

Abstract
In plants, flowering is the most important transition from vegetative to reproductive growth. The flowering patterns of monocots and eudicots are distinctly different, but few studies have described the evolutionary patterns of the flowering genes in them. In this study, we analysed the evolutionary pattern, duplication and expression level of these genes. The main results were as follows: (i) characterization of flowering genes in monocots and eudicots, including the identification of family-specific, orthologous and collinear genes; (ii) full characterization of CONSTANS-like genes in Brassica rapa (BraCOL genes), the key flowering genes; (iii) exploration of the evolution of COL genes in plant kingdom and construction of the evolutionary pattern of COL genes; (iv) comparative analysis of CO and FT genes between Brassicaceae and Grass, which identified several family-specific amino acids, and revealed that CO and FT protein structures were similar in B. rapa and Arabidopsis but different in rice; and (v) expression analysis of photoperiod pathway-related genes in B. rapa under different photoperiod treatments by RT-qPCR. This analysis will provide resources for understanding the flowering mechanisms and evolutionary pattern of COL genes. In addition, this genome-wide comparative study of COL genes may also provide clues for evolution of other flowering genes.
In plants, successful sexual reproduction and seed development depend on flowering at the appropriate time1. The transition from vegetative to reproductive growth is one of the most important developmental switches in the plant life cycle2,3. A diverse range of endogenous and environmental signals regulates this transition, and these signals are integrated into a single decision—to flower or not. The interactions between proteins that transduce and integrate these signals promote or inhibit the transition to flowering4,5. In Arabidopsis, most key flowering genes have been identified and functionally characterized1,6,7. Genetic and molecular studies have revealed that many genes are involved and can be assigned to distinct regulatory pathways, including the vernalization, photoperiod, gibberellin, autonomous, ambient temperature and aging pathways1,6. Interestingly, it has been discovered that many homologous genes of Arabidopsis regulate flowering in other species8,9.
The identified flowering genes are involved in a network of six regulatory pathways (Supplementary Fig. S1). These pathways converge to regulate a set of ‘floral integrator’ genes that integrate the outputs of the various pathways and directly activate the identity genes of the floral meristem1,3,10. These integrator genes include SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), FLOWERING LOCUS T (FT), TWIN SISTER OF FT (TSF), FLOWERING LOCUS C (FLC) and LEAFY (LFY)6. TSF is the closest homolog of FT in Arabidopsis, and their mRNA levels present similar patterns of diurnal oscillation and response to photoperiod. TSF promotes flowering largely redundantly with FT but makes a distinct contribution under SD conditions11. The expression of these floral promoters leads to further up-regulation of LFY and other floral-identity genes, such as APETALA 1 (AP1), and then promotes flowering12,13. Changing the seasonal timing of flowering is a major goal of plant breeding, which may allow the production of novel varieties adapted to local climatic and environmental conditions5. The day length can control several plant processes, including flowering time, formation of bud dormancy and production of storage organs. Arabidopsis, a model eudicot, is a facultative long-day (LD) plant because its flowering is promoted by long days and delayed by short days, whereas rice is a typical short-day (SD) monocot. In all seed crops, floral transition is a key developmental switch that determines the production of dry matter3,5.
The photoperiod regulates flowering mainly by the CONSTANS (CO) and FT genes14,15,16,17,18. CO is a phloem-specific transcription activator of FT that promotes flowering by up-regulating the expression of FT and SOC1 genes19,20,21,22,23,24. These genes are up-regulated in LD plants, resulting in rapid flowering. The CO-like genes delay flowering, while FT-like genes promote flowering in SD plants1,25. Previous analyses have demonstrated that the timing of the formation of the FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 (FKF1) and GIGANTEA (GI) gene complex determines the timing of daytime CO gene expression in LD plants26. The role of the FKF1-GI complex is to remove CYCLING DOF FACTORs (CDFs), which are transcriptional repressors of CO27. At the posttranscriptional level, CO is degraded in the dark by CONSTITUTIVE PHOTOMORPHOGENIC 1 (COP1) and destabilized in the morning by phytochrome B (PHYB). However, CO is promoted by CRY and phytochrome A (PHYA)28,29. Therefore, the stability of CO based on light and the circadian clock is the core of day-length-sensing mechanisms21,28,29. These regulatory mechanisms ensure that CO activates FT transcription only during long days. The expression and translation of FT occur in the leaves, and the translated protein translocates to the meristem, where it activates floral development. The FT protein is a major florigen and is synthesized in the leaf vasculature18,20,30,31. The FT protein then travels to the shoot apical meristem to initiate the expression of floral-identity genes32,33. Previous analyses have shown that the CO/FT module is highly conserved in most angiosperms34. CO and FT orthologs are found in LD and SD plants. In rice, Hd1 and Hd3a are the orthologs of Arabidopsis CO and FT, respectively25,35. In addition, RICE FLOWERING LOCUS T 1 (RFT1) is the closest homologue of Hd3a and is thus also an ortholog of Arabidopsis CO36. The CO/FT module plays an important role in flowering-time regulation37,38. The functions of the key genes in the pathways controlling photoperiodic flowering are the same in both Arabidopsis and rice. However, the regulation of flowering by Hd1 is different under long-day conditions. Under LD conditions, CO promotes FT expression in Arabidopsis, but Hd1 represses Hd3a in rice. Under SD conditions, Hd1 promotes flowering and up-regulates Hd3a, which differs from the function of Arabidopsis CO25,35,39. The time-keeping mechanism with a periodicity of 24 hours is known as the circadian clock, which confers diurnal patterns on gene expression. It has three interlocked feed-back loops: the central, morning and evening loops. The central loop contains the transcription factors LATE ELONGATED HYPOCOTYL (LHY) and CIRCADIAN CLOCK ASSOCIATED 1 (CCA1), which repress the transcription of TIMING OF CAB 1 (TOC1). TOC1 encodes a pseudo-response regulator and activates the transcription of LHY and CCA1. Similar loops exist in the morning and evening, and these include PRR7/PRR9 and GI. The circadian clock ensures that CO transcription peaks late in the day, and GI enhances this peak under LD conditions6.
Until now, several studies have investigated the CO and FT genes in plants, and their evolution in different species has been investigated17,37,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54. However, there remain several questions that need to be answered. For example, the event/protein that changes Hd1 from a repressor of rice flowering under LD conditions to an activator of flowering under SD conditions has not yet been identified. It is also unclear why Arabidopsis CO and rice Hd1 exert opposite effects on flowering under LD conditions. Owing to the increasing number of sequenced genomes from different species, it is currently possible and necessary to systematically analyse these genes. Chinese cabbage is one of the most important vegetables worldwide. Given its significant economic value and close relationship to Arabidopsis, the Chinese cabbage (Chiifu-401-42) genome has been sequenced and assembled55. Chinese cabbage is a LD biennial plant that flowers in the spring after undergoing vernalization in the winter. However, despite the important role of flowering genes in growth regulation, few studies have systematically investigated these genes in Chinese cabbage. Thus, the investigation of flowering-related genes in the whole Chinese cabbage genome is timely. In this study, we comprehensively identified the flowering genes of this species through comparative genomic analysis. Seventy-three common gene families were identified among 11 species using OrthoMCL56. The evolutionary divergence of the genetic factors underlying the differences in morphological and physiological features between monocots and eudicots is of great interest. Grasses, including the most important cereal crops, are typically monocots. However, few studies have comprehensively compared the evolutionary history of flowering genes at the whole-genome level between these two groups. In this study, we compared the gene number, gene duplication and class-specific genes involved in the flowering time of one basal angiosperm, four grasses and six eudicots. Furthermore, we also determined the evolutionary pattern and history of the COL genes in the plant kingdom.
Results and Discussion
Characterization of flowering genes in monocots and eudicots
We systematically collected all flowering-related genes of Arabidopsis according to previous reports1,3,6. Based on previously reported methods57,58, the homogeneous candidate flowering genes between Arabidopsis and other species were identified by BLAST (E-value <1 × 10−10, Identity>40%) (Fig. 1a, Supplementary Table S1). There were more flowering genes in B. rapa (798) than in the other ten species, and A. trichopoda had the least flowering genes (273) (Supplementary Table S2). This result may indicate that B. rapa underwent genome triplication, whereas A. trichopoda did not. In monocots, we noted that Z. mays had more flowering genes (630) compared with the other three monocots because it had undergone an additional whole-genome duplication (WGD) event. In eudicots, although B. rapa had the most flowering genes, only four pathways were found to have more genes than those found in the other five eudicots.
Figure 1. The comparative analysis of flowering-related genes in angiosperms.
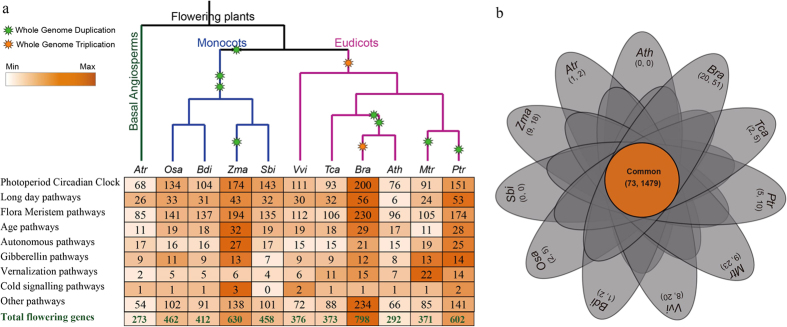
(a) The number of genes in each flowering-related pathway in six eudicots, four monocots and one basal angiosperm. Information regarding genome duplication or triplication was obtained from the Plant Genome Duplication Database (PGDD)70. (b) The Venn diagram shows the number of common and specific gene families and genes in 11 angiosperms. The first number in the brackets represents the number of gene families, and the second number represents the number of genes. The abbreviations represent the species as follows: Ath, Arabidopsis thaliana; Bra, Brassica rapa; Tca, Theobroma cacao; Ptr, Populus trichocarpa; Mtr, Medicago truncatula; Vvi, Vitis vinifera; Bdi, Brachypodium distachyon; Osa, Oryza sativa; Sbi, Sorghum bicolor; Zma, Zea mays; Atr, Amborella trichopoda.
We identified common and specific flowering genes in these 11 angiosperms according to a previous report58. Seventy-three subfamilies were common to the 11 species, and these contained 1479 genes (Fig. 1b). B. rapa contained the highest number of specific subfamilies (20), followed by M. truncatula (9) and Z. mays (9). The monocots contained 68 specific subfamilies (319 genes), and the eudicots had eight specific subfamilies (72 genes). However, A. trichopoda, a basal angiosperm species, only contained one specific subfamily, which includes two genes (Supplementary Fig. S2a). To further analyse species-specific genes in eudicots and monocots, four monocots and six eudicots were analysed. The results revealed that B. distachyon, rice and Z. mays contained several specific genes, whereas no specific gene was identified in S. bicolor (Supplementary Fig. S2b). Similarly, we did not find any specific genes in Arabidopsis (Supplementary Fig. S2c).
To analyse the duplication events of flowering genes, we identified the duplicated type for each flowering gene and found that B. rapa and P. trichocarpa contained more flowering genes that had undergone a WGD or segmental duplication, namely 78.20% and 76.08%, respectively (Supplementary Table S2). Interestingly, we noted that the percentage of flowering genes that had undergone a WGD or segmental duplication in all the species (except A. trichopoda) was greater than that obtained at the whole-genome level (Supplementary Table S3). This phenomenon indicated that the flowering genes expanded during WGD events. Because no genome duplication event occurred during the evolution of A. trichopoda, it contained very few flowering genes that had undergone a WGD or segmental duplication (0.73%), and most of its flowering genes were dispersed (88.28%).
Detection of collinear orthologs is very important for understanding gene evolution in different species. The ratio of collinear ortholog pairs to all ortholog pairs was calculated (Table 1, Supplementary Table S4). This ratio can reveal how gene orders are conserved or how frequently chromosomes are rearranged between species59. The results showed that the gene order is better conserved within monocots and eudicots than between monocots and eudicots. However, we also found several exceptions, such as B. rapa and M. truncatula. Although these species are eudicots, the ratios of collinear ortholog pairs to all ortholog pairs obtained for their flowering genes (7.09%) and their whole genomes (4.84%) were very low, which demonstrated that the genes were rearranged between these two species. In addition, we obtained a markedly higher ratio between S. bicolor and V. vinifera for the flowering genes (48.66%) than for the whole genome (7.01%). This phenomenon indicated that the order of the flowering genes was better conserved than the order of the whole genome between these two species. In contrast, a markedly higher ratio was found between M. truncatula and rice for the whole genome (47.58%) than for the flowering genes (2.43%), which suggested that the flowering genes were rearranged between these two species. A. trichopoda was found to exhibit a higher level of collinearity with the eudicots than with the monocots, suggesting that its gene order most closely resembles that of the eudicot ancestral genome because of a lack of genome duplication60. However, very low ratios were obtained between A. trichopoda and B. rapa for the flowering genes and the whole genome possibly because Chinese cabbage underwent a recent WGT event that resulted in rearrangement of its genes55. All of these phenomena may be related to genome duplication during the evolutionary history of the different species.
Table 1. The numbers located below the diagonal represent the percentage of collinear ortholog pairs (collinear ortholog pairs number/number of all ortholog pairs) in the genomes of 11 angiosperms. The numbers located above the diagonal represent the percentage of collinear ortholog pairs in the flowering genes of 11 angiosperms.
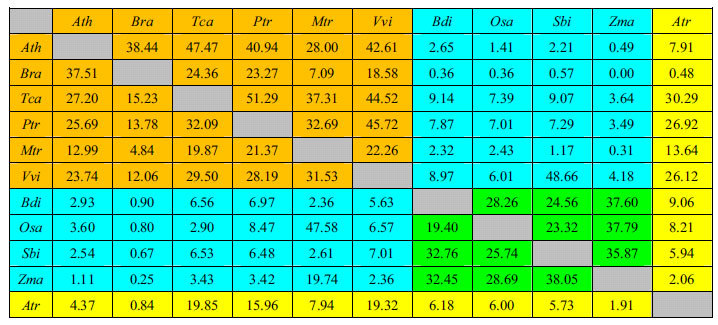
Note: The abbreviations represent the species as follows: Ath, Arabidopsis thaliana; Bra, Brassica rapa; Tca, Theobroma cacao; Ptr, Populus trichocarpa; Mtr, Medicago truncatula; Vvi, Vitis vinifera; Bdi, Brachypodium distachyon; Osa, Oryza sativa; Sbi, Sorghum bicolor; Zma, Zea mays; Atr, Amborella trichopoda. The orange colour represents the percentage of collinear ortholog pairs within eudicot species, the green colour represents the percentage of collinear ortholog pairs within monocot species, the blue colour represents the percentage of collinear ortholog pairs between eudicot and monocot species, and the yellow colour represents the percentage of collinear ortholog pairs between Amborella trichopoda and other angiosperm species.
Comparative analysis of COL genes in the whole Chinese cabbage genome
CO promotes the induction of flowering in Arabidopsis under long photoperiods and is one of the most important COL transcription factor families. Twenty-five BraCOL genes were identified in Chinese cabbage. All the BraCOL genes contained one to two B-box domains located in the N-terminus but had only one CCT domain, which comprised approximately 45 amino acids, was located in the C-terminus of the proteins, and was more conserved than the B-box domain. In addition, 34 COL genes identified in Arabidopsis and rice were selected for comparative analysis. The six most conserved motifs in the BraCOL genes were detected by MEME (Supplementary Fig. S3). Most of the BraCOL genes had motifs 2, 3, and 6, which correspond to highly conserved B-box domains. However, no motif 3 was detected in the group B proteins because these only contained one B-box domain. Motifs 1 and 4 were almost always found in all the BraCOL genes, and these constituted the CCT domain. In addition, the results from gene structure analyses were mostly consistent with those obtained from the analysis of gene motifs, and the members of the same branch usually shared a common intron or exon pattern (Supplementary Fig. S3).
To classify BraCOL genes, 25 BraCOL genes and 17 AthCOL genes were used to construct a phylogenetic tree (Supplementary Fig. S4). The BraCOL genes were clearly grouped into three groups according to their bootstrap values for phylogenetic relationship and classification in Arabidopsis61. Most of the BraCOL genes belonged to group C (11 genes), followed by groups A (eight genes) and B (six genes) (Supplementary Table S5). Genes with high sequence similarity generally have similar functions across different species. Thus, the construction of the phylogenetic tree of BraCOL genes based on Arabidopsis aids the determination of the functions of BraCOL genes. The phylogenetic analysis provides a solid foundation for future functional studies involving comparisons with AthCOL genes.
Most BraCOL genes could be mapped onto ten chromosomes of Chinese cabbage with a non-random distribution (Fig. 2a). However, BraCOL14 is located in Scaffold000467. Chromosome 2 contained more BraCOL genes (four genes) than the other chromosomes, whereas chromosomes 3 and 4 only contained one BraCOL gene. Arabidopsis has undergone two whole-genome duplications (WGD: α and β) and one whole-genome triplication event (WGT: γ). B. rapa shares this evolutionary history with Arabidopsis but has undergone an additional WGT event. Therefore, the B. rapa genome was further divided into three differentially fractionated subgenomes, namely the least fractionated (LF), medium fractionated (MF1), and most fractionated (MF2) subgenomes55. In this study, 24 BraCOL genes were mapped onto chromosomes fractionated into these three subgenomes: nine (38%) in LF, eight (33%) in MF1, and seven (29%) in MF2 (Fig. 2a). In addition, we reconstructed the B. rapa genome containing 24 conserved chromosomal blocks (labelled A–X) according to previous reports. The colour coding of these blocks was based on their positions in a proposed ancestral karyotype (AK1-8)62. Most of the BraCOL genes belonged to AK3 (29%), followed by AK1 and AK6 (21%), whereas no BraCOL gene was identified in AK7 (Fig. 2a). In Chinese cabbage, we identified 14 duplicated BraCOL genes, which were located in synteny regions (Supplementary Fig. S5). All of these duplicated genes were divided into six groups (Fig. 2a). Four of these groups each contained two duplicated genes, whereas two groups contained three duplicated genes. The groups contained only twoduplicated genes, which may be explained by the gene loss that accompanied genome triploidization in Chinese cabbage. Furthermore, the types and divergence time of the duplicated genes were estimated by calculating the numbers of synonymous substitutions (Ks). The Ks, Ka, and Ka/Ks values for seven Chinese cabbage and two Arabidopsis duplication pairs were calculated (Table 2). All the duplicated gene pairs belonged to genes that had undergone a WGD or segmental duplication, and all the duplicated COL gene pairs had a Ka/Ks ratio less than 1, indicating the purifying selection of these genes. The calculation of the divergence time of the duplicated COL genes revealed that it spanned 8.49 to 28.88 MYA (Table 2). The divergence time of BraCOL9b and BraCOL9c was 8.49 MYA, which indicates that their divergence occurred during the Brassica triplication events (5 ~ 9 MYA)63. However, the values obtained for the two duplication pairs in Arabidopsis and one duplication pair (BraCOL2- BraCOL1a) were greater than 20 MYA, which indicates that these duplications occurred during the α-tetraploidy event (20–40 MYA)63. The divergence time obtained for the other six duplication pairs ranged from 11.33 to 13.79 MYA, indicating that these duplications occurred during the divergence of Chinese cabbage and Arabidopsis (9.6–16.1 MYA)55. In addition, we also calculated the divergence times for the 14 duplication pairs between Chinese cabbage and Arabidopsis (Table 2) and found that it ranged from 10.37 to 30.04, indicating that these genes underwent differentiation before their triplication events63.
Figure 2. The features of the COL genes in B. rapa.

(a) The distribution of BraCOL genes on ten chromosomes. The 8 ancestral blocks and three subgenomes, including the least fractionated (LF), medium fractionated (MF1), and most fractionated (MF2) subgenomes, were plotted as described by Cheng et al. (2013). AK represents the ancestral karyotype. The orange lines connect the duplicated BraCOL genes. (b) The following three maps were constructed based on the orthologous and paralogous pair positions: Chinese cabbage (A01–A10) and Arabidopsis chromosome (Chr1–Chr5) maps (left), Chinese cabbage (A01–A10) and rice chromosome (Chr1–Chr12) maps (middle), and Arabidopsis (Chr1–Chr5) and rice chromosome (Chr1–Chr12) maps (right). The red lines represent the orthologous pairs between Chinese cabbage and Arabidopsis, the purple lines represent the orthologous pairs between Chinese cabbage and rice, and the light blue lines represent the orthologous pairs between Arabidopsis and rice. The blue, green, and orange lines represent the paralogous pairs in Chinese cabbage, Arabidopsis, and rice, respectively.
Table 2. Calculation of Ka/Ks and the divergence time of the duplicated COL gene pairs in Chinese cabbage and Arabidopsis.
| Duplicated gene pairs | Ka | Ks | Ka/Ks | P-Value(Fisher) | Duplication type | Purify selection | Time (MYA) |
|---|---|---|---|---|---|---|---|
| BraCOL2-BraCOL1a | 0.24 | 0.63 | 0.37 | 1.01E–09 | WGD or segmental | Yes | 21.14 |
| BraCOL9b-BraCOL9c | 0.05 | 0.25 | 0.18 | 6.68E–14 | WGD or segmental | Yes | 8.49 |
| BraCOL6b-BraCOL6a | 0.12 | 0.40 | 0.30 | 1.39E–13 | WGD or segmental | Yes | 13.23 |
| BraCOL9c-BraCOL9a | 0.04 | 0.41 | 0.10 | 1.18E–28 | WGD or segmental | Yes | 13.79 |
| BraCOL4b-BraCOL4a | 0.05 | 0.34 | 0.14 | 1.26E–21 | WGD or segmental | Yes | 11.39 |
| BraCOL16b-BraCOL7 | 0.20 | 0.38 | 0.54 | 8.07E–05 | WGD or segmental | Yes | 12.63 |
| BraCOL16b-BraCOL16a | 0.08 | 0.36 | 0.22 | 1.81E–17 | WGD or segmental | Yes | 11.97 |
| BraCOL15a-BraCOL15b | 0.12 | 0.34 | 0.35 | 1.34E–23 | WGD or segmental | Yes | 11.33 |
| AthCOL2-AthCO | 0.17 | 0.61 | 0.29 | 1.23E–15 | WGD or segmental | Yes | 20.33 |
| AthCOL9-AthCOL10 | 0.18 | 0.87 | 0.21 | 3.35E–27 | WGD or segmental | Yes | 28.88 |
| AthCOL2-BraCOL2 | 0.11 | 0.31 | 0.34 | 2.67E–08 | WGD or segmental | Yes | 10.37 |
| AthCOL2-BraCOL1b | 0.25 | 0.70 | 0.35 | 5.92E–12 | WGD or segmental | Yes | 23.4 |
| AthCOL11-BraCOL12 | 0.28 | 0.90 | 0.31 | 1.38E–15 | WGD or segmental | Yes | 30.04 |
| AthCO-BraCOL2 | 0.21 | 0.69 | 0.30 | 4.81E–14 | WGD or segmental | Yes | 23.17 |
| AthCOL10-BraCOL10b | 0.10 | 0.52 | 0.19 | 1.11E–22 | WGD or segmental | Yes | 17.29 |
| AthCOL10-BraCOL10a | 0.12 | 0.61 | 0.20 | 1.24E–23 | WGD or segmental | Yes | 20.38 |
| AthCOL4-BraCOL4a | 0.04 | 0.79 | 0.05 | 3.84E–56 | WGD or segmental | Yes | 26.21 |
| AthCOL6-BraCOL6b | 0.12 | 0.40 | 0.30 | 5.03E–13 | WGD or segmental | Yes | 13.46 |
| AthCOL8-BraCOL8 | 0.15 | 0.35 | 0.43 | 7.58E–06 | WGD or segmental | Yes | 11.52 |
| AthCOL6-BraCOL6a | 0.10 | 0.37 | 0.27 | 2.03E–14 | WGD or segmental | Yes | 12.38 |
| AthCOL16-BraCOL7 | 0.23 | 0.66 | 0.35 | 7.07E–14 | WGD or segmental | Yes | 21.9 |
| AthCOL15-BraCOL15a | 0.16 | 0.73 | 0.23 | 5.14E–26 | WGD or segmental | Yes | 24.24 |
| AthCOL16-BraCOL16a | 0.08 | 0.46 | 0.18 | 4.96E–25 | WGD or segmental | Yes | 15.3 |
| AthCOL9-BraCOL9b | 0.07 | 0.39 | 0.18 | 9.26E–20 | WGD or segmental | Yes | 12.85 |
A comparative analysis was performed to identify orthologous and paralogous COL genes for the assessment of triplication in Chinese cabbage (Fig. 2b, Supplementary Table S6). Among all the COL genes, 27 orthologous gene pairs were identified between Chinese cabbage and Arabidopsis. Conversely, only 21 orthologous gene pairs were found between Chinese cabbage and rice, and 13 orthologous gene pairs were identified between Arabidopsis and rice. The number of orthologous genes between Chinese cabbage and Arabidopsis was greater than that between Chinese cabbage and rice, which is consistent with the close relationship between Chinese cabbage and Arabidopsis. This analysis showed that a more distant genetic relationship is associated with fewer orthologous genes between the two species. Our analysis may provide a new resource for comparing the relationships among different species. Additionally, nine and six paralogous COL gene pairs were identified in Chinese cabbage and rice, respectively, whereas only one paralogous COL gene pair was found in Arabidopsis. Among the nine COL gene pairs of Chinese cabbage, five pairs were also duplicated, which demonstrated that these five paralogous pairs were also located in the synteny region.
The evolutionary pattern and origin of COL genes in plants
The COL gene family members are subdivided into three classes, called groups A to C. A previous report showed that these groups evolved before the divergence of gymnosperms and angiosperms and indicated that the COL genes in the Brassicaceae family evolved rapidly40. To investigate the evolution of the COL gene family in the plant kingdom, we selected 34 Angiospermae (27 eudicots, six monocots and one basal angiosperm), three Gymnospermae, one Pteridophyta, one Bryophyta and six Chlorophyta species for comparative analysis (Supplementary Table S7). A total of 538 COL genes were identified in all of these species using the Pfam program (Supplementary Table S8). The number of COL genes in most angiosperms was greater than that in the other species, potentially because most angiosperms have undergone genome duplication, which leads to gene expansion. Surprisingly, Physcomitrella patens contained 17 COL genes, which is a greater number than those found in the other lower plants. We constructed phylogenetic trees of the COL genes to analyse the evolutionary relationships of these species (Supplementary Fig. S6). The results showed that the COL genes of Chlorophyta were divided into two groups and separated from those of the other species. However, the COL genes of the Bryophyta, Pteridophyta and Gymnospermae species were mixed with those of the Angiospermae species and divided into three groups. In addition, all the COL genes of the Angiospermae species were divided into three groups but were separated based on the monocot and eudicot classification (Supplementary Fig. S7), which may be due to the sequence differentiation obtained after the divergence of monocots and eudicots.
In general, the group A genes have two zinc finger B-boxes, whereas the group B and C genes have one B-box, and the group C genes have an additional diverged zinc finger. However, Chlorophyta only contained two groups of COL genes (G1 and G2), which had one and two B-box domains (Supplementary Fig. S8). In Chlorophyta, a relatively lower plant, the COL genes were not completely separated based on their number of B-box domains. All the COL genes in the analysed Bryophyta, Pteridophyta, Gymnospermae and basal Angiospermae species can be divided into three groups defined using the above-mentioned classification (Supplementary Fig. S8). These results lead one to question whether the one B-box domain in group B originated from G1 or G2. To answer this question, we constructed a phylogenetic tree of these COL genes (Supplementary Fig. S9a). The results showed that G1 had a closer relationship with group B than G2. In addition, the six most conserved motifs were detected in these genes, and all group B and G1 genes contained motif 1. However, only a few G2 genes contained motif 1, whereas the other G2 genes contained motif 5 (Supplementary Fig. S9a,b). Furthermore, we also calculated the genetic distance among these three groups and found that the group B genes presented the shortest genetic distance, followed by G1 and G2. The genetic distance between the group B and G1 genes was shorter than that between the group B and G2 genes (Supplementary Fig. S9c). All of these results indicated that group B had a close relationship with G1 and may have originated from G1.
To analyse the relationships of the B-box domains in Bryophyta, Pteridophyta, Gymnospermae and basal Angiospermae, we extracted the sequence of the B-box domains. The group A genes have two B-box domains, denoted Ab1 and Ab2, and the same result was obtained for the group C genes (Cb1 and Cb2). Group B contained only one B-box domain, named Bb. The phylogenetic tree showed that these B-box sequences were clearly divided into five classes (Fig. 3a). The results showed that the divergence in the Cb2 domain was greater than that found for the other four B-box domains. The genetic distance between Cb2 and the other four B-box domains was very large (Fig. 3b). This phenomenon could be verified by the alignment of multiple B-box sequences, and there was less conservation in Cb2 than in the other four B-box domains (Fig. 3c). We also constructed a phylogenetic tree for these four types of species using the sequences of their CCT domains, and the resulting tree was divided into three groups (Supplementary Fig. S10a). The MEME results showed that the CCT domain was more conserved than the B-box domain. Most CCT domains contained motif 1, and only three CCT domains of the group B COL genes exhibited a slight divergence (Supplementary Fig. S10a,b). The genetic distance analysis showed that group A was the most conserved among the three groups (Supplementary Fig. S10c). Although the CCT domain of the three groups was markedly conserved, we also identified several group-specific amino acids, including those in positions 25, 29, 31, and 34 in the multiple sequence alignment (Supplementary Fig. S10d). In addition, the phylogenetic tree also showed that the genes in groups A and B exhibited a close relationship (Supplementary Fig. S10e).
Figure 3. The analysis of the B-box domains in Bryophyta, Pteridophyta, Gymnospermae and basal Angiospermae.

Ab1 and Ab2 represent the two B-box domains of group A, Cb1 and Cb2 represent the two B-box domains of group C, and Bb represents the one B-box domain of group B. (a) The phylogenetic tree of these five B-box sequences. (b) The genetic distance among the five B-box sequences. (c) The multiple sequence alignment of the five B-box domains. The abbreviations represent the species as follows: Atr, Amborella trichopoda; Pta, Pinus taeda; Psi, Picea sitchensis; Pab, Picea abies; Smo, Selaginella moellendorffii; Ppa, Physcomitrella patens.
In angiosperms, we found that several COL genes were assigned to group A or group C according to their classification in Arabidopsis but had only one B-box domain, which is not consistent with the traditional definition. This phenomenon was only observed in some angiosperms. We then questioned why these genes that were classified as group A or C genes contain only one B-box domain; i.e., why they were assigned to group A or C and not to group B, which generally consists of the genes with only one B-box domain. To answer this question, we constructed a phylogenetic tree using the B-box domain sequences of several angiosperms, including B. rapa, Arabidopsis and rice (Supplementary Fig. S11a). The group A or C genes only contained one B-box domain, which we denoted Ab or Cb, respectively. The phylogenetic tree showed that all the Ab and Ab2 sequences clustered together and that all the Cb and Cb1 sequences were assigned to a group. To further analyse the relationship between these B-box domains, we calculated the genetic distances among them (Supplementary Fig. S11b). The results indicated that the genetic distance between Ab and Ab2 was shorter than that between Ab and Ab1 and that between Ab and Bb, and the genetic distance between Cb and Cb1 was shorter than that between Cb and Cb2 and that between Cb and Bb. This result was also consistent with the phylogenetic tree.
Based on these results, we speculated that the Ab1 domain of several COL genes in group A was lost as a result of genome or gene duplication. However, the Cb2 domain of several COL genes in group C was lost during species evolution. This speculation is consistent with the theory of angiosperm genome duplication and loss. Therefore, we constructed the pattern underlying the evolutionary history of COL genes in the plant kingdom based on our findings (Fig. 4). Furthermore, we also calculated the percentage of B-box loss in group A or group C genes among these species (Table 3) and found that the percentages of B-box loss in groups A and C ranged from 0 to 17.65% (rice) and from 0 to 33.33% (Panicum virgatum), respectively. This phenomenon indicated that the percentage of B-box loss differed between the Angiosperm species due to their different evolutionary mechanisms.
Figure 4. The evolutionary history pattern of COL genes in the plant kingdom.
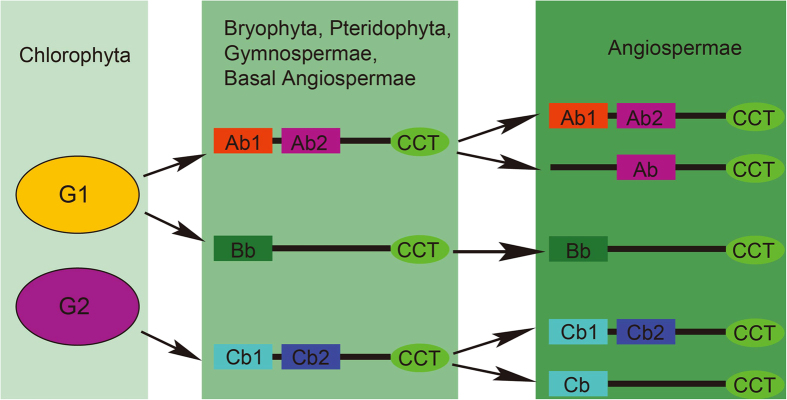
The tick and cross marks indicate that group B originated from G1. Ab and Cb represent the B-box domains of group A and group C genes that contained only one B-box domain.
Table 3. The statistics of the B-box domain lost in group A and group C COL genes of angiosperm species.
| Group A | Group B | Group C | Total | Percentage* | Percentage* | |||
|---|---|---|---|---|---|---|---|---|
| Species | Two B-box | One B-box | One B-box | Two B-box | One B-box | Gene Number | Group A-lost | Group C-lost |
| Aco | 2 | 0 | 1 | 5 | 0 | 8 | 0.00 | 0.00 |
| Aly | 6 | 0 | 4 | 5 | 3 | 18 | 0.00 | 16.67 |
| Ath | 6 | 0 | 4 | 7 | 0 | 17 | 0.00 | 0.00 |
| Bdi | 5 | 2 | 3 | 2 | 3 | 15 | 13.33 | 20.00 |
| Bra | 7 | 1 | 6 | 8 | 3 | 25 | 4.00 | 12.00 |
| Ccl | 3 | 1 | 2 | 8 | 0 | 14 | 7.14 | 0.00 |
| Cpa | 3 | 0 | 2 | 5 | 0 | 10 | 0.00 | 0.00 |
| Cru | 6 | 0 | 4 | 5 | 1 | 16 | 0.00 | 6.25 |
| Csa | 4 | 1 | 3 | 5 | 0 | 13 | 7.69 | 0.00 |
| Csi | 3 | 1 | 2 | 12 | 0 | 18 | 5.56 | 0.00 |
| Egr | 3 | 0 | 2 | 5 | 0 | 10 | 0.00 | 0.00 |
| Fve | 3 | 0 | 2 | 2 | 1 | 8 | 0.00 | 12.50 |
| Gma | 8 | 0 | 6 | 12 | 0 | 26 | 0.00 | 0.00 |
| Gra | 6 | 2 | 3 | 12 | 0 | 23 | 8.70 | 0.00 |
| Lus | 2 | 2 | 2 | 8 | 1 | 15 | 13.33 | 6.67 |
| Mdo | 6 | 0 | 5 | 6 | 0 | 17 | 0.00 | 0.00 |
| Mes | 7 | 0 | 3 | 7 | 1 | 18 | 0.00 | 5.56 |
| Mgu | 3 | 1 | 3 | 4 | 1 | 12 | 8.33 | 8.33 |
| Mtr | 4 | 0 | 1 | 4 | 1 | 10 | 0.00 | 10.00 |
| Osa | 4 | 3 | 3 | 2 | 5 | 17 | 17.65 | 29.41 |
| Ppe | 3 | 0 | 2 | 5 | 0 | 10 | 0.00 | 0.00 |
| Ptr | 6 | 0 | 3 | 7 | 3 | 19 | 0.00 | 15.79 |
| Pvi | 6 | 3 | 2 | 1 | 6 | 18 | 16.67 | 33.33 |
| Pvu | 4 | 0 | 4 | 6 | 0 | 14 | 0.00 | 0.00 |
| Rco | 3 | 0 | 2 | 4 | 1 | 10 | 0.00 | 10.00 |
| Sbi | 5 | 1 | 3 | 3 | 4 | 16 | 6.25 | 25.00 |
| Sit | 4 | 1 | 5 | 4 | 1 | 15 | 6.67 | 6.67 |
| Sly | 5 | 0 | 3 | 5 | 0 | 13 | 0.00 | 0.00 |
| Stu | 5 | 0 | 2 | 9 | 0 | 16 | 0.00 | 0.00 |
| Tca | 4 | 0 | 2 | 6 | 0 | 12 | 0.00 | 0.00 |
| Tha | 4 | 0 | 4 | 7 | 1 | 16 | 0.00 | 6.25 |
| Vvi | 2 | 0 | 1 | 3 | 0 | 6 | 0.00 | 0.00 |
| Zma | 5 | 2 | 5 | 4 | 1 | 17 | 11.76 | 5.88 |
Note: The abbreviations represent the species as follows: Aco, Aquilegia coerulea; Aly, Arabidopsis lyrata; Ath, Arabidopsis thaliana; Bdi, Brachypodium distachyon; Bra, Brassica rapa; Ccl, Citrus clementina; Cpa, Carica papaya; Cru, Capsella rubella; Csa, Cucumis sativus; Csi, Citrus sinensis; Egr, Eucalyptus grandis; Fve, Fragaria vesca; Gma, Glycine max; Gra, Gossypium raimondii; Lus, Linum usitatissimum; Mdo, Malus domestica; Mes, Manihot esculenta; Mgu, Mimulus guttatus; Mtr, Medicago truncatula; Osa, Oryza sativa; Ppe, Prunus persica; Ptr, Populus trichocarpa; Pvi, Panicum virgatum; Pvu, Phaseolus vulgaris; Rco, Ricinus communis; Sbi, Sorghum bicolor; Sit, Setaria italica; Sly, Solanum lycopersicum; Stu, Solanum tuberosum; Tca, Theobroma cacao; Tha, Thellungiella halophila; Vvi, Vitis vinifera; Zma, Zea mays.
*The percentage of B-box loss in Group A or Group C = Number of genes that contained only one B-box domain in Group A or Group C/All the COL genes in the species.
Comparative analysis of CO and FT genes between Brassicaceae and Grass species
CO is a central regulator of the photoperiod pathway because it triggers the production of the mobile florigen hormone FT, which induces flower differentiation64. It is an important member of the COL gene family, and 17, 16, 9 and 23 COL genes have been identified in Arabidopsis, rice, barley and cotton, respectively17,52. The flowering transition is regulated by FT and TERMINAL FLOWER 1 (TFL1). FT promotes the transition to reproductive development and flowering, whereas TFL1 represses this transition53. The common ancestor of the FT and TFL1 subfamilies functionally diverged from the MOTHER OF FT AND TFL1 (MFT) subfamily to activate and repress flowering, respectively54. The Brassicaceae species are typically eudicots, whereas grass is a typical monocot. Therefore, we surveyed the differences in CO and FT between Brassicaceae and Grass species and compared the protein, full gene and promoter sequences of five Brassicaceae species and three Grass species to uncover the factors affecting this regulatory mechanism.
The multiple sequences of CO revealed 48 family-specific amino acids between Brassicaceae and Grass (Fig. 5a). Among these, 13 sites were located in the two B-box domains, and three sites were located in the CCT domain. The phylogenetic trees constructed based on the protein and gene sequences exhibited similar topology (Fig. 5b). The sequences were divided into two groups, one corresponded to the sequences found in Brassicaceae species, and the other comprised the sequences in Grass species. This phenomenon indicated that CO was highly conserved in Brassicaceae and in Grass. The genetic distance calculated between the two sequence types confirmed this finding (Fig. 5c). The genetic distance between Brassicaceae and Grass was greater than that obtained between Brassicaceae species and that obtained between Grass species. Furthermore, we predicted a 3D model of the CO proteins in B. rapa, Arabidopsis and rice. The confidence of the protein structure prediction reached 97% in these three species. Twelve family-specific amino acids were located in the two B-box domains on the predicted protein structure (Fig. 6). Overall, the structures found for CO proteins in B. rapa and Arabidopsis were similar, but these were quite different from that found in rice CO proteins. The mutation sensitivity of sites 8, 10, 11 and 12 was higher than that obtained for other sites in Arabidopsis and B. rapa. However, only site 8 exhibited a higher mutation sensitivity than the other sites in rice. In addition, sites 2, 3, 5, 10 and 11 were associated with hydrophilic amino acids in B. rapa and Arabidopsis and with hydrophobic amino acids in rice. Therefore, these amino acids may be more likely to affect the function of the CO proteins.
Figure 5. The comparative analysis of CO genes between Brassicaceae and Grass species.

(a) The multiple sequence alignment of CO genes. The orange solid rectangle represents the five Brassicaceae species, and the green solid rectangle represents the three Grass species. The red hollow rectangle indicates the family-specific amino acids. (b) The phylogenetic trees were constructed using the CO protein and gene sequences. The numbers on the branches of the phylogenetic tree represent the bootstrap supports provided by the NJ, ME and ML methods. (c) The genetic distance in Brassicaceae and Grass was estimated using the CO protein and gene sequences.
Figure 6. The protein structure of CO in Arabidopsis, Chinese cabbage and rice.
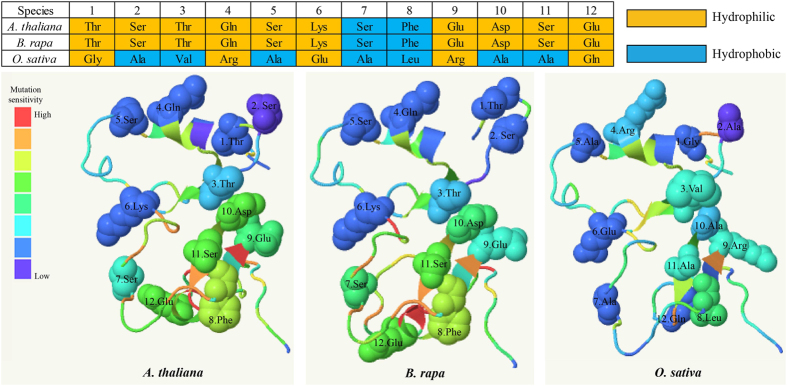
The nine family-specific amino acids located in the two B-box domains are marked on the protein structure. The orange solid rectangle represents the hydrophilic amino acids, and the blue solid rectangle indicates the hydrophobic amino acids.
The FT genes in five Brassicaceae and three Grass species were investigated in this study (Supplementary Table S9). The genes with the most sequence similarity with Arabidopsis FT gene were defined as the FT genes in each Brassicaceae and Grass species. These genes were representative members of each species FT family. For example, LOC_Os06g06320 gene corresponded to Hd3a in rice, and was chosen for the FT comparative analysis. The multiple sequences of FT revealed 27 family-specific amino acids between Brassicaceae and Grass (Supplementary Fig. S12a). The phylogenetic trees constructed based on the protein and gene sequences had similar topology (Supplementary Fig. S12b). We also predicted the 3D model of FT proteins in B. rapa, Arabidopsis and rice. The confidence of the protein structure prediction reached 100% in these three species. Twenty-two family-specific amino acids were located in the predicted protein structures (Supplementary Fig. S13). The mutation sensitivity of sites 17 and 19 was higher than that obtained for the other 17 sites in Arabidopsis. However, with the exception of sites 19 and 22, site 12 also exhibited a high mutation sensitivity in B. rapa. In rice, the mutation sensitivity of sites 12, 15 and 22 was higher than that of the other 19 sites. Furthermore, sites 1, 4, 15 and 22 were associated with hydrophobic amino acids in B. rapa and Arabidopsis and with hydrophilic amino acids in rice. Sites 7 and 16 were associated with hydrophilic amino acids in B. rapa and Arabidopsis but with hydrophobic amino acids in rice. Therefore, these amino acids may be more likely to affect FT protein function.
Expression analysis of photoperiod pathway- and circadian clock-related genes
The flowering of Arabidopsis is promoted by long days and is repressed by short days. Chinese cabbage is a member of the Brassica genus and is one of most important vegetables cultivated worldwide. It has experienced thousands of years of cultivation and artificial selection. The genome has undergone triplication events since its divergence from Arabidopsis; however, high degrees of sequence similarity and genome structure conservation remain between the two species. The flowering genes in Arabidopsis have been well studied, making Arabidopsis a viable reference species for comparative genomic studies. Variations in the number of flowering genes due to genome triplication may contribute to the broad range of flowering time plasticities observed in Brassica species.
To investigate the expression pattern of the regulatory network of photoperiod pathway- and circadian clock-related genes in B. rapa, we measured the expression of these genes under different photoperiod treatments by RT-qPCR65. We compared the expression levels of these genes under LD and SD conditions over a four-week period (Supplementary Table S10). The results showed that most genes were up-regulated at night during the first week and down-regulated during the third week (Supplementary Fig. S14). The expression levels of these genes during the day and night differed, which indicated that they were very sensitive to light or were controlled by the circadian clock. In addition, we also surveyed the expression levels of these genes at different times over a period of one day under LD and SD conditions. Compared with Arabidopsis, several genes were duplicated in B. rapa, but only one duplicated gene in each of the duplicated pairs exhibited high expression. For example, there were three FKF1 genes in B. rapa, but only the Bra038831-FKF1 gene was highly expressed, whereas the other two FKF1 genes were less strongly expressed (Fig. 7b). A similar phenomenon was found for other duplicated gene pairs, such as CDF1, CO, FT, LHY, TOC1 and COP1 (Fig. 7a–d, Supplementary Fig. S15). The expression of the two PRR7 genes was lower than the expression of the two PRR9 genes in B. rapa. The PRR7 genes presented similar expression trends under LD conditions, whereas the opposite expression trend was observed under SD conditions (Supplementary Fig. S15a,b). Under LD and SD conditions, the expression trend for TOC1 contrasted with that obtained for LHY genes (Supplementary Fig. S15c,d). Bra005541-COP1 and the two CRY1 genes exhibited similar expression trends, whereas the Bra021818-COP1 gene was hardly expressed (Supplementary Fig. S15e,f). The expression of the PHYB gene was higher than the expression levels of the PHYA genes under LD and SD conditions (Supplementary Fig. S15g,h). The expression trend detected for Bra038831-FKF1 and Bra010082-CDF1 was the opposite of that obtained under LD conditions, whereas their expression levels were similar between 4 h to 16 h under SD conditions (Fig. 7b,c). The expression trends obtained for Bra023541-CO and Bra022475-FT were the same under LD conditions, whereas their expression trends were the opposite between 8 h and 24 h under SD conditions (Fig. 7d,e). Interestingly, all three duplicated SOC1 genes were expressed, whereas their expression trends were reversed under LD and SD conditions (Supplementary Fig. S15i,j).
Figure 7. The analysis of the expression of photoperiod pathway and circadian clock-related genes by RT-qPCR.

The bars correspond to the means ± S.D. from three independent experiments. D and N represent day and night, respectively. The RNA expression level relative to the actin gene was calculated using the 2−ΔΔCT method and was thenlog2-transformed. (a) The relative expression of CDF1 and FKF1 genes in Chinese cabbage under LD conditions over a one-day period. (b) The relative expression of CDF1 and FKF1 genes in Chinese cabbage under SD conditions over a one-day period. (c) The relative expression of CO and FT genes in Chinese cabbage under LD conditions over a one-day period. (d) The relative expression of CO and FT genes in Chinese cabbage under SD conditions over a one-day period.
Conclusion
In this study, we analysed the evolutionary pattern, gene duplication, and expression level of the key genes involved in the photoperiod pathway. We compared the flowering genes in monocots and eudicots, and this analysis included the identification of the family-specific genes as well as the orthologous and collinear genes. The BraCOL genes in the Chinese cabbage genome were identified and subjected to a systematic analysis. Furthermore, the evolution and origin of the COL genes in the plant kingdom were analysed, and the evolutionary pattern of the COL genes was determined. In addition, we compared the CO and FT genes between Brassicaceae and Grass species. Moreover, the expression of photoperiod pathway- and the circadian clock-related genes in B. rapa under different photoperiod treatments were analysed by RT-qPCR. Because the photoperiod pathway- and circadian clock-related genes in B. rapa have not been previously studied, the expression analysis presented in this article provides a solid foundation for future functional studies. In conclusion, this study provides useful resources for future studies on the structure and function of flowering genes and for identifying and characterizing flowering genes in other species. In addition, this study may also facilitate our understanding of the effect of polyploidy during the evolution of flowering genes.
Methods
Retrieval of Sequences
The Arabidopsis sequences were retrieved from TAIR (http://www.arabidopsis.org/), and the sequences of rice were retrieved from RGAP (http://rice.plantbiology.msu.edu/)66. The B. rapa sequences were downloaded from BRAD (http://brassicadb.org/brad/)67. The genome sequences of A. trichopoda were downloaded from the Amborella Genome Database (http://amborella.huck.psu.edu/)60. In addition, the protein sequences of Gymnospermae species, including P. taeda, P. sitchensis and P. abies, were downloaded from the Forest Tree Genome database (http://dendrome.ucdavis.edu/treegenes/)68, and the sequences of the other 38 species used in this study were downloaded from JGI (http://www.phytozome.net/)69. All of these species are representative species of the different branches in the plant phylogenetic tree. The homologous flowering genes in other species were identified through comparison with Arabidopsis. First, BLASTP searches were performed against the rice protein sequences using an E-value threshold of 1 × 10−10. The top-ranked rice hit was used for BLASTP searches of the Arabidopsis proteins to confirm homologies. Starting with both Arabidopsis and rice homologues, BLASTP searches were performed against the proteins of other species (e-value <1 × 10−10, identity >40%). The putative orthologous flowering genes were confirmed by reciprocal BLASTP searches of the Arabidopsis and rice protein sequence datasets (Fig. 1a, Supplementary Table S1). Information regarding genome duplication or triplication was obtained from the Plant Genome Duplication Database (PGDD)70. A flow chart of this study was drawn using Microsoft Visio (Supplementary Fig. S16).
Identification of gene synteny and duplicated CO genes
BLAST and the Multiple Collinearity Scan toolkit (MCScanX) were used for gene synteny analysis according to previous reports59,71. The duplicated CO genes were identified using MCScanX. First, the whole-genome protein sequences from Chinese cabbage, Arabidopsis, and rice were searched against themselves using BLASTP with an E-value cut-off of 1 × 10−10 and identity >75%72. MCScanX was then used for detecting synteny regions according to a previous report59. We then identified the duplicated CO genes from these duplicated regions. The Venn diagrams were drawn using the R program.
Phylogenetic analysis of COL and FT family genes
Phylogenetic analyses were conducted using MEGA v5.073. Neighbour-joining (NJ), maximum-likelihood (ML) and minimum-evolution (ME) trees were constructed with a bootstrap value of 1,000 replications to assess the reliability of the resulting trees. For analysis of the COL genes, the B-box and CCT domains of the protein sequences were used to construct phylogenetic trees for the different species. In addition, the full amino acid and DNA sequences of the CO and FT genes were used to construct a tree for the analysis of the relationship between Brassicaceae and Grass species. The genetic distance used in this study was also calculated with MEGA v5.0.
Characterization of genes of the COL and FT families
The Pfam database was used to identify the genes belonging to the COL and FT families74. The Pfam accession number PF06203.9 was used for identifying the CCT domain, and. PF00643.19 was used for identification of the zf-B_box domain. The proteins containing these two conserved domains were defined as COL genes. PF01161.15 was used for identifying genes of the FT family. The genes identified by Pfam were further verified using SMART75. Conserved motifs were identified using MEME76, and the gene structures were obtained by GSDS (http://gsds.cbi.pku.edu.cn/)77. The genes with the most sequence similarity with Arabidopsis CO or FT genes were defined as the CO or FT genes in each Brassicaceae and Grass species, respectively. The full amino-acid and DNA sequences of the CO and FT genes were used to identify the family-specific sites in Brassicaceae and Grass species. The sequences were aligned using ClustalX 2.078. The CO and FT protein structures were predicted using the Phyre2 program79.
Identification of orthologous and paralogous COL genes
The orthologous and paralogous COL genes in Chinese cabbage, Arabidopsis, and rice were identified using OrthoMCl56. The relationships between orthologous and paralogous genes among the three species were plotted using Circos80. The position of the COL gene of Chinese cabbage was marked on a chromosome using an in-house-developed Perl script.
Calculation of the Ka/Ks and dating of the duplication events
To estimate the divergence of the duplicated COL genes, the sequences of the duplicated pairs of COL genes were aligned using ClustalW2. We calculated the synonymous rate (Ks), non-synonymous rate (Ka), and evolutionary constraint (Ka/Ks) between the duplicated pairs of COL genes based on the alignments of their coding sequences obtained using the method developed by Nei and Gojobori, which is incorporated in the KaKs_calculator81. The divergence time was calculated using the formula T = Ks/2R, where Ks refers to the synonymous substitutions per site, and R is the rate of divergence of nuclear genes from plants. R was considered to equal 1.5 × 10−8 synonymous substitutions per site per year for dicotyledonous plants82.
Plant materials, growth conditions, and photoperiod treatments
Seeds of the Chinese cabbage cultivar Chiifu-401–42 were grown in pots containing a soil:vermiculite mixture (3:1) in a controlled-environment growth chamber programmed for 16/8 h at 24/18 °C for day/night and a relative humidity of 60–65%. Seedlings at the five-leaf stage were transferred to growth chambers and exposed to different photoperiods. The photoperiod treatments were defined as short-day (8 hours of light/16 hours of dark) and long-day (16 hours of light/8 hours of dark), and the other conditions were not changed. After treatment for one week, we collected leaf samples at 6/20 h under the SD conditions, and at 12/22 h under the LD conditions using 0 h as the dawn. Samples were collected once a week for four weeks, when flowering occurred. In addition, we also obtained samples every 4 hours to survey the pattern of gene expression over a one-day period after two weeks of treatment. All the leaf samples were frozen in liquid nitrogen and stored at −70 °C.
RNA isolation and RT-qPCR analyses
The RNA from the leaves was isolated using an RNA kit (Tiangen, Beijing, China) according to the manufacturer’s instructions. Using the PrimeScript™ RT Reagent Kit (TaKaRa, Kyoto, Japan), 1 μg of RNA was then used to synthesize cDNA for RT-qPCR, which was performed in a 20 μl reaction volume. The cDNA reaction mixture was diluted 1:10 with EASY Dilution for RT-qPCR, and 2 μl of this mixture was used as the template in the 20 μl PCR reactions. The actin gene (Bra028615) of Chinese cabbage was used as an internal control to normalize the expression levels of the target genes. Specific primers were designed according to the corresponding gene sequences using Primer 5.0 (Supplementary Table S11). The RT-qPCR assays were performed using three biological and technical replicates. Each reaction was performed in 20 μl reaction mixtures, including SYBR Premix Ex Taq (2×) (TaKaRa, Kyoto, Japan), gene-specific primers, and the diluted cDNA sample as the template. The RT-qPCR assay was performed according to a previous report83. The gene expression level relative to that of the actin gene was calculated as 2−ΔΔCT according to a previous analysis84. The gene expression patterns of each tissue were analysed using Cluster3.0, and the expression values were log2-transformed. Heat maps were then constructed using Tree View (http://jtreeview.sourceforge.net/) for visualization of the hierarchical clustering results.
Additional Information
How to cite this article: Song, X. et al. Comprehensive analysis of the flowering genes in Chinese cabbage and examination of evolutionary pattern of CO-like genes in plant kingdom. Sci. Rep. 5, 14631; doi: 10.1038/srep14631 (2015).
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 31330067, 31301782), National Program on Key Basic Research Projects (The 973 Program: 2012CB113900), National High Technology Research and Development Program of China (863 Program, No. 2012AA100101), and China Postdoctoral Science Foundation (2014M550294).
Footnotes
Author Contributions The study was conceived by X.H. and X.S. X.S., W.D. and P.W. contributed to data collection and bioinformatics analysis. Z.H., G.L. and T.L. performed the experiments. X.S., W.D., X.H. and Y.L. participated in preparing and writing the manuscript. All authors contributed to revising the manuscript. All authors had read and approved the final manuscript.
References
- Putterill J., Laurie R. & Macknight R. It’s time to flower: the genetic control of flowering time. Bioessays 26, 363–73 (2004). [DOI] [PubMed] [Google Scholar]
- Mouhu K. et al. Identification of flowering genes in strawberry, a perennial SD plant. BMC Plant Biol 9, 122 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth A. & Schmid M. Regulation of flowering time: all roads lead to Rome. Cellular and Molecular Life Sciences 68, 2013–2037 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres F. & Coupland G. The genetic basis of flowering responses to seasonal cues. Nature Reviews Genetics 13, 627–639 (2012). [DOI] [PubMed] [Google Scholar]
- Jung C. & Muller A. E. Flowering time control and applications in plant breeding. Trends in Plant Science 14, 563–573 (2009). [DOI] [PubMed] [Google Scholar]
- Fornara F., de Montaigu A. & Coupland G. SnapShot: Control of flowering in Arabidopsis. Cell 141, 550, 550 e1–2 (2010). [DOI] [PubMed] [Google Scholar]
- Levy Y. Y. & Dean C. The transition to flowering. Plant Cell 10, 1973–90 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenup A., Peacock W. J., Dennis E. S. & Trevaskis B. The molecular biology of seasonal flowering-responses in Arabidopsis and the cereals. Ann Bot 103, 1165–72 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen C. H., Jensen C. S. & Petersen K. Similar genetic switch systems might integrate the floral inductive pathways in dicots and monocots. Trends in Plant Science 9, 105–107 (2004). [DOI] [PubMed] [Google Scholar]
- Amasino R. Seasonal and developmental timing of flowering. Plant Journal 61, 1001–1013 (2010). [DOI] [PubMed] [Google Scholar]
- Yamaguchi A., Kobayashi Y., Goto K., Abe M. & Araki T. TWIN SISTER OF FT (TSF) acts as a floral pathway integrator redundantly with FT. Plant Cell Physiol 46, 1175–89 (2005). [DOI] [PubMed] [Google Scholar]
- Liljegren S. J., Gustafson-Brown C., Pinyopich A., Ditta G. S. & Yanofsky M. F. Interactions among APETALA1, LEAFY, and TERMINAL FLOWER1 specify meristem fate. Plant Cell 11, 1007–18 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazquez M. A., Soowal L. N., Lee I. & Weigel D. LEAFY expression and flower initiation in Arabidopsis. Development 124, 3835–44 (1997). [DOI] [PubMed] [Google Scholar]
- Putterill J., Robson F., Lee K., Simon R. & Coupland G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell 80, 847–57 (1995). [DOI] [PubMed] [Google Scholar]
- Kobayashi Y., Kaya H., Goto K., Iwabuchi M. & Araki T. A pair of related genes with antagonistic roles in mediating flowering signals. Science 286, 1960–2 (1999). [DOI] [PubMed] [Google Scholar]
- Kardailsky I. et al. Activation tagging of the floral inducjer FT. Science 286, 1962–5 (1999). [DOI] [PubMed] [Google Scholar]
- Griffiths S., Dunford R. P., Coupland G. & Laurie D. A. The evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiol 131, 1855–67 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbesier L. et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–3 (2007). [DOI] [PubMed] [Google Scholar]
- Samach A. et al. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 288, 1613–6 (2000). [DOI] [PubMed] [Google Scholar]
- An H. L. et al. CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Development 131, 3615–3626 (2004). [DOI] [PubMed] [Google Scholar]
- Suarez-Lopez P. et al. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 410, 1116–1120 (2001). [DOI] [PubMed] [Google Scholar]
- Wigge P. A. Integration of spatial and temporal information during floral induction in Arabidopsis (vol. 309, pg 1056, 2005). Science 312, 1600–1600 (2006). [DOI] [PubMed] [Google Scholar]
- Tiwari S. B. et al. The flowering time regulator CONSTANS is recruited to the FLOWERING LOCUS T promoter via a unique cis-element. New Phytologist 187, 57–66 (2010). [DOI] [PubMed] [Google Scholar]
- Yoo S. K. et al. CONSTANS activates SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 through FLOWERING LOCUS T to promote flowering in Arabidopsis. Plant Physiol 139, 770–8 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima S. et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol 43, 1096–105 (2002). [DOI] [PubMed] [Google Scholar]
- Sawa M., Nusinow D. A., Kay S. A. & Imaizumi T. FKF1 and GIGANTEA complex formation is required for day-length measurement in Arabidopsis. Science 318, 261–265 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi T., Schultz T. F., Harmon F. G., Ho L. A. & Kay S. A. FKF1 F-box protein mediates cyclic degradation of a repressor of CONSTANS in Arabidopsis. Science 309, 293–7 (2005). [DOI] [PubMed] [Google Scholar]
- Valverde F. et al. Photoreceptor regulation of CONSTANS protein in photoperiodic flowering. Science 303, 1003–6 (2004). [DOI] [PubMed] [Google Scholar]
- Yanovsky M. J. & Kay S. A. Molecular basis of seasonal time measurement in Arabidopsis. Nature 419, 308–12 (2002). [DOI] [PubMed] [Google Scholar]
- Jaeger K. E. & Wigge P. A. FT protein acts as a long-range signal in Arabidopsis. Curr Biol 17, 1050–4 (2007). [DOI] [PubMed] [Google Scholar]
- Mathieu J., Warthmann N., Kuttner F. & Schmid M. Export of FT protein from phloem companion cells is sufficient for floral induction in Arabidopsis. Curr Biol 17, 1055–60 (2007). [DOI] [PubMed] [Google Scholar]
- Abe M. et al. FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309, 1052–1056 (2005). [DOI] [PubMed] [Google Scholar]
- Wigge P. A. et al. Integration of spatial and temporal information during floral induction in Arabidopsis. Science 309, 1056–1059 (2005). [DOI] [PubMed] [Google Scholar]
- Song Y. H., Ito S. & Imaizumi T. Similarities in the circadian clock and photoperiodism in plants. Current Opinion in Plant Biology 13, 594–603 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano M. et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12, 2473–2484 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiya R., Ikegami A., Tamaki S., Yokoi S. & Shimamoto K. Hd3a and RFT1 are essential for flowering in rice. Development 135, 767–74 (2008). [DOI] [PubMed] [Google Scholar]
- Bohlenius H. et al. CO/FT regulatory module controls timing of flowering and seasonal growth cessation in trees. Science 312, 1040–1043 (2006). [DOI] [PubMed] [Google Scholar]
- Distelfeld A., Li C. & Dubcovsky J. Regulation of flowering in temperate cereals. Current Opinion in Plant Biology 12, 178–184 (2009). [DOI] [PubMed] [Google Scholar]
- Hayama R., Yokoi S., Tamaki S., Yano M. & Shimamoto K. Adaptation of photoperiodic control pathways produces short-day flowering in rice. Nature 422, 719–22 (2003). [DOI] [PubMed] [Google Scholar]
- Lagercrantz U. & Axelsson T. Rapid evolution of the family of CONSTANS LIKE genes in plants. Mol Biol Evol 17, 1499–507 (2000). [DOI] [PubMed] [Google Scholar]
- Lee R., Baldwin S., Kenel F., McCallum J. & Macknight R. FLOWERING LOCUS T genes control onion bulb formation and flowering. Nat Commun 4, 2884 (2013). [DOI] [PubMed] [Google Scholar]
- Faure S., Higgins J., Turner A. & Laurie D. A. The FLOWERING LOCUS T-like gene family in barley (Hordeum vulgare). Genetics 176, 599–609 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon Y. H., Chae S., Jung J. Y. & An G. Expressed sequence tags of radish flower buds and characterization of a CONSTANS LIKE 1 gene. Mol Cells 8, 452–8 (1998). [PubMed] [Google Scholar]
- Liu J., Yu J., McIntosh L., Kende H. & Zeevaart J. A. Isolation of a CONSTANS ortholog from Pharbitis nil and its role in flowering. Plant Physiol 125, 1821–30 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M., Ichikawa K. & Aoki S. Photoperiod-regulated expression of the PpCOL1 gene encoding a homolog of CO/COL proteins in the moss Physcomitrella patens. Biochem Biophys Res Commun 324, 1296–301 (2004). [DOI] [PubMed] [Google Scholar]
- Hsu C. Y., Liu Y., Luthe D. S. & Yuceer C. Poplar FT2 shortens the juvenile phase and promotes seasonal flowering. Plant Cell 18, 1846–61 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin P. A. et al. An antagonistic pair of FT homologs mediates the control of flowering time in sugar beet. Science 330, 1397–400 (2010). [DOI] [PubMed] [Google Scholar]
- Navarro C. et al. Control of flowering and storage organ formation in potato by FLOWERING LOCUS T. Nature 478, 119–22 (2011). [DOI] [PubMed] [Google Scholar]
- Campoli C., Drosse B., Searle I., Coupland G. & von Korff M. Functional characterisation of HvCO1, the barley (Hordeum vulgare) flowering time ortholog of CONSTANS. Plant J 69, 868–80 (2012). [DOI] [PubMed] [Google Scholar]
- Gonzalez-Schain N. D., Diaz-Mendoza M., Zurczak M. & Suarez-Lopez P. Potato CONSTANS is involved in photoperiodic tuberization in a graft-transmissible manner. Plant J 70, 678–90 (2012). [DOI] [PubMed] [Google Scholar]
- Nakano Y., Higuchi Y., Yoshida Y. & Hisamatsu T. Environmental responses of the FT/TFL1 gene family and their involvement in flower induction in Fragaria × ananassa. Journal of Plant Physiology 177, 60–66 (2015). [DOI] [PubMed] [Google Scholar]
- Zhang R. et al. Molecular evolution and phylogenetic analysis of eight COL superfamily genes in group I related to photoperiodic regulation of flowering time in wild and domesticated cotton (Gossypium) species. PLoS One 10, e0118669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickland D. P. & Hanzawa Y. The FLOWERING LOCUS T/TERMINAL FLOWER 1 Gene Family: Functional Evolution and Molecular Mechanisms. Mol Plant, 10.1016/j.molp.2015.01.007 (2015). [DOI] [PubMed] [Google Scholar]
- Wang Z. et al. Functional evolution of phosphatidylethanolamine binding proteins in soybean and Arabidopsis. Plant Cell 27, 323–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43, 1035–9 (2011). [DOI] [PubMed] [Google Scholar]
- Li L., Stoeckert C. J. Jr. & Roos D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13, 2178–89 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou P. et al. Preferential retention of circadian clock genes during diploidization following whole genome triplication in Brassica rapa. Plant Cell 24, 2415–26 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q. et al. The draft genome of sweet orange (Citrus sinensis). Nat Genet 45, 59–66 (2013). [DOI] [PubMed] [Google Scholar]
- Wang Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res 40, e49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert V. A. et al. The Amborella Genome and the Evolution of Flowering Plants. Science 342, 1467−+ (2013). [DOI] [PubMed] [Google Scholar]
- Khanna R. et al. The Arabidopsis B-box zinc finger family. Plant Cell 21, 3416–20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F. et al. Deciphering the diploid ancestral genome of the Mesohexaploid Brassica rapa. Plant Cell 25, 1541–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse M. R. et al. Origin, inheritance, and gene regulatory consequences of genome dominance in polyploids. Proc Natl Acad Sci USA 111, 5283–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde F. CONSTANS and the evolutionary origin of photoperiodic timing of flowering. J Exp Bot 62, 2453–63 (2011). [DOI] [PubMed] [Google Scholar]
- Bustin S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55, 611–22 (2009). [DOI] [PubMed] [Google Scholar]
- Kawahara Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng F. et al. BRAD, the genetics and genomics database for Brassica plants. Bmc Plant Biology 11, 136 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn J. L., Lee J. M., Tearse B. R. & Neale D. B. TreeGenes: A forest tree genome database. Int J Plant Genomics 2008, 412875 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodstein D. M. et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40, D1178–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. H., Tang H., Wang X. & Paterson A. H. PGDD: a database of gene and genome duplication in plants. Nucleic Acids Res 41, D1152–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X. et al. Genome-wide identification, classification and expression analysis of the heat shock transcription factor family in Chinese cabbage. Mol Genet Genomics 289, 541–551 (2014). [DOI] [PubMed] [Google Scholar]
- Tamura K. et al. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Molecular Biology and Evolution 28, 2731–2739 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I., Doerks T. & Bork P. SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40, D302–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. L. et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B. et al. GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F. & Higgins D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25, 4876–82 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley L. A. & Sternberg M. J. E. Protein structure prediction on the Web: a case study using the Phyre server. Nature Protocols 4, 363–371 (2009). [DOI] [PubMed] [Google Scholar]
- Krzywinski M. et al. Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D., Zhang Y., Zhang Z., Zhu J. & Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8, 77–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M. A., Haubold B. & Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae). Mol Biol Evol 17, 1483–98 (2000). [DOI] [PubMed] [Google Scholar]
- Song X. M. et al. Genome-wide analysis of the bHLH transcription factor family in Chinese cabbage (Brassica rapa ssp. pekinensis). Mol Genet Genomics 289, 77–91 (2014). [DOI] [PubMed] [Google Scholar]
- Pfaffl M. W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.