

Significance
Virophages are viruses that hijack the replication machinery of giant viruses for their own replication. Virophages negatively impact giant virus replication and improve the survival chances of eukaryotic cells infected by giant viruses. In this study, we identified segments of the Bigelowiella natans genome that originate from virophages and giant viruses, revealing genomic footprints of battles between these viral entities that occurred in this unicellular alga. Interestingly, genes of virophage origin are transcribed, suggesting that they are functional. We hypothesize that virophage integration may be beneficial to both the virophage and B. natans by increasing the chances for the virophage to coinfect the cell with a giant virus prey and by defending the host cell against fatal giant virus infections.
Keywords: virophage, nucleocytoplasmic large DNA virus, microbial community, endogenous virus, Maverick/polinton
Abstract
Virophages are recently discovered double-stranded DNA virus satellites that prey on giant viruses (nucleocytoplasmic large DNA viruses; NCLDVs), which are themselves parasites of unicellular eukaryotes. This coupled parasitism can result in the indirect control of eukaryotic cell mortality by virophages. However, the details of such tripartite relationships remain largely unexplored. We have discovered ∼300 predicted genes of putative virophage origin in the nuclear genome of the unicellular alga Bigelowiella natans. Physical clustering of these genes indicates that virophage genomes are integrated into the B. natans genome. Virophage inserts show high levels of similarity and synteny between each other, indicating that they are closely related. Virophage genes are transcribed not only in the sequenced B. natans strain but also in other Bigelowiella isolates, suggesting that transcriptionally active virophage inserts are widespread in Bigelowiella populations. Evidence that B. natans is also a host to NCLDV members is provided by the identification of NCLDV inserts in its genome. These putative large DNA viruses may be infected by B. natans virophages. We also identify four repeated elements sharing structural and genetic similarities with transpovirons—a class of mobile elements first discovered in giant viruses—that were probably independently inserted in the B. natans genome. We argue that endogenized provirophages may be beneficial to both the virophage and B. natans by (i) increasing the chances for the virophage to coinfect the host cell with an NCLDV prey and (ii) defending the host cell against fatal NCLDV infections.
Sputnik was first described in 2008 as a new class of small icosahedral viruses with an ∼20-kb circular double-stranded DNA genome (1). Sputnik is a satellite virus, because its replication depends upon proteins produced by the nucleocytoplasmic large DNA virus [NCLDV; also giant virus or proposed order Megavirales (2)] Acanthamoeba polyphaga Mimivirus (APMV; Mimiviridae) and replicates in APMV viral factories. Sputnik was shown to inhibit replication of its helper virus and thus acted as a parasite of that virus. In analogy to the term bacteriophage it was called a virophage, but this designation has been challenged (3). Three additional virophages infecting members of the Mimiviridae, e.g., Sputnik 2, Rio Negro, and Zamilon, were subsequently reported (4–6). Virophages that prey on giant viruses that infect heterotrophic nanoflagellates and microalgae have also been discovered, including Organic Lake virophage 1 [OLV1 (7)], Mavirus (8), and a virophage of the Phaeocystis globosa virus (PgVV) (9), yet the classification of the latter as a virophage sensu stricto is uncertain. In addition, complete or near-complete virophage genomes have been assembled from environmental DNA: Yellowstone Lake virophages 1–7 (YSLV1–7) and Ace Lake Mavirus (ALM) (10, 11).
Overall, virophage genomes have similar sizes (∼18–28 kb) and low G+C content (∼27–39%) and are related to Sputnik by genetic and structural homologies (12). Among the 20–34 protein-coding sequences predicted in virophage genomes, the putative core gene set comprises six genes encoding the FtsK-HerA family DNA-packaging ATPase (ATPase), primase-superfamily 3 (S3) helicase, cysteine protease (PRO), and zinc-ribbon domain (ZnR) as well as major and minor capsid proteins (MCPs and mCPs, respectively) (12). In addition, genes encoding two different families of integrases have been identified in several virophages: A putative rve integrase was found in Mavirus and ALM (8, 10), whereas Sputnik encodes a putative tyrosine integrase (1). Among virophage genes, only PRO, ATPase, MCP, and mCP support the monophyly of virophages, whereas the remaining gene complement shows complex phylogenies suggestive of gene replacement (12).
Remarkably, phylogenetic analysis of the Mavirus rve integrase indicated that it is mostly related to homologs from eukaryotic mobile elements of the Maverick/polinton (MP) family (8). The polintons are widely distributed in diverse protists and animals and were initially classified as transposable elements (TEs) (13, 14). However, convincing arguments support the hypothesis that polintons encode capsid proteins and might be bona fide viruses (15). Because Mavirus was reported to display further synapomorphy with a putative MP from the slime mold Polysphondylium pallidum, it was hypothesized that MPs may have originated from ancient Mavirus relatives that would have acquired the capability of intragenomic transposition (8). However, this hypothesis was recently challenged by Yutin et al. (12). A critical prerequisite for such an evolutionary scenario is the integration of virophage DNA in the genome of a eukaryotic host that would permit vertical transmission and adaptation to an intracellular parasitic lifestyle. However, although Sputnik 2 was shown to integrate into the genome of its Mimivirus host (4), evidence of virophage insertions in eukaryotic genomes is lacking.
The coinfection of host cells by NCLDVs and virophages has been shown to limit the production of NCLDV particles, accompanied by greater survival of the eukaryotic host (1, 5, 6, 8, 16). At the community level, these parasitic relationships result in complex interplays between virophages, NCLDVs, and eukaryotic hosts. As a result, virophages indirectly positively regulate the population size of eukaryotic hosts. At the global scale, these interactions may have significant impacts on biogeochemical cycles. For instance, in the marine environment, the co-occurrence of giant viruses and virophages in the context of algal blooms may influence the overall carbon flux as proposed for Antarctic lakes (7). Nevertheless, such tripartite community networks remain poorly explored except on theoretical grounds (17, 18).
The ecological prominence and diversity of virophages are largely unknown and wait for the isolation and sequencing of new specimens. Recently, we demonstrated the value of searching integrated viral DNA in the genomes of potential eukaryotic hosts to identify new members of the NCLDVs (19). Here we analyzed the nuclear genome assemblies of 1,153 fully sequenced eukaryotes and report the identification of integrated virophage elements in the genome of the Chlorarachniophyte Bigelowiella natans (supergroup Rhizaria). This discovery led to the prediction that this alga is also the host of viruses that are members of the NCLDVs. In support of this prediction, we also identified inserts of likely NCLDV origin. We investigated the transcriptional activity of the B. natans genome using RNA-sequencing (seq) data and show that virophage-like genes are actively transcribed in different B. natans strains whereas NCLDV-like genes tend to be silent—albeit with notable exceptions. Finally, we identified repeated genetic elements that have structural and genetic similarities to transpovirons, a distinct class of mobile genetic elements associated with giant viruses that were first discovered in members of the Mimiviridae (4). We discuss the biological relevance of integrated, actively transcribed virophages and propose a model for the mode of virophage–NCLDV coinfection. Altogether, our results contribute to the understanding of the genetic interactions occurring within microbial communities between eukaryotes, virophages, and NCLDVs.
Results
Integrated Virophage-Like Elements in the Algal Genome.
A previous comparative analysis of fully sequenced virophage genomes revealed six core proteins or protein domains that are universally conserved, including S3 helicase, zinc-ribbon domain, major capsid protein, minor capsid protein, DNA-packaging ATPase, and a cysteine protease (12). The four latter proteins were shown to produce consistent monophyletic clades that contrasted virophages from polintons, a class of repeated elements related to virophages. We therefore used these proteins as markers of DNA inserts of putative virophage origin in eukaryotic genome sequences. In practice, we searched 1,153 predicted proteomes of protists, fungi, and basal metazoans for homologs of the four virophage markers using BLASTP. The proteome of B. natans was the only one to exhibit homologs for each of the four virophage core protein families. No homolog for any of the virophage markers was identified in the other proteomes using predefined family-specific score thresholds.
To better delineate the subset of B. natans proteins that have a potential virophage origin, we used a score plot approach. BLAST scores obtained between B. natans predicted proteins and their best virophage matches were plotted against the respective BLAST scores obtained between the B. natans predicted proteins and their best cellular matches (Fig. 1). Blue dots below the diagonal identify B. natans proteins that have higher similarity to a virophage protein than to a homolog in a cellular organism. Overall, 103 B. natans proteins had a match within a virophage proteome, of which 64 had a higher score with virophages than with cellular organisms. Furthermore, examination of the physical location of the virophage-like protein genes revealed that they tend to cluster in specific loci in the genome assembly, revealing large regions of possible virophage origin.
Fig. 1.

Similarity plot of B. natans proteins against virophage/NCLDV and cellular best hits. Circles represent relative BLASTP scores of B. natans proteins aligned against their best cellular hits in the NR database (y axis) and their best viral hits among NCLDVs or virophages. When no cellular hit was recorded whereas a viral hit was obtained, the cellular score was set to zero. BLAST scores were normalized by dividing them by the score of the alignment of the query sequence against itself. Circles are colored according to the origin of the best overall scoring hit (yellow, cellular organisms; blue, virophages; green, NCLDVs).
A total of 38 virophage-like elements (VLEs) ranging from 100 base pairs (bp) to 33.3 kb were detected by nucleotide alignments in the B. natans genome assembly. Many VLEs correspond to truncated copies of larger elements, suggesting recurrent insertions followed by degradation. However, the number of VLEs may be misestimated because some of them lie at the end of contigs, suggesting that VLEs are difficult to assemble. None of the VLEs were located on the nucleomorph chromosomes, mitochondrial genome, or chloroplast genome. The cumulated size of VLE sequences reaches 327 kb. The VLEs were highly similar between each other—that is, nucleotide identities averaged 91.3%—indicating that they belong to the same family of closely related elements. However, sequence conservation was occasionally interrupted by unique sequences containing one or more genes as shown in Fig. S1, revealing insertion or deletion events that occurred subsequent to their divergence. Some VLEs may be unable to produce viable virophages (i.e., unable to complete a full replication cycle) because important genes may have been lost following integration. Alternatively, the difference in gene content between VLEs may reflect the genetic diversity of virophages before their integration. Six of the identified VLEs contained terminal inverted repeats (TIRs) of 2.0–2.6 kb at their two extremities. TIRs were also described at the extremities of the PgVV virophage-like element [associated with the virus PgV-16T infecting P. globosa (9)] and polintons, and are common among poxviruses, chloroviruses, and asfarviruses (7). In B. natans, each TIR contained at least two putative ORFs. Other VLEs contained a single TIR copy at one of their extremities, most probably because these elements were truncated.
Fig. S1.

Schematic representation of the identified virophage-like elements. Homologous ORFs (>90 codons) are colored with the same color across virophage-like elements. TIRs are represented by yellow semiarrows. Shaded areas between virophage-like elements indicate regions of high nucleotide similarity.
As shown in Fig. 2A, the G+C content of VLEs (36.4% on average) was markedly lower than the background G+C content of the host genome (44.9% on average). Such a difference in G+C content suggests that the VLEs have been acquired horizontally in the relatively recent past.
Fig. 2.

Schematic representation of the largest virophage-like element of B. natans. (A) G+C content curve in the region of the virophage-like element on scaffold 2. (B) Gene map of the linear virophage-like element. Genes displayed on the outer and inner rims of the outermost track are coded on the direct (Watson) and reverse (Crick) strand, respectively. Purple and green genes correspond to core virophage genes and genes with annotated functions, respectively. The middle track indicates the number of RNA-seq reads mapped to the regions (logarithmic scale). The inner track represents the G+C skew, with positive values in red and negative values in blue. The genome coordinates (in kb) are indicated in the innermost track of the map.
Virophage-Like Element Genes.
Overall, 298 ORFs (>90 codons) were predicted out of the 38 VLEs and organized into 54 gene families (Dataset S1). The largest element of 33.3 kb was identified on scaffold 2 (positions 1,655,224–1,688,550; Fig. 2B) and contained 27 predicted ORFs representing 25 distinct gene families listed in Table 1. Functional annotation could be predicted for only 14 of the pan-VLE gene families. Furthermore, 39 gene families did not have a significant match in sequence databases (BLASTP E value < 0.01). Such a small fraction of predicted function is a trademark of virophages that generally contain a majority of orphan genes (1, 7, 8). Also, the functions encoded by the B. natans VLEs are reminiscent of bona fide virophages. Homologs could be identified for each of the six core genes that are ubiquitously conserved in complete virophage genomes (12). Importantly, these include the major and minor capsid protein genes that are among the viral hallmark proteins distinguishing viruses from other types of mobile elements. In addition, the VLEs encode two key proteins involved in virion maturation, namely the protease and the packaging ATPase. The core virophage zinc-ribbon domain is fused with a GIY-YIG nuclease domain like in Mavirus and OLV; noticeably, the corresponding protein (Bn119_7) is encoded by only one of the VLEs on scaffold 119, suggesting that the other VLEs have lost the gene at some point in their evolution. Furthermore, the VLEs were found to encode an rve family integrase closely related to the one encoded in Mavirus and ALM. This protein might have catalyzed the integration of the original virophage-like DNA into the algal genome. The VLEs also contain five gene families homologous to sequences that have a more patchy distribution among virophages, including genes for lipase, ribonucleotide reductase small subunit, B family DNA polymerase, and two ORFs with unknown function.
Table 1.
Virophage-like genes
| ORF no. | Putative function | RPKM | Percentile expression,* % | Best hit† |
| Scaffold 2 largest elements | ||||
| Bn2_1 | Orphan protein | 0.2 | 3.9 | |
| Bn2_2 | Kelch and kinase domain protein | 16.7 | 74.5 | Dendroctonus ponderosae 478256302 (2e-32) |
| Bn2_3 | Orphan protein | 6.3 | 55.1 | |
| Bn2_4 | Unknown virophage protein | 1.8 | 27.5 | Zamilon 563399747 (2e-04) |
| Bn2_5 | Adhesin-like protein | 1.0 | 19.0 | Escherichia coli 693111543 (9e-13) |
| Bn2_7 | Orphan protein | 0.9 | 16.7 | |
| Bn2_8 | Orphan protein | 0.0 | 0.0 | |
| Bn2_9 | DNA polymerase B | 0.9 | 17.2 | Mavirus 326439151 (2e-09) |
| Bn2_10 | Orphan protein | 0.5 | 10.4 | |
| Bn2_11 | S3 helicase | 0.8 | 15.1 | YSL5 701905635 (2e-28) |
| Bn2_13 | Orphan protein | 604.1 | 99.2 | |
| Bn2_14 | Orphan protein | 125.7 | 95.6 | |
| Bn2_15 | Orphan protein | 194.4 | 97.2 | |
| Bn2_17 | Orphan protein | 21.8 | 78.7 | |
| Bn2_18 | DnaJ domain protein | 24.4 | 80.4 | Ostreococcus lucimarinus virus 313843979 (2e-39) |
| Bn2_19 | DNA-packaging ATPase | 14.2 | 71.8 | OLV 322510450 (7e-25) |
| Bn2_20 | Orphan protein | 9.4 | 63.8 | |
| Bn2_21 | Cysteine protease | 1.5 | 24.5 | OLV 322510453 (5e-08) |
| Bn2_22 | Lipase | 5.1 | 50.5 | Mavirus 326439161 (6e-04) |
| Bn2_23 | Orphan protein | 4.2 | 46.1 | |
| Bn2_25 | Minor capsid protein | 4.2 | 45.8 | OLV 322510454 (2e-11) |
| Bn2_26 | Major capsid protein | 3.9 | 44.0 | OLV 322510455 (3e-16) |
| Bn2_27 | Unknown protein | 1.4 | 24.0 | Guillardia theta 551643434 (4e-16) |
| Bn2_28 | Ribonucleotide reductase small subunit | 1.6 | 25.9 | Cafeteria roenbergensis virus 310831442 (3e-80) |
| Bn2_29 | rve integrase | 2.6 | 35.3 | Dictyostelium fasciculatum 470248944 (3e-39) |
| Bn2_31 | Kelch domain protein | 33.4 | 84.5 | Strongylocentrotus purpuratus 390342441 (6e-35) |
| Bn2_33 | Orphan protein | 0.2 | 5.2 | |
| Other remarkable ORFs found in smaller elements | ||||
| Bn119_7 | ZnR and GIY-YIG domains | 3.6 | 42 | Phytophthora sojae 695382398 (3e-05) |
| Bn161_7 | Ankyrin domain protein | 93.4 | 94 | Amoebophilus asiaticus 501449850 (3e-34) |
| Bn92_2 | Unknown phage protein | 0.0 | 1 | Synechococcus phage 472343273 (1e-05) |
| Bn92_3 | Ankyrin domain protein | 0.1 | 4 | Pseudogymnoascus pannorum 682412062 (3e-08) |
| Bn187_9 | Unknown phage protein | 20.4 | 78 | Vibrio phage 510792797 (5e-06) |
| Bn84_33 | Unknown virophage protein | 3.0 | 39 | YSLV5 701905611 (2e-07) |
Percentile rank calculated over all B. natans genes.
Species name and GenBank identifier (BLAST E value).
Each TIR region encompasses two ORFs, resulting in two pairs of duplicated genes in complete VLEs. The outward duplicated ORFs within TIRs lack functional annotation. On the other hand, the inward duplicated ORFs encode Kelch-repeat domains most similar to eukaryotic and poxvirus homologs (Fig. S2A) that may be involved in protein–protein interaction. Interestingly, these ORFs terminate outside the TIR regions, so that the N termini differ between the two protein copies. One of the N termini (Bn2_31) encodes a domain with no match in sequence databases, whereas the N-terminal region of the other protein (Bn2_2) encodes a protein kinase (PK) domain that is indirectly related to those encoded in various giant viruses (Fig. S2B). Sequenced virophages have been shown to contain genes that are closely related to homologs from their respective giant virus hosts (1, 7, 8, 12). These apparent host-derived genes encode different repetitive proteins (distinct forms of collagen-like repeats in Sputnik and OLV, and FNIP repeats in Mavirus) that could be implicated in the interaction of the virophages with their giant virus hosts (12). The VLEs encode two gene families that are most similar to giant virus homologs (i.e., Bn2_18 and Bn2_28), but not one encodes repetitive proteins. However, the Kelch domain proteins (i.e., Bn2_2 and Bn2_31), ankyrin domain protein (Bn161_7), and adhesion-like protein (Bn2_5) are proteins with repetitive domains likely implicated in the interaction of the virophages with their giant virus hosts; however, they have no significant match in sequenced giant viruses.
Fig. S2.
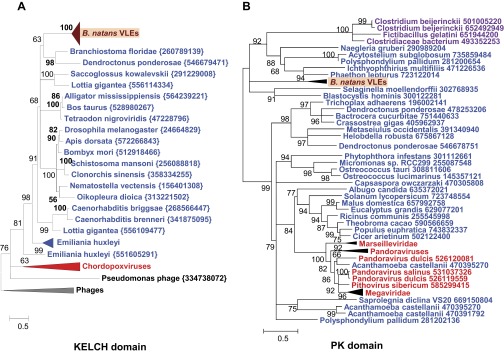



Phylogenetic reconstructions of virophage-like proteins. (A) ML tree of KELCH domains; (B) ML tree of protein kinase (PK) domains contained in KELCH proteins; (C) ML tree of lipases; (D) ML tree of S3 helicase domains; (E) ML tree of rve integrases; (F) ML tree of Bn2_27 homologs; (G) ML tree of DNA polymerase type B family; (H) ML tree of GIY-YIG nuclease domains; (I) ML tree of Bn2_18 homologs; and (J) ML tree of ribonucleotide reductase small subunits. Virophage, NCLDV, prokaryotic, and eukaryotic sequence names are shown in brown, red, mauve, and blue, respectively. Sequences marked with an asterisk were taken from Repbase (38). For other sequences, species name abbreviation and GenBank accession number are indicated. For clarity, subtrees containing sequences originating from the same taxonomic clade were condensed (colored triangles). Statistical supports for branches in percent (approximate likelihood-ratio test) are shown beside nodes (only branch supports >50%). Branches with statistical support <50% were collapsed. The scale bars represent the number of substitutions per amino acid site. env, marine metagenome. Species: Aciph, Acinetobacter phage; Acypi, Acyrthosiphon pisum; Bacam, Bacillus amyloliquefaciens; Bacth, Bacillus thuringiensis; Capow, Capsaspora owczarzaki; Debha, Debaryomyces hansenii; Deigo, Deinococcus gobiensis; Dicfa, Dictyostelium fasciculatum; Ectsi, Ectocarpus siliculosus virus 1; Grama, Granulicella mallensis; Guith, Guillardia theta; Klula, Kluyveromyces lactis; Lackl, Lachancea kluyveri; Marse, Marseillevirus; Micpu, Micromonas pusilla virus PL1; Monbr, Monosiga brevicollis MX1; Parbu, Paramecium bursaria Chlorella virus NY2A; Physo, Phytophthora sojae; Popla, Polysphondylium pallidum PN500; Psyto, Psychroflexus torquis; Steph, Stenotrophomonas phage S1; Tlr, Tetrahymena thermophile; Trica, Tribolium castaneum; Tursa, Turicibacter sanguinis. The numbers of validated amino acid positions in cleaned alignments are 340 (A), 237 (B), 233 (C), 333 (D), 304 (E), 93 (F), 472 (G), 92 (H), 158 (I), and 994 (J).
Origin of Virophage-Like Elements.
We performed a phylogenetic analysis of the virophage-like proteins that had sufficient evolutionary conservation with homologs in sequence databases. For each protein family, the B. natans paralogs aggregated in a single clade containing short branches, further confirming the very close relationships between the different VLEs. The packaging ATPase and protease were previously shown to support the monophyly of virophages (12). The corresponding B. natans proteins cluster within the virophage clade (Fig. 3 A and B). In both these trees, the B. natans proteins group with Sputnik, OLV, and YSLVs, whereas Mavirus and ALM cluster in a sister clade. The phylogenetic tree of the major capsid proteins is compatible with the scenario of a closer evolutionary proximity of the B. natans VLEs to OLV, YSLVs, and Sputnik than to the ALM-Mavirus clade (Fig. 3C). The same conclusion can be drawn from the phylogenetic reconstruction of the virophage lipase (Fig. S2C). Moreover, the fast-evolving minor capsid protein was conserved between Sputnik, OLV, Zamilon, YSLVs, and the B. natans elements, whereas the Mavirus and ALM homologs were comparatively too diverged to allow accurate phylogenetic reconstruction, further pointing to a more distant relationship with the latter (Fig. 3D). Altogether, these features suggest that the B. natans virophage elements share a common ancestor with Sputnik-OLV-YSLVs. However, the exact timing of speciation events within this subtree could not be established, owing to incongruences between the phylogenetic reconstructions.
Fig. 3.

Maximum-likelihood phylogenetic trees of conserved virophage proteins. (A) Packaging ATPase. (B) Maturation protease. (C) Major capsid protein. (D) Minor capsid protein. Branches with bootstrap support (expected-likelihood weights) less than 50% were collapsed. Sequences marked with an asterisk were taken from Repbase (38). For other sequences, the species name abbreviation and GenBank accession number are indicated; env, marine metagenome. Species: Amsmo, Amsacta moorei entomopoxvirus “L”; Caebr, Caenorhabditis brenneri; Caere, Caenorhabditis remanei; Dicfa, Dictyostelium fasciculatum; Giala, Giardia lamblia; Melsa, Melanoplus sanguinipes entomopoxvirus; Salsp, Salpingoeca rosetta; Tetvi, Tetraselmis viridis virus; uncvi, uncultured virus. Taxa: Ea, Amoebozoa; El, Opisthokonta; u2, Entomopoxvirinae. Dark red, virophages; blue, (predicted) polintons and related elements; light red, NCLDV; gray, unassigned environmental sequences. The numbers of validated amino acid positions in cleaned alignments are 210 (A), 166 (B), 548 (C), and 408 (D).
The phylogenetic trees for the other B. natans virophage-like proteins disclosed more complex evolutionary scenarios. It was suggested that gene replacements and horizontal gene transfers might explain the entangled phylogenetic relationships between virophages, large DNA viruses, and certain classes of mobile elements related to virophages such as polintons (12). An elegant example is the ORFs encoding a superfamily 3 helicase domain that exists in all fully sequenced virophages (core gene) but fails to support monophyly of the latter, revealing multiple origins of the virophage genes (Fig. S2D). Nevertheless, the corresponding B. natans proteins cluster with the YSLV3 homolog, suggesting that they originate from a common virophage ancestor. B. natans genes encoding rve family integrase (Fig. S2E), an unknown protein family represented by Bn2_27 (Fig. S2F), DNA polymerase (Fig. S2G), and GIY-YIG nuclease domains (Fig. S2H) exhibit preferential phylogenetic affinities, albeit with moderate bootstrap support with eukaryotic homologs encoded by polinton-related elements. These mixed clades are nested within larger clades containing virophages and environmental sequences, suggesting that gene acquisitions or replacements most likely occurred in the polinton-like elements. In contrast, the phylogenetic trees of the unknown protein family represented by Bn2_18 (Fig. S2I), the ribonucleotide reductase small subunit (Fig. S2J), and the Kelch protein family (i.e., PK and Kelch domains have distinct origins; Fig. S2 A and B) support scenarios of gene acquisition from different sources (bacteria, eukaryotes, or dsDNA viruses). Thus, the VLE genes reveal a mosaic origin that is typical of bona fide virophages (1, 7, 8). Altogether, the structure, gene content, and phylogenetic affinities of VLEs provide substantial evidence that they represent remains of integrated virophage genomes.
VLE Genes Are Transcribed.
To investigate the transcriptional activity of the B. natans virophage-like elements, we analyzed a previously published RNA-seq dataset generated from cultivated B. natans cells (20). A total of 45.3 million Illumina paired-end reads were aligned onto the B. natans genome assembly (Dataset S2), of which 116,671 mapped within one of the virophage-like regions (Dataset S1). Two hundred seventy-eight out of the 302 predicted virophage-like ORFs had at least one read mapped to it. The levels of transcription between B. natans genes were compared by the mean of the RPKM metric (reads per kilobase per million mapped reads). Ninety-three virophage-like ORFs had RPKM values >5, which ranks them in the top half of the most-transcribed genes in B. natans, including 10 genes that figure in the top 10% (i.e., RPKM >50). Interestingly, these highly transcribed virophage-like ORFs encode one major and one minor capsid protein, one DNA-packaging ATPase, one rve integrase, two Kelch domain proteins, and three families of orphan proteins. Other virophage core genes are generally transcribed at low to moderate levels (i.e., the majority of them have RPKM values ranking between the 20th and 36th percentiles), yet 6 of the 10 ATPase gene copies have substantial transcription levels, as indicated by RPKM values ranking between the 50th and 90th percentiles. Interestingly, we identified transcript sequences closely related to the VLEs in assembled RNA-seq datasets generated from various B. natans isolates that were sequenced as part of the Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP) (21) (Dataset S3). This suggests that the same virophage elements are present and transcribed in other B. natans strains, possibly because they were inherited vertically from a common ancestor.
This observation questions the biological significance of the expression of virophage-like genes. One possibility is that the integrations of virophage genomes were accidental events, and that the residual transcriptional activity reflects the fortuitous recognition of regulatory signals of the virophage genes by the cellular host transcription complex. According to this scenario, the expression of the virophage genes has no expected biological effect (neutral) and would disappear as the virophage sequences decay by accumulating random mutations. Alternatively, the observed transcriptional activity might reflect an adaptive strategy that benefits the cellular host population, the virophage, or both. Experimental evidence indicates that virophages have a positive effect on the host-cell population. Mavirus interferes with Cafeteria roenbergensis virus propagation and increases the survival of the host-cell population (8). Sputnik causes a 70% decrease in infective Mamavirus particles and a threefold decrease in amoeba cell lysis (1); it also delays or abolishes replication of Marseillevirus (22). A model of population dynamics suggests that the presence of virophages reduces overall mortality of the host algal cell after a bloom (7). Hence, eukaryotes that are susceptible to infection by giant viruses will gain a selective advantage if they can stably associate themselves with virophages (8). An analogous hypothesis was briefly exposed by Katzourakis and Aswad (23) to explain the possible emergence of Maverick/polinton elements from hypothetical integrated virophage genomes in eukaryotes. Under this scenario, the hijacked virophage genes evolve under negative selection in the new eukaryotic genome environment.
Another possible explanation, which is not mutually exclusive with the former, relates to the adaptive strategy of virophages to increase the frequency of coinfection with a host virus. Different modes of virophage entry into the cell have been evaluated on theoretical grounds (18). However, these scenarios exclusively consider the simultaneous entry of the virophage and the host virus, either independently or in a paired mode [i.e., the virophage adheres to the virus before infection; see Taylor et al. (18)]. Indirect evidence supports the paired-entry mode for Sputnik (1, 22), whereas an independent-entry mode has been observed for Mavirus (8). The transcribed B. natans virophage elements bring out a third hypothetical mode that we have dubbed the delayed-entry mode (Fig. 4). According to this scenario, the virophage is expected to enter the cell first by an unknown mechanism; its DNA reaches the cell nucleus, integrates into the cellular genome, and remains latent until superinfection by a virus reactivates and rescues the virophage, allowing its replication in the virus factory. This hypothetical scenario gets support from the observation that the B. natans virophage elements encode integrases, which suggests that genome integration is an active process rather than an incidental event. To go further into the delayed-entry scenario, it is possible that the transcription of virophage genes leads to the production of sentinel proteins that are able to detect infection of the host cell by another virus and transduce a signal triggering virophage reactivation. There is an obvious advantage of the delayed-entry mode over the independent-entry mode when the simultaneous independent entry of both the virus and virophage is a rare event due to, for example, high dilution of the virus particles in the environment. Furthermore, the integrated virophage can be passed on to the next generations of host cells, contributing to its spread and multiplication. In the form of an integrated provirophage, the virophage can potentially wait for virus superinfection during long periods of time, whose length depends on the rate at which random mutations inactivate the endogenous virophage element.
Fig. 4.
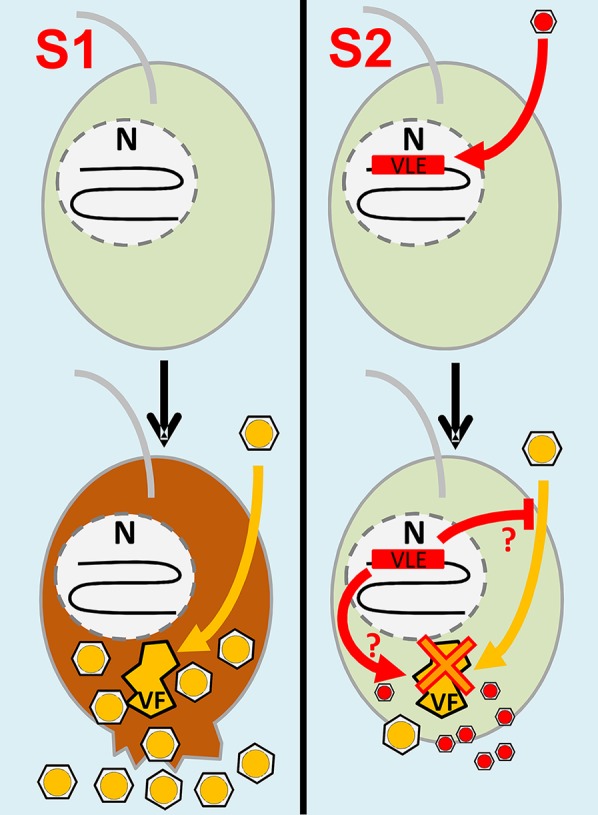
Scenarios of infection of B. natans cells by NCLDVs and virophages. Scenario (S)1 starts with B. natans cells devoid of VLE insertions. An NCLDV particle (yellow hexagon) or its DNA enters the cell and establishes viral factories (VFs) that produce new NCLDV particles. The infection causes cell lysis and death accompanied by release of NCLDV particles. Scenario 2 begins with B. natans cells carrying VLEs in the form of functional provirophages integrated in the nuclear genome (N). VLEs may have been produced by independent entry of a virophage followed by active DNA integration in the host nuclear genome (delayed-entry mode). Upon NCLDV infection, expressed VLE proteins may inhibit virus penetration or trigger reactivation and excision of the provirophage, which in turn inhibits NCLDV replication and takes advantage of the viral factories for its own replication. As a result, a limited number of NCLDV particles are created compared with S1, leading to increased rates of cell survival. Potentially, new virophages and a limited number of NCLDV particles are released in the environment through exocytosis or another unknown mechanism that does not kill the host cell.
Putative NCLDV Insertions.
All characterized virophages so far have been shown to infect members of the Mimiviridae (NCLDVs). Thus, although no large DNA virus of B. natans has been identified so far, the discovery of virophage sequences in B. natans suggests that this alga is the prey of giant viruses that are themselves infected by virophages. DNA fragments that are relics of integrated NCLDV genomes have been discovered in various eukaryotic genomes (24–26), revealing footprints of past interaction between viruses and hosts (19). We therefore searched the B. natans genome for sequences related to NCLDVs using the BLAST score plot approach. As shown in Fig. 1, a total of 36 B. natans predicted proteins have better alignment scores with homologs in NCLDVs than in cellular organisms (Dataset S4). The closer relationship of these proteins with giant virus homologs was confirmed by phylogenetic reconstruction (for examples, see Fig. S3). This phylogenetic affinity may reflect horizontal gene transfer between viruses and eukaryotic hosts, the polarity of which (virus to host or host to virus) cannot always be determined with certainty. However, some proteins are homologous to typical NCLDV core genes, including major capsid protein and packaging ATPase (distantly related to the virophage ones), which have most likely been acquired by the B. natans host (Fig. S3 A and B). Interestingly, some NCLDV-like genes are physically clustered in discrete regions of the B. natans genome assembly (Fig. S4). For example, we identified a 165-kb region of scaffold 10 (position ∼340–505 kb) that is transcriptionally silent according to our analysis of the RNA-seq dataset (Dataset S4). Such a large untranscribed region is unique in the B. natans genome. This region contains 83 predicted genes, of which 9 have obvious NCLDV affinities, including genes for DNA-packaging ATPase, exonuclease, major capsid protein, and transcription factor. Of the remaining genes, only 7 have best matches in eukaryotes (albeit with low similarity) and 4 have best matches with homologs in bacteria and phages. The large majority of predicted genes (63/83 = 76%) are orphans, a characteristic shared with giant virus genomes. Thus, it is likely that this large DNA stretch is the remnant of an integrated NCLDV genome similar to those previously observed in various eukaryotic genomes (19, 24–27). Inserts of likely NCLDV origin identified in the moss genome were also reported as transcriptionally silent (19). These hypothesized viral inserts probably behave like neutrally evolving nonfunctional DNA. The G+C content of the NCLDV-like region (45.5%) is similar to the background G+C content of the B. natans genome (44.8%), which prevents precisely identifying the insert boundaries.
Fig. S3.

Phylogenetic reconstructions of NCLDV-like proteins. (A) ML tree of capsid proteins; (B) ML tree of DNA-packaging ATPases; and (C) ML tree of DNA polymerases (DNAPs). Same legend as Fig. S2. The numbers of validated amino acid positions in cleaned alignments are 370 (A), 229 (B), and 755 (C).
Fig. S4.

B. natans scaffolds containing virophage- or NCLDV-like inserts. The blue graphs represent the number of transcriptome reads mapped onto contigs. GC content variation in 5-kb sliding windows along the contig sequence is depicted by a yellow-to-red color scale, with yellow indicating low GC content (minimum 30%) and red indicating high GC content (maximum 60%). The distance ruler is expressed in kilobases. Cyan rectangles represent virophage inserts; green circles represent NCLDV-like genes.
Outside of the large NCLDV-like insert, some B. natans genes with viral phylogenetic affinity were found to be transcribed (Dataset S4). Six genes have transcription levels among the top 50% of B. natans genes. These genes may have been inherited from the B. natans ancestor and captured by large DNA viruses from eukaryotic hosts that are closely related to B. natans. This scenario can explain the preferential affinity between B. natans proteins and NCLDV homologs, whereas the corresponding genes actually have a eukaryotic origin. Alternatively, the corresponding genes may have been recruited in the metabolism of a Bigelowiella ancestor subsequent to their acquisition from viral donors, perhaps now fulfilling new cellular functions. For example, B. natans has one copy for each of the four regular B family eukaryotic DNA polymerase catalytic subunits alpha, delta, epsilon, and zeta. However, the alga possesses two extra delta DNA polymerases that group in phylogenetic positions compatible with distinct viral origins (Fig. S3C). The two viral-like DNA polymerase catalytic subunits are transcribed (i.e., 43th and 53th percentiles) at higher levels than those of the four regular eukaryotic isoforms (25th–40th percentiles, respectively). Furthermore, they have a large number of introns (i.e., 19 and 30 introns, respectively), whereas viral genes are generally devoid of introns or, in rare cases, only contain a small number of them. The two extra DNA polymerases have no identifiable orthologs in other sequenced eukaryotes, except in the transcriptomes of other Bigelowiella isolates (Dataset S4). We can therefore not exclude a scenario in which the corresponding genes have been captured from viral donors and were progressively “eukaryogenized” through the accumulation of introns.
Putative Transpoviron Insertions.
Four of the six NCLDV-like DNA-packaging ATPase genes are carried by four closely related repeated genetic elements (18.8–21.8 kb in length) flanked by TIRs 300–900 bp in length (Fig. 5). These repeated elements contain between 17 and 22 ORFs (Dataset S5), but some of the original genes seem to have accumulated internal stop codons and frameshifts, resulting in truncated translation. Very few proteins encoded by the repeated elements exhibit detectable similarity in public databases. They include a homolog of the Aureococcus anophagefferens virus protein AaV202, a homolog of the P. globosa virus virophage protein PgvV_00016, a homolog of the Mavirus virophage protein MV06, and a homolog of a hypothetical protein of the fungus Batrachochytrium dendrobatidis. Interestingly, two additional proteins are homologous to core transpoviron proteins, including the C-terminal superfamily I helicase domain protein and C2H2 Zn-finger protein. Transpovirons form a distinct class of mobile genetic elements (6.5–7.5 kb) associated with Mimiviridae (4). The genes for helicase and C2H2 protein are adjacent in the B. natans repeated elements and Mimivirus-associated transpovirons. Furthermore, Mimivirus-associated transpovirons are flanked by TIRs (∼530 bp). Thus, the B. natans repeated elements and transpovirons share structural and genetic similarities, suggesting that the B. natans elements might belong to the transpoviron family. However, the putative integrated B. natans transpovirons are substantially bigger in size relative to their Mimivirus counterparts, possibly due to the incorporation of foreign genes of diverse origins, including NCLDVs and virophages. Some of the integrated transpoviron genes show evidence of transcription, including six genes that have transcription levels in the B. natans top 50%. In addition, homologous transcripts were identified in the transcriptome data of the other Bigelowiella isolates (Dataset S6).
Fig. 5.

Organization of putative inserted transpoviron genomes. The position and strand orientation of each ORF (>90 codons) are indicated by an arrow. Homologous ORFs across transpoviron-like elements are indicated with the same color. ORFs with no homologs in the other transpovirons are shown in black. TIRs are represented by yellow semiarrows. Shaded areas between elements indicate regions of high nucleotide similarity.
Conclusion
The first discovered virophages have the form of small icosahedral virion particles and have been shown to infect giant viruses. However, integrated virophages have been found more recently in a Mimivirus genome (4). Here we show for the first time, to our knowledge, that virophage genomes can also integrate in a eukaryotic genome. This finding led us to predict that B. natans might be the prey for NCLDVs. Additional integrated DNA fragments that most probably originate from NCLDV genomes provide data showing that B. natans or its recent ancestor had physical contacts with NCLDV members. Furthermore, we also identified repeated genetic elements that resemble transpovirons associated with Mimivirus. Thus, the B. natans genome appears to have recorded genetic footprints of molecular “battles” between virophages, transpovirons, and giant viruses. B. natans belongs to the Chlorarachniophytes, a group of unicellular marine algae that acquired a plastid by secondary endosymbiosis involving engulfment of a green alga by a eukaryotic heterotroph host (28). They are typically mixotrophic, ingesting bacteria and smaller protists as well as conducting photosynthesis. B. natans is the only species of the supergroup Rhizaria for which a complete genome sequence is available (29). During endosymbiosis, hundreds of genes of green origin have been transferred toward the host genome in a process called endosymbiotic gene transfer (29). The acquisition of DNA from giant viruses and transpovirons as well as from virophages through horizontal gene transfer represents an additional component in the melting pot of genes composing the B. natans nuclear genome.
One of the most intriguing findings of our analysis is that integrated virophage genes are highly transcribed, suggesting that they are biologically functional. We speculated on three potential adaptive scenarios to explain this observation. Certainly the most interesting of them is the possibility that both the cellular host and virophage take advantage of the integration strategy, by providing the cell with a defense mechanism against giant viruses and providing the virophage with a mechanism to increase the rate of coinfection with a viral prey (i.e., delayed-entry mode S2, schematized in Fig. 4). From the perspective of the cellular host, it is tempting to speculate that integrated virophages can act as molecular weapons against viral pathogens, conferring a sort of immunity transmissible from generation to generation. Sputnik and Zamilon strains have been shown to have a broad host spectrum among the Mimiviridae (5, 16), and Sputnik even affects the replication of Marseillevirus, which is distantly related to the Mimiviridae (22). By extrapolation, we can further hypothesize that the putative defense function triggered by integrated virophages can be efficient over a diversity of viral pathogens.
It is currently difficult to estimate the prevalence and biological significance of virophage insertion in eukaryotes. At first glance, the fact that only B. natans showed virophage genes out of 1,153 screened eukaryotic genomes might suggest that genomic integration of virophages is highly unusual. However, there is a historical bias among sequenced eukaryotes, which include a majority of model organisms, crop plants, fungi, animals, and pathogens (30), all of which are apparently not infected by giant viruses and, hence, virophages. As a result, the organisms tested here included relatively few potential hosts of giant viruses and virophages [i.e., known hosts are amoebas and microalgae (1, 5, 8); Dataset S6]. This bias could explain the apparent low prevalence of virophage insertions detected in our study. In contrast, giant viruses and virophages are readily identified in environmental metagenomes (7, 10, 11, 31), suggesting that these viral entities are common in natural ecosystems. Thus, the question of the prevalence and biological significance of virophage insertions can only be reasonably addressed when more genomes of putative hosts are sequenced.
Methods
Sequence Analysis.
Annotations and sequences of 1,153 eukaryotic genome assemblies representing various protists, fungi, and basal metazoans were downloaded from GenBank. Dataset S6 lists the investigated species together with their GenBank accession numbers, including members of the Alveolate (103), Amoebozoa (35), Apusozoa (1), Chlorophyta (12), Choanoflagellida (2), Cryptophyta (1), Euglenozoa (49), Fonticula (1), Fornicata (6), Fungi (858), Haptophyta (1), Heterolobosea (2), Parabassalia (1), Metazoa (8), Opisthokonta (2), Rhizaria (3), Rhodophyta (4), Stramenopiles (63), and Streptophyta (2). We also downloaded the genome annotations of nine sequenced virophages, including Mavirus (GenBank accession no. GCF_000890715), Sputnik (GenBank accession no. EU606015), Zamilon (GenBank accession no. NC_022990), ALM (GenBank accession no. KC556923), OLV (GenBank accession no. HQ704801), YSLV1 (GenBank accession no. KC556924), YSLV2 (GenBank accession no. KC556925), YSLV3 (GenBank accession no. KC556926), and YSLV4 (GenBank accession no. KC556922).
For each virophage, the major capsid protein, minor capsid protein, DNA-packaging ATPase, and cysteine protease were aligned against the predicted eukaryotic proteomes using BLASTP. Experience showed that computational annotation of eukaryotic genomes can be inefficient in predicting genes of viral origin, because they have become pseudogenes, often resulting in truncation or in-frame stop codons and/or because they can have very distinct GC content relative to the host genome and no introns. Therefore, we also aligned the virophage markers against the translated products of ORFs (>90 codons) lying between predicted genes. Based on prior analysis of the distribution of BLASTP scores against the nonredundant (NR) database, we applied family-specific score thresholds to avoid false detection of remote homologs that are of cellular origin or nonhomologs with similar low-complexity sequences. The score threshold was set as the minimal BLASTP score between any two virophage proteins in the family. Scores obtained between any virophage proteins and cellular homologs were always lower. These score thresholds were 44.7 for proteases, 46.6 for ATPases, 41.2 for mCPs, and 45.1 for MCPs.
The genome assembly of B. natans CCMP2755 exhibited homologs for each of the four marker proteins with BLASTP E value >1E-5, except proteases, for which we used an E-value threshold of 1E-3, because this protein is less conserved among virophages. To identify additional candidate viral-like protein genes in B. natans, we performed a BLAST-score plot analysis previously described in Maumus et al. (19). Briefly, the full complement of B. natans predicted proteins together with intergenic ORFs was used to probe the National Center for Biotechnology Information database using BLASTP (E value < 1E-5). For each protein query, the alignment scores with the best cellular hit and the best virophage or NCLDV hit were recorded. When no cellular hit was recorded whereas a viral hit was obtained, the cellular score was set to zero. BLAST scores were then normalized by the score of the alignment of the query sequence against itself (i.e., self-score), resulting in relative scores expressed in percent of self-score. Nonviral hit scores are plotted against viral hit scores in Fig. 1.
Identification and Delineation of Individual VLEs.
The physical location of the virophage-like protein genes identified by BLASTP revealed that they tend to cluster in specific loci in the genome assembly, unveiling large regions of possible virophage origin. Six of these regions were bordered by long inverted repeats on each side, which coincide with sharp changes in GC content (e.g., Fig. 2). We made the assumption that the long inverted repeats mark the beginning and the end of VLEs. We extracted the nucleotide sequences of these putative complete VLEs from the genome assembly and used BLASTN to align the VLEs back to the genome assembly to identify additional truncated VLEs. Adjacent BLASTN matches that had an E value <1E-25 and a minimal length of 100 pb were assembled to identify a total of 38 VLEs up to 33.3 kb in length. Every candidate VLE was checked and validated manually.
Phylogenetic Analysis.
Construction of adequate homologous protein sets for phylogenetic analysis was performed using the BLAST-EXPLORER website (32). Homologous proteins were aligned using MUSCLE (33), and amino acid positions in multiple alignments containing >30% gaps were removed. We used this criterion for alignment cleaning to keep coherence with the pioneering study of Yutin et al. (12), which produced a comprehensive phylogenetic study of virophage genes. Maximum-likelihood (ML) phylogenetic reconstruction was performed using the PhyML program (34). Before phylogenetic reconstruction, the best-fitting substitution model for each sequence dataset was determined using the ProtTest program (35). Sequences, alignments, ProtTest outputs, and phylogenetic trees are available in Dataset S7.
RNA-Seq Analysis.
To analyze the transcriptional activity of the B. natans genome, we downloaded an RNA-seq dataset (ID MMETSP0045) from the CAMERA database (36). This dataset contained 61.6 million Illumina paired reads generated from polyadenylated RNA extracted from B. natans CCMP2755 cells grown in f/2-Si media for a month under a 12-h:12-h light:dark cycle at room temperature (20). Reads were aligned onto the reference genome sequence using Bowtie 2 (37) with default parameters. Due to the high nucleotide similarity between VLEs, 63% of the reads that mapped onto a VLE also had valid alignments in at least another VLE. For these cases the origin of the read is ambiguous, and we only picked one of the alignments at random for read-count purposes. In addition, we downloaded assembled RNA-seq datasets (contigs) of other Bigelowiella isolates publicly available in the CAMERA database: MMETSP1052 (B. natans CCMP623), MMETSP1054 (B. natans CCMP1259), MMETSP1055 (B. natans CCMP1258.1), MMETSP1358 (B. natans CCMP1242), and MMETSP1359 (Bigelowiella longifila CCMP242). These datasets were generated as part of the Marine Microbial Eukaryote Transcriptome Sequencing Project (21). Homologs of virophage-like encoded proteins were searched in the transcribed sequences using TBLASTN.
Supplementary Material
Acknowledgments
We thank Yongjie Wang for providing the YSLV and ALM sequences ahead of publication. We thank Jean Michel Claverie and Deborah Byrne for critical reading of the manuscript. The Information Génomique et Structurale laboratory is partially supported by the CNRS and Aix-Marseille University. We acknowledge the use of the Paca-Bioinfo platform, supported by Infrastructures en Biologie Santé et Agronomie and France-Génomique (ANR-10-INBS-0009).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 11750.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1506469112/-/DCSupplemental.
References
- 1.La Scola B, et al. The virophage as a unique parasite of the giant mimivirus. Nature. 2008;455(7209):100–104. doi: 10.1038/nature07218. [DOI] [PubMed] [Google Scholar]
- 2.Colson P, et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch Virol. 2013;158(12):2517–2521. doi: 10.1007/s00705-013-1768-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krupovic M, Cvirkaite-Krupovic V. Virophages or satellite viruses? Nat Rev Microbiol. 2011;9(11):762–763. doi: 10.1038/nrmicro2676. [DOI] [PubMed] [Google Scholar]
- 4.Desnues C, et al. Provirophages and transpovirons as the diverse mobilome of giant viruses. Proc Natl Acad Sci USA. 2012;109(44):18078–18083. doi: 10.1073/pnas.1208835109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaia M, et al. Zamilon, a novel virophage with Mimiviridae host specificity. PLoS One. 2014;9(4):e94923. doi: 10.1371/journal.pone.0094923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campos RK, et al. Samba virus: A novel mimivirus from a giant rain forest, the Brazilian Amazon. Virol J. 2014;11:95. doi: 10.1186/1743-422X-11-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yau S, et al. Virophage control of antarctic algal host–virus dynamics. Proc Natl Acad Sci USA. 2011;108(15):6163–6168. doi: 10.1073/pnas.1018221108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fischer MG, Suttle CA. A virophage at the origin of large DNA transposons. Science. 2011;332(6026):231–234. doi: 10.1126/science.1199412. [DOI] [PubMed] [Google Scholar]
- 9.Santini S, et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc Natl Acad Sci USA. 2013;110(26):10800–10805. doi: 10.1073/pnas.1303251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou J, et al. Diversity of virophages in metagenomic data sets. J Virol. 2013;87(8):4225–4236. doi: 10.1128/JVI.03398-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou J, et al. Three novel virophage genomes discovered from Yellowstone Lake metagenomes. J Virol. 2015;89(2):1278–1285. doi: 10.1128/JVI.03039-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yutin N, Raoult D, Koonin EV. Virophages, polintons, and transpovirons: A complex evolutionary network of diverse selfish genetic elements with different reproduction strategies. Virol J. 2013;10:158. doi: 10.1186/1743-422X-10-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapitonov VV, Jurka J. Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci USA. 2006;103(12):4540–4545. doi: 10.1073/pnas.0600833103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pritham EJ. Genes in conflict: The biology of selfish genetic elements. Am J Hum Biol. 2006;18(5):727–728. [Google Scholar]
- 15.Krupovic M, Bamford DH, Koonin EV. Conservation of major and minor jelly-roll capsid proteins in Polinton (Maverick) transposons suggests that they are bona fide viruses. Biol Direct. 2014;9:6. doi: 10.1186/1745-6150-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaia M, et al. Broad spectrum of mimiviridae virophage allows its isolation using a mimivirus reporter. PLoS One. 2013;8(4):e61912. doi: 10.1371/journal.pone.0061912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wodarz D. Evolutionary dynamics of giant viruses and their virophages. Ecol Evol. 2013;3(7):2103–2115. doi: 10.1002/ece3.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor BP, Cortez MH, Weitz JS. The virus of my virus is my friend: Ecological effects of virophage with alternative modes of coinfection. J Theor Biol. 2014;354:124–136. doi: 10.1016/j.jtbi.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Maumus F, Epert A, Nogué F, Blanc G. Plant genomes enclose footprints of past infections by giant virus relatives. Nat Commun. 2014;5:4268. doi: 10.1038/ncomms5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanifuji G, Onodera NT, Moore CE, Archibald JM. Reduced nuclear genomes maintain high gene transcription levels. Mol Biol Evol. 2014;31(3):625–635. doi: 10.1093/molbev/mst254. [DOI] [PubMed] [Google Scholar]
- 21.Keeling PJ, et al. The Marine Microbial Eukaryote Transcriptome Sequencing Project (MMETSP): Illuminating the functional diversity of eukaryotic life in the oceans through transcriptome sequencing. PLoS Biol. 2014;12(6):e1001889. doi: 10.1371/journal.pbio.1001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desnues C, Raoult D. Inside the lifestyle of the virophage. Intervirology. 2010;53(5):293–303. doi: 10.1159/000312914. [DOI] [PubMed] [Google Scholar]
- 23.Katzourakis A, Aswad A. The origins of giant viruses, virophages and their relatives in host genomes. BMC Biol. 2014;12:51. doi: 10.1186/s12915-014-0051-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, et al. Endogenous viral elements in algal genomes. Acta Oceanol Sin. 2014;33(2):102–107. [Google Scholar]
- 25.Filée J. Multiple occurrences of giant virus core genes acquired by eukaryotic genomes: The visible part of the iceberg? Virology. 2014;466-467:53–59. doi: 10.1016/j.virol.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Sharma V, Colson P, Giorgi R, Pontarotti P, Raoult D. DNA-dependent RNA polymerase detects hidden giant viruses in published databanks. Genome Biol Evol. 2014;6(7):1603–1610. doi: 10.1093/gbe/evu128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delaroque N, Boland W. The genome of the brown alga Ectocarpus siliculosus contains a series of viral DNA pieces, suggesting an ancient association with large dsDNA viruses. BMC Evol Biol. 2008;8:110. doi: 10.1186/1471-2148-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keeling PJ. The number, speed, and impact of plastid endosymbioses in eukaryotic evolution. Annu Rev Plant Biol. 2013;64:583–607. doi: 10.1146/annurev-arplant-050312-120144. [DOI] [PubMed] [Google Scholar]
- 29.Curtis BA, et al. Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature. 2012;492(7427):59–65. doi: 10.1038/nature11681. [DOI] [PubMed] [Google Scholar]
- 30.del Campo J, et al. The others: Our biased perspective of eukaryotic genomes. Trends Ecol Evol. 2014;29(5):252–259. doi: 10.1016/j.tree.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yutin N, Kapitonov VV, Koonin EV. A new family of hybrid virophages from an animal gut metagenome. Biol Direct. 2015;10:19. doi: 10.1186/s13062-015-0054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dereeper A, Audic S, Claverie J-M, Blanc G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol Biol. 2010;10:8. doi: 10.1186/1471-2148-10-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guindon S, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 35.Abascal F, Zardoya R, Posada D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics. 2005;21(9):2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 36.Sun S, et al. Community cyberinfrastructure for Advanced Microbial Ecology Research and Analysis: The CAMERA resource. Nucleic Acids Res. 2011;39(Database issue):D546–D551. doi: 10.1093/nar/gkq1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jurka J, et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 2005;110(1–4):462–467. doi: 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.