

Significance
Nearly all cervical, anal, vulvar, and penile cancer and up to half of oropharyngeal cancers are driven by the E6 and E7 oncoproteins of human papilloma virus (HPV). Therapeutic vaccination against these HPV proteins can slow disease progression in animal models and in patients, but is rarely curative. We demonstrate that coadministration of agonist antibodies targeting the T-cell costimulatory receptor 4-1BB and an intranasal HPV E6/E7 peptide vaccine promoted durable regression in 100% of animals bearing HPV+ TC-1 tumors established in the female reproductive tract. The efficacy of 4-1BB in this system was unique among immune checkpoint antibodies and provides a paradigm for enhancement of therapeutic cancer vaccines with costimulatory agonist antibodies.
Keywords: HPV, immunotherapy, cancer vaccine, 4-1BB, checkpoint blockade
Abstract
Antibody modulation of T-cell coinhibitory (e.g., CTLA-4) or costimulatory (e.g., 4-1BB) receptors promotes clinical responses to a variety of cancers. Therapeutic cancer vaccination, in contrast, has produced limited clinical benefit and no curative therapies. The E6 and E7 oncoproteins of human papilloma virus (HPV) drive the majority of genital cancers, and many oropharyngeal tumors. We discovered 15–19 amino acid peptides from HPV-16 E6/E7 for which induction of T-cell immunity correlates with disease-free survival in patients treated for high-grade cervical neoplasia. We report here that intranasal vaccination with these peptides and the adjuvant alpha-galactosylceramide elicits systemic and mucosal T-cell responses leading to reduced HPV+ TC-1 tumor growth and prolonged survival in mice. We hypothesized that the inability of these T cells to fully reject established tumors resulted from suppression in the tumor microenvironment which could be ameliorated through checkpoint modulation. Combining this E6/E7 peptide vaccine with checkpoint blockade produced only modest benefit; however, coadministration with a 4-1BB agonist antibody promoted durable regression of established genital TC-1 tumors. Relative to other therapies tested, this combination of vaccine and α4-1BB promoted the highest CD8+ versus regulatory FoxP3+ T-cell ratios, elicited 2- to 5-fold higher infiltration by E7-specific CTL, and evoked higher densities of highly cytotoxic TcEO (T cytotoxic Eomesodermin) CD8 (>70-fold) and ThEO (T helper Eomesodermin) CD4 (>17-fold) T cells. These findings have immediate clinical relevance both in terms of the direct clinical utility of the vaccine studied and in illustrating the potential of 4-1BB antibody to convert therapeutic E6/E7 vaccines already in clinical trials into curative therapies.
Cervical cancer is the second most common malignancy in women worldwide and continues to cause significant morbidity and mortality in the developing world, where screening and prevention programs remain rudimentary (1). The E6 and E7 oncoproteins of human papilloma virus (HPV) drive cervical cancer formation and are critical for maintenance of the transformed state (2). Beyond cervical cancer, HPV infection underlies 40% or greater of cases of oropharyngeal, anal, penile, vaginal, and vulvar cancers (3).
The immunologically foreign nature of the HPV E6 and E7 proteins, coupled with their critical role in maintaining the oncogenic state, makes them ideal target antigens for therapeutic cancer vaccination. Whereas peptide-, protein-, viral- and DNA-based vaccines targeting E6 and E7 have been studied both preclinically and in clinical trials, most fail to induce regression of established HPV+ tumors (4). To some degree, all of these vaccines succeed in eliciting peripheral E6/E7-specific T-cell responses; however, not all are proven to generate T cells capable of trafficking to the genital mucosa where cervical cancer develops. Whereas a number of these vaccines extend survival, few can induce regression of established cervical cancer, suggesting that the T cells they elicit lack the capacity to overcome the suppressive tumor microenvironment and eradicate bulky disease (5).
Within the tumor microenvironment, tumor and stromal cells engage coinhibitory receptors on T cells to attenuate potentially tumoricidal immunity (6). We have previously shown that combination blockade of two of these T-cell checkpoint receptors, CTLA-4 and PD-1, promotes synergistic tumor rejection of murine melanoma (7). In a recently completed phase I clinical trial, more than 50% of metastatic melanoma patients receiving this combination therapy achieved objective clinical responses with the majority experiencing more than 85% reduction in tumor burden (8). Activation of T-cell costimulatory receptors using agonist antibodies, alone or in combination with checkpoint blockade, can also extend T-cell survival and effector function within the microenvironment and promote tumor rejection (9). Among costimulatory receptors, we have found that the tumor necrosis factor receptor superfamily member 4-1BB has a unique capacity to activate both T cells and antigen presenting cells (APCs), which fosters generation of an exquisitely cytotoxic T-cell phenotype termed ThEO/TcEO (T helper/cytotoxic Eomesodermin) (10). We hypothesized that through antibody modulation of T-cell costimulation, the E6/E7-specific T cells generated by therapeutic HPV vaccination could be empowered to expand in magnitude, infiltrate the tumor, and maintain effector function in the suppressive microenvironment.
Previously, we identified 15–19 amino acid peptides derived from HPV-16 E6 and E7, which were the target of measurable T-cell responses in 19/22 patients who remained recurrence-free following treatment for high grade cervical intraepithelial neoplasia (CIN) versus in 0/10 patients who relapsed (11). Subsequently, we found these peptides are also presented on the C57BL/6 mouse background. We have formulated these peptides and the adjuvant alpha-galactosylceramide (αGalCer) into an intranasal (i.n.) vaccine, which evokes both mucosal and systemic E6/E7 T-cell responses (12). In the syngeneic C57BL/6 HPV E6/E7-positive TC-1 tumor model, this vaccine alone significantly delayed the growth of 6-d established tumors and extended survival by more than 20 d. Addition of 4-1BB agonist antibodies, but not blockade of either CTLA-4 or PD-1, converted this therapeutic E6/E7 peptide vaccine into a curative therapy capable of inducing tumor regression, and complete elimination of the majority of s.c. implanted and all vaginally implanted TC-1 tumors. Multiple mechanisms underlie this therapeutic cooperativity including profound enhancement of tumor infiltration by antigen-specific effector T cells and generation of highly cytotoxic ThEO/TcEO phenotype T cells. These observations suggest an optimal T-cell costimulatory antibody partner for therapeutic HPV vaccination capable of converting weakly therapeutic responses into potentially curative ones. Further, the therapeutic and mechanistic insights gained may illuminate future studies seeking to combine other therapeutic vaccines for cancer and infectious disease with T-cell costimulatory modulation.
Results
Intranasal Vaccination with HPV E6/E7 Peptides in Combination with αGalCer Slows the Growth of Preimplanted HPV+ Tumors.
HPV E6/E7-driven TC-1 tumors are an established murine model of cervical cancer (13). To test the efficacy of i.n. vaccination with the E6/E7 peptides identified in our prior study coupled with αGalCer adjuvant, we s.c. implanted 2 × 105 TC-1 cells and waited 6 d for tumors to establish before beginning vaccine administration (11). Mice were vaccinated on days 6, 12, 24, and 32 and followed for tumor growth and survival over 45 d. Compared with untreated animals, αGalCer adjuvant or E6/E7 peptides alone failed to extend median survival as has previously been reported (Fig. 1A) (14, 15); however, the intranasal vaccination of peptides in combination with αGalCer significantly delayed outgrowth of TC-1 tumors and extended overall survival in this system (Fig. 1 B and C).
Fig. 1.

Intranasal vaccination with HPV E6/E7 peptides in combination with α-galactosylceramide (αGalCer) slows the growth of preimplanted HPV+ TC-1 tumors. Mice were challenged s.c. with 2 × 105 TC-1 tumor cells and immunized via the intranasal route with either HPV peptides and αGalCer (vaccine), HPV peptides alone, αGalCer alone, or PBS on days 6, 12, 24, and 32 postimplantation (vertical arrows). The survival (A) and tumor growth (B) of the mice were monitored over time. (A) Survival curves of tumor-bearing mice given different therapeutic treatments. Significance in survival proportions was measured using the log-rank test. P < 0.05. (B) The average tumor size is shown as mean area ± SD for each group of mice. Statistical analyses between different groups were performed using Student’s t test between different treatments and the different levels of significance are indicated by * (P ≤ 0.05). (C) Tumor progression of s.c.-implanted TC-1 tumors in mice given the indicated treatment. Each line indicates an individual mouse. Experiments were performed on four to six mice per group.
Intranasal Vaccination in Combination with Systemic Anti-4-1BB Immunotherapy Promotes Regression of Established s.c. HPV+ Tumors.
To evaluate whether the therapeutic efficacy of the HPV peptide vaccine could be enhanced with coadministration of immunotherapeutic antibodies, we combined the intranasal administration of HPV peptide/αGalCer vaccine with i.p. administration of either immune checkpoint blocking (αCTLA and αPD-1) or costimulatory TNF-receptor agonist (α4-1BB) antibodies. We implanted 2 × 105 TC-1 cells s.c. on the right flank and vaccinated mice with E6/E7 peptide + αGalCer i.n. on days 5 and 11 and the indicated antibody i.p. on days 5, 8, and 11. Antibody therapy alone modestly extended survival relative to untreated animals; however, these differences did not reach statistical significance (Fig. 2A). Whereas αCTLA-4 and α4-1BB monotherapy was able to delay tumor growth in some animals, PD-1 blockade was largely ineffective (Fig. 2B).
Fig. 2.

Combination therapy with an HPV E6/E7 peptide vaccine and T-cell costimulatory modulating antibodies. Mice were challenged s.c. with 2 × 105 TC-1 tumor cells before being immunized intranasally twice (days 5 and 11 posttumor challenge) with the HPV peptide vaccine (HPV peptides and αGalCer) and αCTLA-4, αPD-1, or α4-1BB (days 5, 8, and 11 posttumor challenge). Control animals received PBS, vaccine, or monotherapy alone. Tumor growth (measured in square millimeters) and animal survival were monitored over time. (A) Survival curves of tumor-bearing mice receiving the indicated treatment. (B) Tumor progression in mice receiving various monotherapies or in combination with the HPV peptide vaccine. (C) Three weeks post–complete-tumor regression, mice (n = 3) treated with vaccine in combination with α4-1BB monotherapy (from B) were rechallenged with 2 × 105 TC-1 cells. Tumor growth is presented as an average area ± SD for untreated (n = 5), vaccine-treated (n = 5), and vaccine + α4-1BB monotherapy-treated (n = 3) animals. Mice were monitored for 60 d postrechallenge for evidence of tumor growth. The persistence of E7 antigen-specific CD8+ T lymphocytes was determined in the peripheral blood mononuclear cells of mice receiving the vaccine either alone (the three longest-surviving vaccine-alone animals were used) or with α4-1BB monotherapy by staining with fluorescently labeled E749-57/Kb tetramer and antibodies to CD44 and CD8, and expressed as a percentage of CD8+ T lymphocytes from different time points (Inset). (D) In a different set of experiments, mice treated i.n. with peptide vaccine with i.p. α4-1BB were depleted of CD8 or CD4 T cells 1 d pre- and 1 d posttumor challenge (2.5 × 105 TC-1 s.c.). Depletion was maintained every 3 d until mice were killed. Tumor growth was measured in each group and the average tumor area for each group was plotted over time (n = 5 mice per group). Data in B were pooled from two independent experiments (n = 5–10 mice per group). Significance in survival proportions in A was determined using a Mantel–Cox test (P < 0.001). Each line in B represents an individual mouse. The average tumor growth in B is shown as mean area ± SD. Circles represent mean ± SD. Statistical significance in D was calculated using a Student’s T test to compare the vaccine + α4-1BB group to the CD8-depleted group. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Significant therapeutic responses, however, were observed when checkpoint modulating antibodies were administered in combination with the HPV peptide vaccine. Unique among the therapies tested, the combination of 4-1BB agonist antibodies and vaccination induced tumor regressions in all animals with 5/8 surviving tumor free and 3/8 relapsing (Fig. 2B). The three relapsing vaccine + α4-1BB–treated mice were killed at day 38 and RT-PCR analysis confirmed that their relapses were not due to loss of E6 or E7 expression, which are essential for maintenance of the transformed state in human cervical cancer (Fig. S1) (16). CTLA-4 blockade with vaccination promoted tumor regression in 2/10 mice; however, none of these regressions were durable. Surprisingly, PD-1 blockade failed to significantly improve the efficacy of this vaccine. Of the other TNF-receptor agonist antibodies tested with the peptide vaccine, αOX-40 and αCD40 produced outcomes similar to αCTLA-4, whereas responses to GITR agonist antibodies resembled those to αPD-1 (Fig. S2). Aside from α4-1BB, however, no other checkpoint modulating antibody tested proved capable of eliciting any durable TC-1 tumor regression. Together, these data reveal a unique and unexpected potential for intranasal vaccination with HPV E6/E7 peptides in combination with α4-1BB immunotherapy to promote complete and sustained immune rejection of established HPV+ tumors.
Fig. S1.
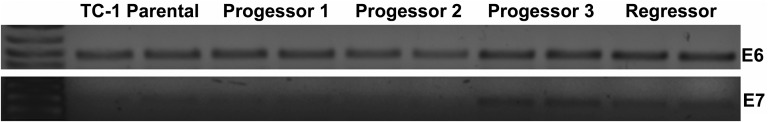
Tumor relapse in vaccine and α4-1BB–treated mice is not due to E6/E7 antigen loss. Tumors were isolated from mice relapsing after initial response to combo therapy (progressor) or in the process of marked regression, suggesting a likely complete response (regressor). Tumor RNA was purified using Trireagent (Sigma), reverse transcribed into DNA using the SuperScript II reverse transcriptase kit (Life Technologies), and then PCR amplified for the full-length E6 and E7 reading frames using HotStar Taq Mastermix (Qiagen). Separate gels were run for E6 and E7 and amplified DNA was visualized using ethidium bromide staining.
Fig. S2.

HPV E6/E7 peptide vaccine in combination with TNF-receptor agonist antibody therapy. Mice were s.c. challenged with 2 × 105 TC-1 tumor cells and immunized intranasally twice (days 5 and 11 posttumor challenge) with the HPV peptide vaccine (HPV peptides and αGalCer) in addition to (A) αCD-40 (FGK4.5), (B) αOX-40 (OX-86), or (C) αGITR (DTA-1) agonist antibodies (days 5, 8, and 11 posttumor challenge). Tumor growth (expressed in square millimeters) was monitored over time. Each line represents an individual mouse (n = 5 mice per group). Two tumor-bearing animals in B were killed before day 30 for nontumor, nontreatment-related causes at the request of the facility veterinarians.
We further validated the durability of the immune response generated by vaccine and α4-1BB combination therapy by rechallenging the mice that had achieved complete tumor regression with an additional 2 × 105 TC-1 cells at 3 wk posttumor regression. All of these animals remained tumor-free for 60 d postrechallenge and maintained detectable, E7-specific CD8 T-cell memory responses (Fig. 2C).
To uncover the relative contribution of the T-cell response in mediating regression in mice administered the vaccine in combination with α4-1BB, animals were challenged s.c. with 2.5 × 105 TC-1 tumors and depleted of either CD8 or CD4 T cells starting 1 d before challenge and concurrently with α-41BB immunotherapy. Depletion of CD4 T cells had little effect on the ability of 4-1BB stimulation to boost the antitumor effect of the peptide vaccine. In line with recently published literature (17), depletion of CD8 T cells, however, completely abrogated any therapeutic benefit imparted by the dual therapies (Fig. 2D). This loss of tumor control following CD8 depletion suggested a critical role for cytotoxic antitumor T-cell responses in mediating regression of HPV+ tumors that we wished to further examine.
Vaccination and α4-1BB Therapy Promotes High-Density Tumor Infiltration by Cytotoxic CD8 T Cells.
To further explore the mechanisms driving the synergy between intranasal vaccination and α4-1BB immunotherapy, we vaccinated mice with preestablished TC-1 tumors and administered immunotherapy, harvested tumors on day 16, and analyzed tumor infiltrating lymphocytes (TILs) by flow cytometry. We have previously shown that improved intratumoral ratios of effector T cells relative to suppressive CD4+FoxP3+ regulatory T cells (Tregs) correlate with greater therapeutic efficacy for T-cell immune checkpoint modulation (7, 18, 19). Combination of the peptide vaccine with α4-1BB therapy synergized in promoting proinflammatory ratios of CD8 versus Tregs within TC-1 tumors (17:1) relative to 4-1BB antibody alone (2.8:1) or vaccine alone (6.5:1; Fig. 3A). The combination of peptide vaccine with CTLA-4 blockade also resulted in elevated CD8 to Treg ratios (10:1), but not to the level achieved with the 4-1BB agonist in combination with peptide vaccine. Relative to vaccine alone, the 4-1BB combination resulted in significantly elevated CD8 frequencies coupled with profoundly diminished Treg frequencies. Whereas the absolute density of Tregs in the tumor did not decrease, the numbers of effector CD8 T cells increased so dramatically that the representation of Tregs as a fraction of TILs fell (Fig. S3C).
Fig. 3.

Immune correlates of protection. Antitumor T-cell responses in the tumor microenvironment of mice treated with vaccine, αCTLA-4, α4-1BB monotherapy, or in combination were characterized by flow cytometry. (A) The CD8/Treg ratio was calculated by dividing the total number of CD8+ CTLs infiltrating the tumor by the total number of CD4+Foxp3+ T-cell infiltrate. Percent of CD8+ T-cell infiltrate was calculated as a percent of total CD3+ T-cell infiltrate. The percent of Treg infiltrate was calculated as a percent of total CD4+ T cells in the tumor fraction. (B) Infiltration (Left and Center) and cytotoxic effector function (Right) of E7-specific CD8 T cells in the tumor infiltrate was also determined by staining lymphocytes with fluorescently labeled E749-57/Kb tetramer and antibodies to CD8 and granzyme B. Data are expressed as a percentage of CD8+ T cells (Left), a population of CD8 T cells in the tumor (Middle), or as a percentage of antigen-specific CD8+ T cells expressing granzyme B (Right). (C) T-cell proliferation (as indicated by percent of cells expressing Ki67) and (D) ICOS expression was also determined for CD8, CD4, Teff, and CD4 Treg cells in the tumor microenvironment and is represented as percent of T cells expressing ICOS and fold increase in %ICOS-positive cells over untreated mice. Circles represent individual mice (3–10 mice per group from two experiments). Bars represent mean ± SD. Statistical significance was calculated using a one-way ANOVA. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Fig. S3.

Gating strategy and representative FACS analysis of tumor infiltrating T-cell populations. (A) Representative gating strategy to analyze CD8+, CD4 Teff, and CD4 Treg populations in the TILs as well as tetramer-specific CD8 responses. (B) Representative gating strategy for analysis of cytotoxic antigen-specific CD8 T-cell responses in TILs. (C) Quantitation of CD8+ T-cell density (Left) CD4 Teff density (Middle) and Treg density (Right) in the tumor microenvironment. (D) Quantitation of CD4 Teff/Treg ratio in the tumor microenvironment calculated by dividing the total number of CD4+Foxp3− T cells by the total number of CD4+Foxp3+ T cells recovered from the TIL fraction. Experiments were performed in duplicate with at least three mice per group. Bars in C and D represent mean ± SD. Statistical significance was calculated using a one-way ANOVA. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (E) TC-1 tumors were treated as indicated, excised at day 16, frozen in OCT, sectioned, and stained for CD8 (Alexa 488), CD4 (V450), FoxP3 (eFluor 570), and CD31 (Alexa 647). Images were acquired using a TCS SP8 laser scanning confocal microscope (Leica) equipped with lasers for fluorescence excitation at 405, 442, 458, 488, 514, 561, and 633 nm. Images were analyzed using ImageJ image analysis software.
Beyond its efficiency at evoking overall tumor infiltration by CD8 T cells, this therapy also increased the fraction of those CD8 T cells composed of HPV E7-specific CTL (Fig. 3B and Fig. S3A). In fact, intranasal vaccination in combination with α4-1BB, but not αCTLA-4, greatly increased the density of antigen-specific CD8 T cells within the tumor microenvironment to over 160 CD8 T cells per square millimeter of tumor (Fig. 3B and Fig. S3C). From a functional standpoint, the fraction of antigen-specific CD8 T cells expressing granzyme B also increased with 4-1BB agonist combination therapy (Fig. 3B and Fig. S3B). Underlying these advantageous CD8-to-Treg ratios, we found that coadministration of α4-1BB and the vaccine drove the highest frequency of both effector CD8 and CD4 T-cell proliferation in the tumor without augmenting Treg expansion compared with the vaccine alone (Fig. 3C).
The vaccine and α4-1BB combination also modestly increased CD4 effector T-cell proliferation resulting in elevated effector-to-Treg ratios in the combination (3:1) versus either vaccine (0.8:1) or antibody (2:1) alone (Fig. 3C and Fig. S3D). The capacity of CTLA-4 antibodies to deplete Tregs from the microenvironment while expanding CD4 effectors promoted the highest CD4 effector-to-Treg ratios (5:1) in combination with the vaccine (Fig. S3 D and E) (20, 21). We found that both vaccine alone and the combination drove T cells to robustly infiltrate the tumor tissue itself, whereas in untreated tumors, CD8 T cells were scant and often appeared contained within, or tethered to blood vessels (Fig. S3E). Taken together these data suggest that the impressive therapeutic responses evidenced during α4-1BB therapy are due, in part, to robust vaccine-induced antigen-specific CD8 T-cell infiltration into the tumor microenvironment, absent a proportional increase in the suppressive Treg population.
Intranasal HPV Peptide Vaccination Generates Inducible T-Cell Costimulator-Expressing CD4 and CD8 T Cells, Which Infiltrate HPV+ Tumors.
We next investigated the phenotypic characteristics of T cells, which may account for the enhanced qualitative and quantitative antitumor effector responses. Whereas expression of the checkpoint receptor CTLA-4 was largely unaltered during the course of treatment, PD-1 expression was significantly elevated in CD8 T cells during the course of vaccination therapy (Fig. S4 A and B). We also observed an unexpected increase in the frequency of inducible T-cell costimulator (ICOS)+ T cells infiltrating the tumor environment (Fig. 3D). Administration of peptide vaccine alone increased the frequency of ICOS+ CD8 T cells greater than fourfold relative to untreated mice. In accordance with recent publications, αCTLA-4 therapy also increased the infiltration of tumors by ICOS+ T cells, although not to a level that reached statistical significance (22, 23). Addition of the peptide vaccine to αCTLA-4 promoted a threefold higher frequency of ICOS+ CD8 T-cell infiltration (28% versus 9%) relative to antibody monotherapy. Additionally, whereas α4-1BB alone failed to induce ICOS on T cells, administration of α4-1BB in combination with the intranasal vaccine increased the percentage of ICOS+ CD8 T cells to ∼20% (Fig. 3D). As observed for CD8 T cells, CD4 T effector cells also expressed ICOS at a higher frequency in mice treated with the intranasal vaccine. In much the same way that αCTLA-4 increased ICOS+ CD8 T-cell infiltrates alone or in combination with vaccination, ICOS+ CD4 effector infiltration also increased, though the magnitude of these increases was less than for CD8 T cells. HPV peptide vaccination also drove substantially increased expression of ICOS by Tregs in the tumor microenvironment. Vaccination, alone or in combination with immunotherapy, induced ICOS expression on ∼80% of Tregs, though the fold increase in ICOS+ Tregs was greatly minor compared with CD8 or effector CD4 T cells (Fig. 3D). Taken together, our results show that intranasal vaccination induces ICOS expression on tumor infiltrating lymphocytes, suggesting that these T cells have adopted a more robust effector phenotype.
Fig. S4.

Phenotypic analysis of tumor infiltrating lymphocytes. The percent of CTLA-4–expressing (A) and PD-1–expressing (B) CD8 T cells (Top), CD4 T effector cells (Middle), and regulatory T cells (Bottom) within the tumor microenvironment. Experiments were performed in duplicate with at least three mice per group. Bars represent mean ± SD. Statistical significance was calculated using a one-way ANOVA. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Treatment with 4-1BB Agonist Antibody Polarizes Tumor-Specific T Cells to the Highly Cytotoxic ThEO/TcEO Phenotype.
Previously, we and others have described the capacity of 4-1BB agonist antibodies to polarize T cells to a potently cytotoxic T-cell phenotype in both the CD4 (ThEO) and CD8 (TcEO) T-cell pools driven by high expression of the T-box transcription factor eomesodermin and typified by surface expression of the coinhibitory receptor KLRG1 (10, 24). Analysis of mice treated with 4-1BB agonist antibody, with or without i.n. HPV E6/E7 peptide vaccination, revealed that ThEO and TcEO cells are present within the tumor microenvironment (Fig. 4A and Fig. S5A), as well as in the spleen (Fig. S5B) and lymph nodes (LNs) (Fig. S5C). Whereas the percentages of ThEO/TcEO cells appear reduced in mice treated with vaccine and α4-1BB compared with α4-1BB alone, the combination treatment dramatically increased the numerical infiltration of CD4 ThEO cells (>17-fold) and CD8 TcEO cells (>70-fold) into TC-1 tumors. Moreover, whereas the ThEO/Treg ratio during vaccine and α4-1BB therapy remained unchanged relative to α4-1BB therapy alone, the TcEO/Treg ratio increased 4-fold with combination therapy (Fig. 4B). Additionally, we confirmed that nearly a quarter of TcEO cells are specific for the dominant E7 peptide incorporated in the vaccine versus <5% with 4-1BB antibody alone (Fig. 4C). These data suggest a model whereby the vaccine promotes high frequencies of E7-specific CD8 T cells, which are then selectively amplified and converted to the cytotoxic TcEO phenotype by the 4-1BB agonist antibody.
Fig. 4.

Anti–4-1BB agonist antibody therapy induces potent ThEO/TcEO T-cell responses against HPV+ tumors. Induction of a ThEO/TcEO T-cell response in the tumor microenvironment was determined by staining isolated lymphocytes with fluorescently labeled antibodies toward eomesodermin and KLRG1. (A) Percent of total CD4 (Left) and CD8 (Right) effector T-cell populations in TIL composed of Eomes+KLRG+ ThEO or TcEO T cells. Density of ThEO (Left) and TcEO (Right) T cells expressed as number of cells per square millimeter of tumor. (B) The ThEO/Treg ratio and TcEO/Treg ratio was calculated by dividing the total number of effector CD4+Eomes+KLRG1+ or CD8+Eomes+KLRG1+ cells, respectively, by the total number of CD4+Foxp3+ T cells collected from the TIL fraction. (C) The antigen-specific CD8+ TcEO cells were analyzed by calculating the fraction of CD8+Eomes+KLRG1+ T cells bound to fluorescently labeled E749-57/Kb tetramer. Experiments were performed in duplicate with at least three mice per group. Bars represent mean ± SD. Statistical significance was calculated using a one-way ANOVA (A) or Mann–Whitney U test (B and C). ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Fig. S5.

Gating strategy and analysis of ThEO/TcEO cells in the spleens and draining lymph nodes of tumor-bearing mice. (A) Representative FACS plots gating on Eomes+KLRG1+ CD4 T cells (Top) or CD8 T cells (Bottom) of mice receiving various immunomodulatory antibody therapies alone or in combination with the HPV peptide vaccine. Quantitation of Eomes+KLRG1+ ThEO (Left) and TcEO (Right) cells in the spleen (B) and draining lymph nodes (C) of tumor-bearing mice. Experiments were performed in duplicate by pooling spleens or lymph nodes from at least three mice per group. Bars represent mean ± SD. Statistical significance was calculated using a one-way ANOVA.
Next, we sought to determine whether ThEO/TcEO cells within the tumor microenvironment elevated expression of ICOS when α4-1BB was administered in combination with the peptide vaccine. Whereas neither ThEO nor TcEO cells expressed ICOS to an appreciable degree during α4-1BB monotherapy, the addition of the HPV vaccine amplified the frequency of ICOS+ ThEO (more than sixfold) and TcEO cells (more than threefold) in the tumor microenvironment (Fig. 5A). These eomesodermin-driven T cells, which have been shown to kill tumors more efficiently than their Tc1/Th1 counterparts, may acquire even higher levels of effector function through coexpression of ICOS, which has been correlated with increased IFN-γ production and cytotoxicity (Fig. 5B) (10, 22).
Fig. 5.

Intranasal vaccination with HPV E6/E7 induces infiltration of ICOS+ ThEO and TcEO cells into the tumor microenvironment. (A) The percent of ICOS expression on Eomes+KLRG1+ TcEO (Left) and ThEO (Right) infiltrating s.c. implanted TC-1 tumors. (B) Cytotoxic potential of ICOS-expressing cells was measured by comparing the mean fluorescence intensity (MFI) of granzyme B in ICOS+ versus ICOS− TcEO or ThEO cells isolated from the TIL fraction. Experiments were performed on at least three mice per group. Bars represent mean ± SD. Statistical significance was calculated using a Student’s T test. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Vaccination Plus α4-1BB Immunotherapy Promotes Complete Regression of Intravaginally Implanted HPV E6/E7-Driven Tumors.
To confirm the efficacy of our peptide vaccine in an HPV+ genital tumor challenge model, we implanted 2 × 104 TC-1 tumor cells expressing firefly luciferase (TC-1–Luc) intravaginally (25). Mice received i.n. vaccination on days 5 and 11 with i.p. antibody injections on days 5, 8, and 11, and were imaged for luciferase expression weekly throughout the study to assess tumor growth. In this system, all vaginally implanted TC-1 tumors were cured in mice receiving intranasal vaccination in combination with α4-1BB immunotherapy (Fig. 6 A and B). All mice in this group showed only background luciferase signal out to day 60 and had no observable tumor upon dissection. As in the s.c. system, CTLA-4 blockade augmented the therapeutic effect of the vaccine, but to a significantly lesser degree than the 4-1BB agonist antibody curing 2/5 mice (Fig. 6 A and B). Within the tumor-bearing female reproductive tracts (FRTs) of these animals, combination therapy with vaccine and α4-1BB evoked the highest level of infiltration by E7-specific CTL (>1.5-fold over vaccine alone) (Fig. 6C). Further, the highly cytotoxic CD4 ThEO and CD8 TcEO cells, which likely support the unique capacity of 4-1BB to promote curative responses in this system, comprised 20% of the tumor-infiltrating CD4 population and ∼30% of the CD8 population (Fig. 6D). CD8 T cells isolated from the draining lymphatics of mice receiving the vaccine and α4-1BB combination produced 10- to 20-fold higher levels of the effector cytokines IFN-γ and TNF-α compared with those from mice receiving either vaccine or α4-1BB alone (Fig. 6E). These combination therapy-generated CD8 T cells were also capable of producing more of their own IL-2 than those isolated from mice receiving either monotherapy. CD4 T cells from the nodes also produced IL-2 and low amounts of TNF-α and IFN-γ in response to restimulation with the vaccine peptides (Fig. S6). IFN-γ levels were elevated relative to mice receiving vaccine alone for all animals receiving either αCTLA-4 or α4-1BB. CTLA-4 blockade alone also evoked IL-17 production, which can be tumor supportive in some contexts (26); however, no mice receiving vaccine and/or α4-1BB exhibited Th17 phenotype T-cell responses. Overall, these results demonstrate that curative immune responses to HPV+ tumors implanted in the genital tract closely mimic those observed in the s.c. model with combination therapy.
Fig. 6.

Vaccination with HPV E6/E7 peptides in combination with α4-1BB leads to regression of intravaginally implanted tumors. Mice were challenged intravaginally with 2 × 104 TC-1 tumor cells expressing firefly luciferase and then immunized intranasally twice (days 5 and 11) with HPV peptides and αGalCer either alone or in combination with αCTLA-4 or α4-1BB therapy (days 5, 8, and 11). Control animals received either PBS or monotherapy alone. Tumor growth and survival of the mice was monitored over time. (A) Representative images of luciferase+ tumor progression over time in mice assigned to different treatment groups. (B) Survival curve (Left) and average tumor growth (Right) as measured by average radiance of luciferase activity of mice treated with peptide vaccine alone or in combination with immunotherapy. Tumor measurements ceased upon the first tumor-related mortality in each group. Survival and tumor growth are from a representative experiment of two independent experiments with three to five mice per group. (C) In a separate experiment, the percent of E7 antigen-specific CD8 T cells infiltrating into vaginal tumors and the associated genital tract was analyzed by isolating TILs and staining with fluorescently labeled E749-57/Kb tetramer and antibodies to CD8. (D) The percent of ThEO/TcEO cells capable of infiltrating intravaginally implanted TC-1 tumors was measured by gating on Eomes+KLRG1+ CD4 Teff or CD8 T cells, respectively. (E) CD8 T cells from the draining lymph node were restimulated with E6/E7 peptide-pulsed DCs and their cytokine production measured by cytometric bead array (BD Biosciences). Experiments were performed in duplicate on at least three mice per group except cytokine production, which was performed with quintuplicate wells derived from five pooled LNs per group. Bars represent mean ± SD. Statistical significance was calculated using a one-way ANOVA. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Fig. S6.
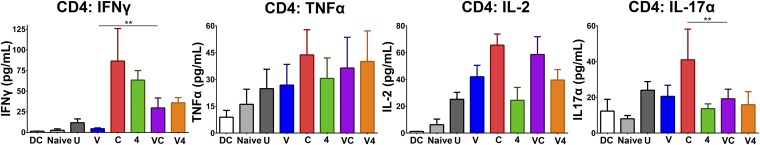
Cytokine production from restimulated CD4 T cells isolated from female reproductive tract implanted TC-1 tumor-draining lymph nodes. CD4 T cells from the draining lymph node were restimulated with E6/E7 peptide-pulsed DCs and their cytokine production was measured by cytometric bead array (BD Biosciences). Experiments were performed with quintuplicate wells derived from five pooled LNs per group. Bars represent mean ± SD. Statistical significance was calculated using a one-way ANOVA. ns, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
Whereas many therapeutic cancer vaccines have proven capable of slowing growth of established tumors, few, if any, can fully overcome the suppressive tumor microenvironment and achieve durable regressions. Here we show that an intranasal HPV E6/E7 peptide vaccine formulated with αGalCer as an adjuvant can promote durable regression of HPV+ TC-1 tumors residing in the skin or in the female reproductive tract when combined with systemic administration of 4-1BB agonist antibodies. Neither antibodies blocking the T-cell immune checkpoint receptors CTLA-4 or PD-1, nor agonist antibodies targeting the TNF receptor family members OX-40, CD40, and GITR could recapitulate the effect of 4-1BB activation in this setting. Reflecting the therapeutic outcome, we found the highest CD8-to-Treg ratios in animals treated with vaccine in combination with 4-1BB agonists. The density of HPV E7-specific T cells in the tumors of these mice was 10-fold greater in animals receiving the vaccine with α4-1BB versus αCTLA-4. The capacity of 4-1BB agonist antibodies to foster such dramatic expansion of the vaccine-induced, tumor-specific CD8 T cells, which efficiently infiltrate and proliferate within these HPV+ tumors, partially explains the ability to drive regression of these tumors in a setting where other types of checkpoint modulation did not.
Previously, we described a potently cytotoxic T-cell phenotype termed ThEO/TcEO to which tumor-infiltrating effector T cells are polarized following treatment with 4-1BB agonist antibodies (10). In combination with the HPV E6/E7 peptide vaccination, 4-1BB agonist antibody treatment promoted accumulation of >17-fold higher densities of ThEO CD4 and >70-fold higher densities of TcEO CD8 T cells within these HPV+ tumors relative to all other therapies tested. Tumors are known to express serine protease inhibitors, which blunt the efficacy of the primary CD8 T-cell lytic effector molecules granzymes A and B; however, TcEO-polarized CD8 cells express high levels of additional granzymes normally limited to natural killer (NK) cells, including granzymes E, D, G, and K, which may account for their heightened killing potential (27). We also observed an unexpected capacity of the vaccine to drive up-regulation of ICOS on T cells, which then infiltrate TC-1 tumors. Whereas CTLA-4 and PD-1 blockade are known to promote ICOS up-regulation, 4-1BB agonist does not (7). Given that ICOS expression has been associated with enhanced T-cell effector function and prolonged survival, we hypothesize that the ICOS+ ThEO and TcEO T cells formed by the combination of vaccination and α4-1BB may have an unparalleled potential to eliminate established tumors (22, 23). Our data suggest that either direct or indirect action of the αGalCer adjuvant at the vaccine site must contribute to ICOS induction as peptides alone or α4-1BB antibody alone have not been reported to have this effect. Among the adjuvant effects of αGalCer, we find that it increases 4-1BB expression by NK T cells raising the possibility of synergy between the antibody and the vaccine during T-cell priming (Fig. S7). Both the mechanism of ICOS induction by this vaccine and the details of its subsequent effect on ThEO phenotype T cells will be the focus of future studies.
Fig. S7.

4-1BB expression on NK T cells is induced by αGalCer. Naïve splenocytes were cultured in complete RPMI with 2 ng/mL αGalCer for 48 h with or without 40 μg/mL anti–4-1BB (clone LOB12.3). Cells were stained for 4-1BB expression using 4-1BB-PE (clone 17B5) and for NK T cells and analyzed by flow cytometry. The mean fluorescence intensity (MFI) of 4-1BB expression in each NK T-cell population is shown.
As most HPV-driven malignancies occur in mucosal tissues, we sought to augment the efficiency of generation of T-cell responses programmed to traffic to and enter these tumor sites by using a mucosally-focused intranasal vaccination strategy consisting of HPV E6 and E7 peptides coupled with the NK T-cell adjuvant αGalCer. In this context, we find that, of a battery of clinically relevant T-cell modulating antibodies, only 4-1BB agonist treatment was capable of converting this vaccine into a curative therapy. Whereas we have yet to experimentally address the breadth of these findings across diverse antigen types, adjuvants, and routes of delivery, recent literature suggests that 4-1BB activation may engage uniquely advantageous biologic pathways for augmenting therapeutic vaccination. Using recombinant 4-1BB ligand and a TLR4 agonist as adjuvant delivered by s.c. injection, Srivastava et al. demonstrated profound enhancement of both therapeutic HPV E7 protein vaccines against cervical cancer, as well as of a survivin-based vaccine targeting lung adenocarcinoma (28).
We expected that PD-1–mediated T-cell suppression might play a role in limiting the efficacy of the vaccine; however, we observed no significant ability of PD-1 blockade to augment vaccine efficacy. The most effective antibodies in our studies were those capable of augmenting T-cell priming and/or amplifying T-cell proliferation either through relieving repression or directly stimulating division. The blockade of PD-1, in contrast, functions primarily through restoration of T-cell effector function and induction of metabolic programs, which increase fitness in the tumor microenvironment (29). The capacity of PD-L1 blockade to augment HPV peptide vaccine responses against TC-1 tumors, although not to a curative degree, has been reported previously by Badou et al. (30). Differences in the T cells generated by vaccination at disparate sites could explain the differences in our findings, as their vaccine was injected into the peritoneum rather than delivered via the nasal mucosa. In terms of their capacity to augment numbers of CD8 T cells with high level effector function, α4-1BB is generally more potent than αCTLA-4, which is generally more potent than αPD-1. In this setting, this suggests that, in the context of vaccination, CD8 numbers are the most limiting factor. PD-1 blockade may have been capable of restoring effector function to exhausted cells, but too few CD8 cells were present, even with blockade, to effect regressions. Similarly, CTLA-4 blockade excels at augmenting effector CD4s and depressing Treg numbers and function, but those benefits appear to be less impactful in this setting compared with CD8 expansion. Human tumors develop over much longer time spans compared with murine transplantable cancer cell lines such as TC-1; however it is possible that in the context of years of tumor development with extended chronic antigen exposure, PD-1 blockade might be more efficacious.
Multiple therapeutic vaccines targeting HPV-driven malignancies, including peptide-based vaccines, are advancing through clinical trials (31). Our preclinical findings from both s.c. and vaginally implanted HPV+ tumors suggest that the efficacy of these ongoing clinical studies could be profoundly enhanced through combination with 4-1BB (CD137) agonist antibodies. Beyond the impact on cervical cancer and other HPV-driven malignancies, these data may also provide insight for designing effective combination vaccine and checkpoint blockade trials for non-HPV cancers.
Methods
Animals.
Six-week-old female C57BL/6 mice were purchased from the National Cancer Institute and Charles River Laboratory. The animals were maintained in a specific pathogen-free environment at the institutional animal facility. All procedures were conducted in accordance with the guidelines established by the University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee. All manipulations were performed on animals anesthetized with a ketamine (100 mg/kg) and xylazine (10 mg/kg) mixture administered by i.p. route. At different time points, posttumor challenge and/or immunization, animals were killed according to institutional guidelines.
Cell Lines and Reagents.
The TC-1-luciferase (TC-1–Luc) tumor cell line is of lung epithelial origin from C57BL/6 mice that was transformed with E6 and E7 oncogenes of HPV-16 as well as the Ras oncogene. This cell line additionally expresses firefly luciferase. This cell line was a kind gift from T.-C. Wu and C. Hung (Johns Hopkins School of Medicine, Baltimore). TC-1 cells were maintained in RPMI 1640 (Thermo Scientific HyClone), supplemented with 10% (vol/vol) heat inactivated FBS (Atlanta Biologicals), 50 units/mL of penicillin–streptomycin (Thermo Scientific HyClone), and 50 µg/mL gentamycin (Lonza BioWhittaker).
The E744–62 peptide, Q19D (QAEPDRAHVYNIVTFCCKCD); E749–57 peptide, R9F (RAHVYNIVTF); E643–57 peptide, Q15L (QLLRREVYDFAFRDL); and E649–58 peptide, V10C (VYDFAFRDLC), which represent murine H2b-restricted epitopes, were purchased from Elim Biopharma and dissolved in 1× PBS at a concentration of 10 mg/mL. αGalCer was purchased from DiagnoCine and dissolved in dimethyl sulfoxide (Sigma) at a concentration of 1 mg/mL. APC-labeled H-2Db epitope E749–57 (RAHYNIVTF)-containing tetramer was procured from the MHC tetramer production facility at Baylor College of Medicine (Houston) and was used for the detection and analysis of peptide-specific CD8 immunity in different tissues by flow cytometry.
Therapeutic Antibodies.
The following agonistic and antagonistic antibodies to various T-cell costimulatory modulating receptors were purchased from BioXcell at <1 endotoxin unit/mg of LPS and tested with and without the E6/E7 peptide vaccine: antibody to CTLA-4 (9H10 at 100 μg per dose), to PD-1 (RMP1-14 at 250 μg per dose), to OX-40 (OX-86 at 100 μg per dose), to GITR (DTA-1 at 350 μg per dose), to 4–1BB (LOB12.3 at 350 μg per dose) and to CD40 (FGK4.5 at 100 μg per dose).
In Vivo s.c. Tumor Experiments.
Mice were injected with a single-cell suspension of 2 × 105 TC-1 cells per animal s.c. on the right flank as described previously (12). Tumor growth was measured using a caliper to determine the diameter: longest surface length (a) and width (b), and the tumor size were expressed as area (a × b). Mice were killed when the area of the tumor reached 300 mm2.
For characterization of tumor infiltrating lymphocytes, TC-1 tumor cells were mixed in a 2:1 ratio with Matrigel (BD Biosciences) and injected at a final volume of 200 μL per animal.
Intravaginal Tumor Challenge.
For intravaginal tumor challenge, 2 × 104 TC-1 cells expressing firefly luciferase were implanted in the vaginal tract of diestrus synchronized 6- to 8-wk-old female C57BL/6 mice according to a previously described protocol (25). Tumor growth was monitored semiweekly using an Xenogen IVIS imaging system.
Vaccination and Immunotherapy.
Five days posttumor challenge, mice were immunized with the HPV E6/E7 peptide vaccine via the i.n. route of immunization. Mice were first anesthetized by i.p. injection of ketamine and xylazine hydrochloride and then administered 100 µg of each peptide with 2 µg of αGalCer intranasally as described previously (32, 33). The animals were maintained with their heads in anteflexion until they regained consciousness. Mice received two immunizations at 5-d intervals (as depicted in Fig. 1 by the vertical arrows pointing downward) and adaptive immune responses in different tissues were determined at various times postimmunization. Immunized animals also received i.p. injection of therapeutic antibodies on days 5, 8, and 11 posttumor challenge. Control animals received i.n. immunization with either PBS or peptides alone or i.p. injection with the therapeutic antibodies alone.
Depletion.
Mice were challenged with TC-1 and treated as above. Once day before tumor challenge, mice were treated i.p. with depleting αCD8 (2.43; 350 μg) or αCD4 (GK1.5; 350 μg) Mice were readministered depleting antibodies 1 d after challenge, and depletion was maintained by administration every 3 days until mice were killed.
Cell Isolation.
On day 16 post–s.c.-tumor challenge, mice were killed, and spleens, tumor draining lymph nodes, and tumors were harvested to characterize cell-mediated antitumor responses. Briefly, tumors were digested in X-Vivo-15 (Lonza) supplemented with Collagenase H (Sigma) and DNase (Roche) and incubated at 37 °C, 5% CO2 for 30 min before being filtered through a 70-μm cell strainer. T cells were enriched through density gradient separation over Histopaque 1119 (Sigma).
Cytokine Production Assays.
Tumor draining lymph nodes were isolated at day 16 as described above. Lymph node cells were pooled from five animals per group and then sorted using TCRβ PE-Cy7 and CD4 eFluor 450 (eBioscience) and CD8 Alexa 488 (Biolegend) antibodies into CD4 and CD8 T-cell pools. A total of 200,000 sorted CD4 or CD8 T cells were restimulated with 40,000 CD11c+ dendritic cells (DCs) pulsed with Q19D, Q15L, R9F, and V10C peptides per well for 48 h. Cytokine production was determined from supernatants using the BD Biosciences TH1/TH2/TH17 cytometric bead array kit.
FACS Analysis.
Samples were fixed using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) and then stained with up to 16 antibodies at a time from Biolegend, BD Biosciences, eBioscience, and Life Technologies. Flow data were collected on a five-laser, 18-color BD Biosciences LSR II cytometer and analyzed using FlowJo version 7.6.5 (Treestar).
Statistical Analysis.
All statistics were calculated using Graphpad Prism version 6 for Windows. Statistical significance was determined using a one-way ANOVA to test for differences between multiple groups or a Mann–Whitney U Test to test for differences between two groups unless otherwise indicated. Statistical significance for survival analysis was analyzed using the Mantel–Cox or log rank test where indicated. Graphs show mean ± SD unless otherwise indicated. P values of <0.05 were considered significant.
Acknowledgments
We thank Dr. T. C. Wu for contribution of the TC-1 tumor cell line. This work was partially supported by MD Anderson Cancer Center Moon Shot Knowledge Gap seed funding, the HPV+ Cancers Pilot Moon Shot, and the Oropharynx Program at The University of Texas MD Anderson Cancer Center and funded in part through the Stiefel Oropharyngeal Research Fund. The Immunology Imaging Core is supported by the S10 RR029552 (NIH) and MD Anderson instrumentation grants.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1514418112/-/DCSupplemental.
References
- 1.Parkin DM, Pisani P, Ferlay J. Estimates of the worldwide incidence of 25 major cancers in 1990. Int J Cancer. 1999;80(6):827–841. doi: 10.1002/(sici)1097-0215(19990315)80:6<827::aid-ijc6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 2.Moody CA, Laimins LA. Human papillomavirus oncoproteins: Pathways to transformation. Nat Rev Cancer. 2010;10(8):550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- 3.Chaturvedi AK. Beyond cervical cancer: Burden of other HPV-related cancers among men and women. J Adolesc Health. 2010;46(4) Suppl:S20–S26. doi: 10.1016/j.jadohealth.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 4.Brinkman JA, et al. Therapeutic vaccination for HPV induced cervical cancers. Dis Markers. 2007;23(4):337–352. doi: 10.1155/2007/245146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: A potential clue to unlock cancer immunotherapy. Cancer J. 2010;16(4):399–403. doi: 10.1097/PPO.0b013e3181eacbd8. [DOI] [PubMed] [Google Scholar]
- 6.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107(9):4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moran AE, Kovacsovics-Bankowski M, Weinberg AD. The TNFRs OX40, 4-1BB, and CD40 as targets for cancer immunotherapy. Curr Opin Immunol. 2013;25(2):230–237. doi: 10.1016/j.coi.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curran MA, et al. Systemic 4-1BB activation induces a novel T cell phenotype driven by high expression of Eomesodermin. J Exp Med. 2013;210(4):743–755. doi: 10.1084/jem.20121190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarkar AK, Tortolero-Luna G, Follen M, Sastry KJ. Inverse correlation of cellular immune responses specific to synthetic peptides from the E6 and E7 oncoproteins of HPV-16 with recurrence of cervical intraepithelial neoplasia in a cross-sectional study. Gynecol Oncol. 2005;99(3) Suppl 1:S251–S261. doi: 10.1016/j.ygyno.2005.07.099. [DOI] [PubMed] [Google Scholar]
- 12.Manuri PR, et al. Intranasal immunization with synthetic peptides corresponding to the E6 and E7 oncoproteins of human papillomavirus type 16 induces systemic and mucosal cellular immune responses and tumor protection. Vaccine. 2007;25(17):3302–3310. doi: 10.1016/j.vaccine.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin KY, et al. Treatment of established tumors with a novel vaccine that enhances major histocompatibility class II presentation of tumor antigen. Cancer Res. 1996;56(1):21–26. [PubMed] [Google Scholar]
- 14.Kim D, Hung CF, Wu TC, Park YM. DNA vaccine with α-galactosylceramide at prime phase enhances anti-tumor immunity after boosting with antigen-expressing dendritic cells. Vaccine. 2010;28(45):7297–7305. doi: 10.1016/j.vaccine.2010.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zwaveling S, et al. Established human papillomavirus type 16-expressing tumors are effectively eradicated following vaccination with long peptides. J Immunol. 2002;169(1):350–358. doi: 10.4049/jimmunol.169.1.350. [DOI] [PubMed] [Google Scholar]
- 16.Rampias T, Sasaki C, Weinberger P, Psyrri A. E6 and e7 gene silencing and transformed phenotype of human papillomavirus 16-positive oropharyngeal cancer cells. J Natl Cancer Inst. 2009;101(6):412–423. doi: 10.1093/jnci/djp017. [DOI] [PubMed] [Google Scholar]
- 17.Sun Y, et al. Intravaginal HPV DNA vaccination with electroporation induces local CD8+ T-cell immune responses and antitumor effects against cervicovaginal tumors. Gene Ther. 2015;22(7):528–535. doi: 10.1038/gt.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One. 2011;6(4):e19499. doi: 10.1371/journal.pone.0019499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quezada SA, Peggs KS, Curran MA, Allison JP. CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest. 2006;116(7):1935–1945. doi: 10.1172/JCI27745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Selby MJ, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013;1(1):32–42. doi: 10.1158/2326-6066.CIR-13-0013. [DOI] [PubMed] [Google Scholar]
- 21.Simpson TR, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013;210(9):1695–1710. doi: 10.1084/jem.20130579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fan X, Quezada SA, Sepulveda MA, Sharma P, Allison JP. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J Exp Med. 2014;211(4):715–725. doi: 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu T, He Q, Sharma P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011;71(16):5445–5454. doi: 10.1158/0008-5472.CAN-11-1138. [DOI] [PubMed] [Google Scholar]
- 24.Song C, et al. Eomesodermin is required for antitumor immunity mediated by 4-1BB-agonist immunotherapy. OncoImmunology. 2014;3(1):e27680. doi: 10.4161/onci.27680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Decrausaz L, et al. A novel mucosal orthotopic murine model of human papillomavirus-associated genital cancers. Int J Cancer. 2011;128(9):2105–2113. doi: 10.1002/ijc.25561. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, et al. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206(7):1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medema JP, et al. Expression of the serpin serine protease inhibitor 6 protects dendritic cells from cytotoxic T lymphocyte-induced apoptosis: Differential modulation by T helper type 1 and type 2 cells. J Exp Med. 2001;194(5):657–667. doi: 10.1084/jem.194.5.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Srivastava AK, et al. SA-4-1BBL and monophosphoryl lipid A constitute an efficacious combination adjuvant for cancer vaccines. Cancer Res. 2014;74(22):6441–6451. doi: 10.1158/0008-5472.CAN-14-1768-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gubin MM, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Badoual C, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013;73(1):128–138. doi: 10.1158/0008-5472.CAN-12-2606. [DOI] [PubMed] [Google Scholar]
- 31.Bergot AS, Kassianos A, Frazer IH, Mittal D. New approaches to immunotherapy for HPV associated cancers. Cancers (Basel) 2011;3(3):3461–3495. doi: 10.3390/cancers3033461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Courtney AN, et al. Alpha-galactosylceramide is an effective mucosal adjuvant for repeated intranasal or oral delivery of HIV peptide antigens. Vaccine. 2009;27(25-26):3335–3341. doi: 10.1016/j.vaccine.2009.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Courtney AN, et al. Intranasal but not intravenous delivery of the adjuvant α-galactosylceramide permits repeated stimulation of natural killer T cells in the lung. Eur J Immunol. 2011;41(11):3312–3322. doi: 10.1002/eji.201041359. [DOI] [PMC free article] [PubMed] [Google Scholar]