818–24. doi: 10.1016/j.bios.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 22.Creager S, Yu CJ, Bamdad C, O'Connor S, MacLean T, Lam E, et al. Electron transfer at electrodes through conjugated “molecular wire” bridges. J Am Chem Soc. 1999;121:1059–64. [Google Scholar]
- 23.Gutiérrez OA, Chavez M, Lissi E. A theoretical approach to some analytical properties of heterogeneous enzymatic assays. Anal Chem. 2004;76:2664–8. doi: 10.1021/ac049885d. [DOI] [PubMed] [Google Scholar]
- 24.Muss HB. Endocrine therapy for advanced breast cancer: a review. Breast Cancer Res Treat. 1992;21:15–26. doi: 10.1007/BF01811960. [DOI] [PubMed] [Google Scholar]
- 25.Santen RJ, Manni A, Harvey H, Redmond C. Endocrine treatment of breast cancer in women. Endocr Rev. 1990;11:221–65. doi: 10.1210/edrv-11-2-221. [DOI] [PubMed] [Google Scholar]
- 26.Lerner LJ, CJ V. Development of antiestrogens and their use in breast cancer: eighth Cain memorial award lecture. Cancer Res. 1990;50:4177–89. [PubMed] [Google Scholar]
- 27.Jordan VC. Alternate antiestrogens and approaches to the prevention of breast cancer. J Cell Biochem Suppl. 1995;22:51–7. doi: 10.1002/jcb.240590808. [DOI] [PubMed] [Google Scholar]
- 28.Morrow M, Jordan VC. Molecular mechanisms of resistance to tamoxifen therapy in breast cancer. Arch Surg. 1993;128:1187–91. doi: 10.1001/archsurg.1993.01420230015002. [DOI] [PubMed] [Google Scholar]
- 29.Clark GM, GH S, Ravdin PM, De Laurentiis M, Osborne CK. Prognostic factors: rationale and methods of analysis and integration. Breast Cancer Res Treat. 1994;32:105–12. doi: 10.1007/BF00666211. [DOI] [PubMed] [Google Scholar]
- 30.Ethier SP. Growth factor synthesis and human breast cancer progression. J Natl Cancer Inst. 1995;87:964–73. doi: 10.1093/jnci/87.13.964. [DOI] [PubMed] [Google Scholar]
- 31.Dickson RB, Lippman ME. Growth factors in breast cancer. Endocr Rev. 1995;16:559–89. doi: 10.1210/edrv-16-5-559. [DOI] [PubMed] [Google Scholar]
- 32.King MC. Breast cancer genes: how many, where and who are they? Nat Genet. 1992;2:89–90. doi: 10.1038/ng1092-89. [DOI] [PubMed] [Google Scholar]
- 33.Nägler DK, Storer AC, Portaro FCV, Carmona E, Juliano L, Ménard R. Major increase in endopeptidase activity of human cathepsin B upon removal of occluding loop contacts. Biochem. 1997;36:12608–15. doi: 10.1021/bi971264+. [DOI] [PubMed] [Google Scholar]
- 34.Cotrin SS, Puzer L, de Souza Judice WA, Juliano L, Carmona AK, Juliano MA. Positional-scanning combinatorial libraries of fluorescence resonance energy transfer peptides to define substrate specificity of carboxydipeptidases: assays with human cathepsin B. Analytical Biochemistry. 2004;335:244–52. doi: 10.1016/j.ab.2004.09.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.