

Abstract
Background
Salicylaldehyde isonicotinoyl hydrazone (SIH) is an iron chelator of the aroylhydrazone class that displays antioxidant or prooxidant effects in different mammalian cell lines. Because the liver is the major site of iron storage, elucidating the effect of SIH on hepatic oxidative metabolism is critical for designing effective hepatic antioxidant therapies.
Methods
Hepatocyte-like HepG2 cells were exposed to SIH or to analogs showing greater stability, such as N′-[1-(2-Hydroxyphenyl)ethyliden]isonicotinoyl hydrazide (HAPI), or devoid of iron chelating properties, such as benzaldehyde isonicotinoyl hydrazone (BIH), and toxicity, oxidative stress and antioxidant (glutathione) metabolism were evaluated.
Results
Autoxidation of Fe2+ in vitro increased in the presence of SIH or HAPI (but not BIH), an effect partially blocked by Fe2+ chelation. Incubation of HepG2 cells with SIH or HAPI (but not BIH) was non-toxic and increased reactive oxygen species (ROS) levels, activated the transcription factor Nrf2, induced the catalytic subunit of γ-glutamate cysteine ligase (Gclc), and increased glutathione concentration. Fe2+ chelation decreased ROS and inhibited Nrf2 activation, and Nrf2 knock-down inhibited the induction of Gclc in the presence of HAPI. Inhibition of γ-glutamate cysteine ligase enzymatic activity inhibited the increase in glutathione caused by HAPI, and increased oxidative stress.
Conclusions
SIH iron chelators display both prooxidant (increasing the autoxidation rate of Fe2+) and antioxidant (activating Nrf2 signaling) effects.
General significance
Activation by SIH iron chelators of a hormetic antioxidant response contributes to its antioxidant properties and modulates the anti- and pro-oxidant balance.
Keywords: Salicylaldehyde isonicotinoyl hydrazone, iron, oxidative stress, antioxidant, glutathione, Nrf2
1. Introduction
Salicylaldehyde isonicotinoyl hydrazone (SIH) is an iron chelator of the aroylhydrazone class that protects against toxicity in mammalian cells exposed to prooxidants in vitro. SIH prevented toxicity in: i) H9c2 rat cardiomyoblasts exposed to catecholamines [1], hydrogen peroxide [2] or tert-butylhydroperoxide [3]; A549 human lung adenocarcinoma cells exposed to hydrogen peroxide/ferrous iron [4]; iii) ARPE-19 human retinal pigment epithelial cells exposed to hydrogen peroxide [5]; and iv) primary rat cardiomyocytes exposed to ferrous sulfate or hydrogen peroxide [6]. SIH readily diffuses into cells by virtue of its hydrophobicity and low molecular weight, binds ferric iron in the labile iron pool (LIP, the transitory pool of cellular chelatable redox-active iron) with high affinity and prevents its reduction to ferrous iron [5,6]. Ferrous iron in the LIP can reduce hydrogen peroxide, molecular oxygen, and lipid hydroperoxides to toxic oxidizing free radicals (including hydroxyl radical, superoxide anion, and lipid alkoxyl radicals, respectively [8], collectively defined as reactive oxygen species (ROS). For this reason, the mechanism of SIH-induced cytoprotection is suggested to be the prevention of the formation of ferrous iron and ROS [5]. Experimental evidence to support this hypothesis includes the observation that SIH decreased the intracellular formation of ROS in H9c2 cells exposed to catecholamines (which reduce ferric to ferrous iron) [1], tert-butyl hydroperoxide [9] or ferric iron [3]. In addition, SIH decreased lipid oxidation in A549 cells exposed to hydrogen peroxide/ferrous iron [4], and in HepG2 human hepatoma cells exposed to cumene hydroperoxide [10].
In contrast, SIH increased oxidative stress in other cultured mammalian cells. Lipid peroxidation was induced in eicosapentaenoic acid-loaded K562 human leukemic cells treated with Fe(SIH)2 [11] and in A549 cells treated with SIH [4]. SIH showed a trend to increase the intracellular production of ROS in H9c2 cells [3,9]. Relatively high concentrations of SIH (≥ 200μM) increased the oxidation of the redox probe dichlorofluorescein in HepG2 cells treated with a peroxyl radical generator (2,2′-azobis(2-amidinopropane)) [10]. Oxidative stress induced by SIH was ascribed to redox cycling of SIH/iron complexes under cellular conditions, and/or mobilization of iron from intracellular stores by SIH, followed by donation of iron to cellular ligands which then redox-cycle [11]. These observations suggest that SIH can alter oxidative stress metabolism and the prooxidant-antioxidant balance depending on multiple factors including the cell type under study, available cellular reductants, chelator/iron relative concentrations and administration protocols (concentration and duration of exposure).
The liver is the major site of iron storage and pathologic iron accumulation, because hepatic LIP levels (that can promote production of ROS) increase when the capacity to maintain iron in storage forms is exceeded [12,13]. Therefore, elucidating the effect of iron chelators such as SIH on hepatic oxidative metabolism is critical for designing effective hepatic antioxidant therapies. The effect of SIH on oxidative stress metabolism in hepatocytes has not been evaluated before. The objective of this work was to investigate the effect of SIH on oxidative stress in HepG2 cells, a model hepatocyte cell line. HepG2 cells were exposed to SIH or analogs showing greater stability or devoid of iron chelating properties, and toxicity, oxidative stress and antioxidant (glutathione) metabolism were evaluated.
2. Materials and Methods
2.1. Chemicals
Fetal bovine serum was from Thermo Scientific Hyclone (Logan, UT). Western blot stripping buffer, Polybrene® and most antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). PCR primers were from Invitrogen (Carlsbad, CA) and Qiagen (Valencia, CA). SIH and analogs were synthesized as described below. Most of the other chemicals used were from Sigma-Aldrich (St Louis, MO).
2.2. Synthesis of SIH and analogs
1H NMR spectra were recorded on a 400 MHz JEOL spectrometer. Chemical shifts (δ) are reported in ppm, and J values are quoted in Hz. HPLC analyses of all synthesized compounds were performed on a Varian Microsorb MV 100 C18 column (Varian, Lake Forest, CA) with a mixture of 10 mM sodium phosphate buffer, 2 mM EDTA pH 6: methanol at a ratio of 55:45 as the mobile phase [3]. Purity of the compounds was assessed as the percentage surface area of the peaks at their UV maximum [14].
SIH
Equimolar quantities (1 mmol) of isoniazide and salicylaldehyde were dissolved in 2 mL of 0.1 M sodium acetate buffer (pH 4.5). The reaction was stirred for 5 min at 100 °C and cooled over ice. The white precipitate was collected via vacuum filtration, washed with water and dried in vacuo [15]. 1H NMR (DMSO-d6): δ 6.932 (1H, dd, J = 7.7, J = 7.7), 6.951 (1H, d, J = 7.8), 7.317 (1H, ddd, J = 1.4, J = 7.7, J = 7.8), 7.607 (1H, dd, J = 1.4, J = 7.7), 7.849 (2H, dd, J = 1.4, J = 4.4), 8.686 (1H, s), 8.801 (2H, dd, J = 1.4, J = 4.4), 11.083 (1H, s), 12.305 (1H, s). HPLC purity: 92% at 288 nm.
N′-[1-(2-Hydroxyphenyl)ethyliden]isonicotinoyl hydrazide (HAPI)
Equimolar quantities (3.9 mmol) of isoniazide and 2-hydroxyacetophenone were dissolved in 10 mL of 48% ethanol and 1 mL of acetic acid. The reaction was stirred under reflux for 9h. After cooling the reaction mixture to room temperature, 10 mL of water was added and the mixture was allowed to crystallize at 4°C. The product was collected by filtration, washed with water, and dried in vacuo [3]. 1H NMR (DMSO-d6): δ 2.458 (3H, s), 6.874 (1H, dd, J = 7.8, J = 7.8), 6.888 (1H, d, J = 8.0), 7.284 (1H, ddd, J = 1.4, J = 7.8, J = 8.0), 7.615 (1H, dd, J = 1.4, J = 7.8), 7.814 (2H, dd, J = 1.4, J = 4.5), 8.759 (2H, dd, J = 1.4, J = 4.5), 11.559 (1H, s), 13.167 (1H, s). HPLC purity: 96% at 288 nm.
Benzaldehyde isonicotinoyl hydrazone (BIH)
Equimolar quantities (1 mmol) of isoniazide and benzaldehyde were dissolved in 2 mL of 0.1 M sodium acetate buffer (pH 4.5). The reaction was stirred for 5 min at 100 °C and cooled over ice. The white precipitate was collected via vacuum filtration, washed with water and dried in vacuo [16]. 1H NMR (CDCl3): δ 7.447 (3H, m), 7.840 (2H, m), 7.895 (2H, dd, J = 1.6, J = 4.6), 8.365 (1H, s), 8.748 (2H, dd, J = 1.6, J = 4.6). HPLC purity: 100% at 301 nm.
2.3. Cell lines and cell culture
Human hepatoma HepG2 cells were used in this study. Cells were grown in Eagle’s minimal essential medium (MEM) containing 10% fetal bovine serum supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere in 5% CO2 at 37°C. Cells were subcultured at a 1:10 ratio once a week. Nrf2 knockdown HepG2 cells were created by lentivirus-mediated gene knockdown. Briefly, HepG2 cells were infected with Nrf2 shRNA (sc-37030-V) or control shRNA (sc-1080080) lentiviral particles (Santa Cruz Biotechnology, Santa Cruz, CA) in the presence of 5 μg/mL Polybrene®, and selected for puromycin resistance as indicated in the instructions provided by the manufacturer.
In selected experiments, rat hepatoma FaO cells were utilized. In this case, cell culture conditions were similar to that in HepG2 cells, except that F12 Ham’s medium was used instead of MEM.
Cells were plated at a density of 50,000 cells/mL in MEM containing 5% fetal bovine serum supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin in a humidified atmosphere in 5% CO2 at 37°C. After 12h of plating, the medium was replaced with MEM supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin without fetal bovine serum. Cells were preincubated with antioxidants or prooxidants for 1h, followed by the addition of SIH, HAPI, or BIH (added as a 500-fold concentrated stock solution in DMSO) at concentrations and time periods indicated in the text. Solvent control assays were performed with cells incubated in the presence of DMSO at 0.2% v/v. After this incubation, cells were used for biochemical analysis.
2.4. Fe2+ autoxidation in vitro
Fe2+ autoxidation was evaluated by following the decrease in Fe2+ and O2 concentration in a reaction mixture containing 2 mL Hepes 20 mM pH 7.0 and 50 μM FeSO4 in the presence or absence of SIH analogs at 100 μM. Ferrous iron concentration was determined by adding 20 uL of the reaction mixture to 180 μL of a 1 mM ferrozine solution at various time points (up to 5 minutes). After mixing, the absorbance at 564 nm was determined and converted into concentration of ferrous iron using an extinction coefficient of 27,900 M−1cm−1 [17]. O2 consumption in the reaction mixture was determined using a Clark-type O2 electrode (Hansatech Oxygraph, Hansatech Instruments Ltd, Norfolk, England).
Generation of hydrogen peroxide during Fe2+ autoxidation in vitro was determined using the Amplex Red Hydrogen Peroxide/Peroxidase Assay Kit from Life Technologies (Grand Island, NY). All of the reactions were performed according to the manufacturer’s protocol except that 20 mM Hepes buffer pH 7.0 was used instead of 50 mM phosphate pH 7.4 buffer (to avoid the interference of phosphate on iron chemistry) [18].
2.5. Cytotoxicity assay
Dead cells were distinguished from live cells by the uptake of propidium iodide (PI) by dead cells caused by loss of plasma membrane integrity. The incorporated probe forms fluorescent adducts with DNA. After the treatments, cells were trypsinized and resuspended in MEM supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin and 5 μg/mL of PI. Cellular fluorescence was measured in the FL3 channel of fluorescence for PI-DNA (red emission at 670±30 nm, with excitation at 488 nm). A minimum of 5,000 events per sample were acquired using an Accuri C6 flow cytometer. For quantification purposes, the percentage of cells displaying high fluorescence (M1%) represents the percentage of nonviable cells [19].
2.6. Intracellular reactive oxygen species determination
Intracellular reactive oxygen species were determined using the hydrophilic probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). DCFH-DA permeates across the cell membrane, is de-esterified by cytosolic esterases to 2′,7′-dichlorodihydrofluorescein (DCFH) which is oxidized to 2′,7′-dichlorofluorescein (DCF) by ROS. Oxidation is associated with an increase in green fluorescence [20]. After the treatment, cells were rinsed with MEM, and incubated in MEM supplemented with 100 units/mL penicillin, 100 μg/mL streptomycin with DCFH-DA at a concentration of 5 μM for 30 min. The cells were then washed with MEM, trypsinized, resuspended in MEM, and evaluated by flow cytometry in the Fl1 channel (green emission at 533±30 nm, with excitation at 488 nm). A minimum of 5,000 events per sample were acquired using an Accuri C6 flow cytometer. For quantification purposes, the average fluorescence in the FL1 channel represents the average level of cellular water-soluble ROS.
2.7. Total glutathione determination
Total glutathione was determined by the enzymatic method of Tietze [21]. At the end of the treatments the cells were washed twice with PBS, and detached by trypsinization in PBS, followed by low-speed centrifugation. The cell pellets were resuspended in potassium phosphate buffer 100 mM, pH 7.4. The protein concentration of the cell suspension was determined using the Bradford reagent (Sigma-Aldrich, St Louis, MO). The cell suspension was mixed 1:1 with 10% trichloroacetic acid to extract cellular glutathione. After centrifugation, total glutathione content was assayed by following the increase in absorbance at 412 nm for 1 min in a cuvette containing 0.1 M sodium phosphate buffer (pH 7.5), 5 μM EDTA, 0.6 mM 5,5′-dithiobis(2-nitrobenzoic acid), 0.2-mM NADPH, 1 unit/mL glutathione reductase, and 10 μL of sample in a final volume of 0.2 mL. The increment in absorbance at 412 nm was converted to glutathione concentration using a standard curve [22].
2.8. γ-Glutamate cysteine ligase (Gclc) mRNA quantification
Total RNA was extracted from cells using the RNeasy kit (Qiagen, Valencia, CA). RNA concentration was determined by UV absorbance at 260 nm using an extinction coefficient in TE buffer (10 mM Tris-HCl pH 7.5, EDTA 1 mM) of 0.025 (μg/ml)−1 cm−1. Purity was evaluated by the ratio of absorbance at 260 and 280 nm (always 1.8). Relative quantification of Gclc mRNA was done by real time- reverse transcriptase (RT) PCR. Real-time RT-PCR was performed by a one-step method with 10 ng of total RNA using QuantiFast SYBR Green RT-PCR Kit (Qiagen, Valencia, CA). The expression levels of the Gclc gene are presented as values normalized against β-actin as housekeeping gene. Specific human primers for Gclc and β-actin were from Invitrogen (Carlsbad, CA) [23]. Specific rat primers for Gclc (Rn_Gclc_1_SG) and β-actin (Rn_Actb_1_SG) were from Qiagen (Valencia, CA).
2.9. Western blots
After washing the cells with PBS, cells were incubated on ice for 15 min in RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) containing protease inhibitor cocktail at a 1:100 dilution (Sigma-Aldrich, St Louis, MO). Lysates were centrifuged at 8,000 × g at 4°C to remove insoluble material, and the protein concentration in whole-cell lysates was measured using the Bradford assay. Nuclear lysates and cytsolic fractions were prepared using a Nuclear Extraction Kit (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer’s protocol. Protein concentration in nuclear lysates and cytosolic fractions was measured using the Bradford assay. Protein samples (30 μg) were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and incubated with primary antibodies against Nrf2 (ab62352, Abcam) or GCLC (sc8030, Santa Cruz Biotechnology, Santa Cruz, CA) and a secondary antibody coupled to horseradish peroxidase. Blots were developed with a chemiluminescent reagent. After stripping the membranes, the blots were reprobed with primary antibodies against beta actin (sc81178, Santa Cruz Biotechnology, Santa Cruz, CA), histone H1 (sc8030, Santa Cruz Biotechnology, Santa Cruz, CA) or lactate dehydrogenase (sc33781, Santa Cruz Biotechnology, Santa Cruz, CA) as controls for equal loading in whole cell lysates, nuclear lysates or cytosolic fractions, respectively. Band intensities were calculated using ImageJ software.
2.10. Nrf2-DNA binding assay
The DNA binding of Nrf2 was measured with a colorimetric Nrf2 Transcription Factor Assay kit (Cayman Chemical Company, Ann Arbor, MI) according to the manufacturer’s instructions. Briefly, nuclear extracts (1–30 μg protein) were incubated in wells of a 96-well plate coated with dsDNA containing the Nrf2 response element. Then, wells were washed and incubated with an antibody against Nrf2. Addition of a secondary antibody conjugated to horseradish peroxidase followed by a developing reagent provided a colorimetric readout at 450 nm.
2.11. Determination of the binding of SIH iron chelators to the LIP
HepG2 cells were loaded with phen green SK diacetate (PG SK-DA) at a concentration of 20 μM for 10 min at 37 °C, in MEM supplemented with 100 units/mL penicillin and 100 μg/mL streptomycin without fetal bovine serum. The cells were then washed with MEM, trypsinized, resuspended in MEM, and evaluated by flow cytometry in the Fl1 channel (green emission at 533±30 nm, with excitation at 488 nm). A minimum of 5,000 events per sample were acquired using an Accuri C6 flow cytometer. PG SK-DA permeates across the cell membrane and is de-esterified by cytosolic esterases to fluorescent PG SK, which chelates intracellular labile iron. The chelation of intracellular labile iron by PG SK quenches its fluorescence. The presence of effective iron-chelating agents leads to the removal of iron from the PG SK-iron complex, and the de-quenching of PG SK fluorescence [24,25].
2.12. Statistics
Data are expressed as mean ± standard error of the mean from two to nine independent experiments run in duplicate. One way analysis of variance with subsequent post hoc comparison by Scheffe was performed using the SigmaStat 2.0 software. A p<0.05 was considered as statistically significant.
3. Results
3.1. Effect of SIH and analogs on viability and intracellular ROS levels in hepatoma cells
Viability and intracellular ROS levels were determined simultaneously in individual cells by flow cytometry using PI and DCFH-DA, respectively. Incubation of HepG2 cells with SIH or HAPI (an analog with greater stability against hydrolysis) [3] at 10 μM for 24h increased ROS levels with respect to control cells, without any decrease in cell viability (Figs. 1A and 1B). HAPI produced a greater increase in DCFH oxidation than SIH. Incubation of HepG2 cells with BIH (an analog devoid of iron chelating properties) [16] under the same time and concentration conditions did not produce any change in ROS levels or viability with respect to control cells (Figs. 1A and B). ROS levels in HepG2 cells incubated with 10 μM HAPI or SIH for shorter periods of time (i.e. 3h or 6h) were not different than that in control cells (data not shown).
Figure 1. Effect of SIH and analogs on viability and intracellular ROS levels in HepG2 cells.

HepG2 cells were incubated for 24h in the presence or absence of SIH analogs, and viability or ROS levels were determined with propidium iodide or DCFH-DA, respectively. In every panel, solvent control refers to cells incubated in 0.2% DMSO. A) ROS levels (as relative DCF fluorescence) in HepG2 cells incubated for 24h with 10 μM SIH, HAPI or BIH. Data are expressed as mean ± standard error of the mean of nine independent experiments. B) Cytotoxicity (as PI positive cells cells) in HepG2 cells incubated for 24h with 10 μM SIH, HAPI or BIH. Data are expressed as mean ± standard error of the mean of three independent experiments. C) ROS levels and cytotoxicity in cells incubated for 24h under increasing concentrations of HAPI (from 1.5 to 50 μM). Data are expressed as mean ± standard error of the mean of four independent experiments. D) ROS levels in HepG2 cells incubated for 24h in the presence or absence of SIH analogs at 10 μM, with or without preincubation for 1 h with anti- or pro-oxidants (DIP or BSO, respectively) at 1 mM. Data are expressed as mean ± standard error of the mean of four independent experiments.* p<0.05, ANOVA, with respect to solvent-control cells.
The increase in ROS levels caused by HAPI was concentration-dependent, with a maximum effect at 10 μM; higher concentrations up to 50 μM did not produce a further increase in ROS levels or loss of cellular viability (Fig. 1C). FaO rat hepatoma cells incubated with 10 μM HAPI under the same conditions as HepG2 cells showed a 1.6±0.1- fold significant increase in DCFH oxidation with respect to control FaO cells, without any change in cell viability (three independent experiments, data not shown). Biological free radical oxidations are mainly initiated by redox reactions between Fe2+ and O2 [26]. 2,2′-dipyridyl (DIP) is an Fe2+ chelator that inhibits its redox activity, therefore acting as a preventive antioxidant [27]. We hypothesized that if Fe2+ redox activity is required for ROS generation by HAPI in HepG2 cells, then co-incubation with HAPI and DIP should decrease ROS generation as assessed by DCFH-DA oxidation. Coincubation of HepG2 cells with HAPI and DIP blocked the increase in DCFH-DA oxidation caused by HAPI alone (Fig. 1D). In turn, pro-oxidant conditions in HepG2 cells were induced by depletion of glutathione with L-buthionine sulfoximine (BSO). BSO induced a significant increase in DCFH-DA oxidation in HepG2 cells; the combination of BSO and HAPI produced a synergistic increase in DCFH-DA oxidation with respect to these agents acting alone (Fig. 1D).
In order to confirm the binding of SIH iron chelators to the LIP in HepG2 cells, the de-quenching by SIH iron chelators of preloaded PG SK was evaluated. PG SK fluorescence was significantly quenched in HepG2 cells exposed to a cell-permeant form of iron (iron-hydroxyquinoline), demonstrating the quenching effect of iron on PG SK fluorescence (Table 1). SIH, HAPI, and DIP (a well-characterized iron chelator that binds ferrous iron in the LIP [28]) significantly increased PG SK fluorescence in HepG2 cells, while BIH was inactive (Table 1). These results confirm the effective binding of SIH iron chelators to the LIP in HepG2 cells.
Table 1.
Binding of SIH iron chelators to the cellular labile iron pool. HepG2 cells were incubated for 1h in the presence of 1:1 ferric iron-hydroxyquinoline (5 μM Fe-HQ), or for 24h in the presence of 1 mM DIP, 50 μM HAPI, 50 μM SIH, 50 μM BIH or solvent (0.2% DMSO). Cells were then loaded with 20 μM PG SK-DA for 10 min. Fluorescence of PG SK was evaluated by flow cytometry. Data are expressed as mean ± standard error of the mean of four independent experiments.
| Treatment | PG SK Fluorescence (AU) |
|---|---|
| Solvent Control | 1.0 ± 0.1 |
| Fe-HQ | 0.3 ± 0.1* |
| DIP | 1.6 ± 0.1* |
| HAPI | 1.5 ± 0.1* |
| SIH | 1.4 ± 0.1* |
| BIH | 1.0 ± 0.1 |
p<0.05, ANOVA, with respect to solvent-control cells.
3.2. Effect of SIH and analogs on ferrous iron autoxidation
The observation that ROS generation in HepG2 cells incubated with HAPI was prevented by an Fe2+ chelator that blocks its redox activity suggested that HAPI promotes Fe2+ autoxidation and generation of ROS as by-products. To test this hypothesis, we evaluated the effect of SIH and analogs on Fe2+ autoxidation in vitro. The autoxidation of Fe2+ in the presence or absence of HAPI is shown in Fig. 2. A rapid decrease in Fe2+ concentration was observed upon addition of 100 μM HAPI to a reaction system containing 20 mM Hepes buffer pH 7.0 and 50 μM FeSO4; this decrease was not observed under the same conditions but in the absence of HAPI (Fig. 2A). A similar decrease in Fe2+ concentration was observed with 100 μM SIH, but not with 100 μM BIH (Fig. 2A). Oxygen consumption in a reaction system containing 20 mM Hepes buffer pH 7.0, 100 μM HAPI (or 100 μM SIH) and 50 μM FeSO4 correlated with Fe2+ disappearance (Fig. 2B), with a stoichiometry at reaction completion of 2:1 Fe2+:O2 in the presence of HAPI (or 2.8:1 in the presence of SIH). In addition, Fe2+-dependent O2 consumption induced by HAPI was significantly blocked by DIP (Fig. 2B). Any stoichiometry lower than 4:1 Fe2+:O2 has been suggested to indicate the generation of partially reduced oxygen species [29]; in particular, a stoichiometry of 2:1 Fe2+:O2 suggests the generation of hydrogen peroxide. The possible generation of hydrogen peroxide in the autoxidation medium was evaluated using two different approaches. First, if hydrogen peroxide is generated as a product of ferrous iron autoxidation, then autoxidation in the presence of catalase should change the stoichiometry from 2:1 Fe2+:O2 to 4:1 Fe2+:O2. Fe2+ autoxidation in the presence of HAPI and catalase decreased O2 consumption by half (with respect to that in the absence of catalase) (Fig. 2B), producing a stoichiometry at reaction completion of 4:1 Fe2+:O2. Second, the generation of H2O2 in the presence of Fe2+ and SIH analogs was evaluated using amplex red/peroxidase. Autoxidation of Fe2+ by itself did not produce significant amounts of hydrogen peroxide (Fig. 2C). Autoxidation of Fe2+ in the presence of HAPI or SIH (but not BIH) increased the generation of hydrogen peroxide, an effect inhibited by catalase (Fig. 2C).
Figure 2. Effect of SIH and analogs on ferrous iron autoxidation.
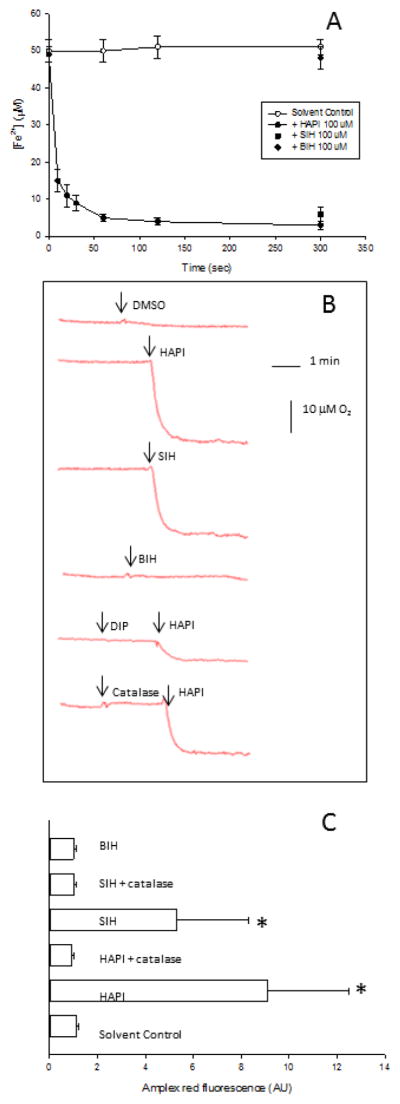
Solutions of Fe2+ (50 μM) were prepared in 20 mM Hepes buffer pH 7.0. (A) The time course of Fe2+ consumption was evaluated in the presence of 100 μM HAPI (black circles), 100 μM SIH (black squares), 100 μM BIH (black diamonds) or 0.2% DMSO (solvent control, white circles). Data are expressed as mean ± standard error of the mean of four independent experiments. (B) The time course of O2 consumption was evaluated after adding 0.2% DMSO (solvent control), 100 μM SIH, 100 μM BIH or 100 μM HAPI in the absence or presence of DIP or catalase. A representative curve out of five is shown for each condition. (C) H2O2 generation was evaluated as amplex red fluorescence after 5 min of incubation in the presence of 0.2% DMSO (solvent control), 100 μM BIH, 100 μM SIH or 100 μM HAPI in the presence or absence of catalase. Data are expressed as mean ± standard error of the mean of three independent experiments.
* p<0.05, ANOVA, with respect to solvent-control cells.
3.3. Effect of SIH and analogs on glutathione metabolism in hepatoma cells
Previous experiments showed that SIH iron chelators induced oxidative stress, which is defined as a state of increased steady-state levels of reactive oxygen species. Oxidative stress can be caused by an increase in the generation rate of prooxidants and/or a decrease in antioxidant levels. In order to evaluate the effect of SIH analogs on cellular antioxidant levels, the concentration of glutathione, the most abundant water-soluble antioxidant in mammalian cells, was evaluated. HAPI and SIH produced a significant increase in total glutathione levels in HepG2 cells, while BIH was ineffective (Fig. 3A). BSO depleted total glutathione in HepG2 cells, an effect that was not reversed by HAPI (Fig. 3A). Sustained increases in glutathione content are controlled primarily through induction of two genes, Gclc (coding for the catalytic subunit of glutamate cysteine ligase or GCLC) and Gclm (coding for the modifier subunit of glutamate cysteine ligase or GCLM), leading to the synthesis of the rate limiting enzyme for GSH synthesis, glutamate cysteine ligase (GCL) [30]. The effect of SIH analogs on the mRNA and protein expression of the catalytic subunit of GCL was evaluated. The expression at the mRNA and protein levels of the catalytic subunit of GCL increased by incubation of HepG2 cells with SIH or HAPI, but not with BIH (Figs. 3 B and C).
Figure 3. Effect of SIH and analogs on glutathione metabolism in HepG2 cells.

HepG2 cells were incubated for 24h in the presence or absence of SIH analogs at 50 μM. In every panel, solvent control refers to cells incubated in 0.2% DMSO. (A) Total glutathione was determined in HepG2 cells by a recycling enzymatic assay. Data are expressed as mean ± standard error of the mean of six independent experiments. (B) Gclc mRNA was determined in RNA extracts by reverse transcriptase-RT PCR. Data are expressed as mean ± standard error of the mean of four independent experiments. (C) GCLC protein levels were determined by western blot in whole cell lysates. Numbers below the representative blots are the relative mean GCLC band intensity normalized by beta-actin (± standard error of the mean of four independent samples). A representative western blot is shown.
* p<0.05, ANOVA, with respect to solvent-control cells.
FaO cells incubated with 50 μM HAPI under the same conditions as HepG2 cells did not show any significant change in total glutathione content or Gclc mRNA levels with respect to FaO control cells (three independent experiments, data not shown).
3.4. Effect of SIH and analogs on Nrf2 metabolism in HepG2 cells
Many studies have demonstrated that factors leading to changes in the glutathione content of cells are mediated predominantly by antioxidant response element (ARE)-dependent expression of the Gcl genes. The ARE is the cis-acting sequence responsible for Nrf2-dependent regulation of gene expression [31]. Nrf2 activation was assessed by nuclear translocation and binding to an ARE oligonucleotide. Translocation of Nrf2 to the nucleus, evaluated by western blotting analysis of Nrf2 in the nuclear and cytosolic fractions, increased by incubation of HepG2 cells with SIH or HAPI, but not BIH (Fig. 4A). The antibody used only detected the biologically relevant species of Nrf2 that migrates between 95 and 110 kDa in SDS-PAGE [32]. The binding activity of Nrf2 to an ARE consensus oligonucleotide, evaluated by ELISA in nuclear extracts, increased by incubation of HepG2 cells with SIH or HAPI, but not BIH (Fig. 4B). An antioxidant such as DIP blocked the effect of HAPI on the activation of Nrf2, while a prooxidant such as BSO did not have an effect on the increased HAPI-dependent Nrf2 activation (Fig. 4B). We hypothesized that if Nrf2 is important for Gclc induction in the presence of SIH analogs, then knocking-down Nrf2 with specific shRNAs should prevent this effect. Nrf2 knockdown cells were generated by transfection of HepG2 cells with lentiviral particles expressing shRNA directed against the Nrf2 gene, followed by puromycin selection. Delivery of shRNA lentiviral constructs into the cells was monitored by transfection of cells with copGFP control lentiviral particles followed by puromycin selection; 99.4% of the transfected cells showed high fluorescence by flow cytometry in the green (Fl1) channel of fluorescence, with respect to cells transfected with control shRNA lentiviral particles (Fig. 5A). In addition, Nrf2 knock-down in cells transfected with Nrf2 shRNA lentiviral particles was confirmed by western blot for Nrf2 in total lysates (Fig. 5A insert). HAPI induced the expression of Gclc at the mRNA and protein levels in control cells; this effect was significantly inhibited in Nrf2 knock-down cells (Figs. 5B and C). Expression of Gclc at the mRNA and protein levels significantly decreased in Nrf2 knock-down cells with respect to control cells, suggesting that basal Gclc expression is Nrf2-dependent in HepG2 cells (Figs. 5B and C).
Figure 4. Effect of SIH and analogs on Nrf2 metabolism in HepG2 cells.

In every panel, solvent control refers to cells incubated in 0.2% DMSO. (A) HepG2 cells were incubated for 24h in the presence or absence of SIH analogs at 50 μM. Nrf2 protein levels were assessed by western blot in nuclear lysates and cytosolic fractions. Numbers below the representative blots are the relative mean Nrf2 band intensity in each compartment normalized by its loading control (± standard error of the mean of three independent samples). (B) HepG2 cells were incubated for 24h in the presence or absence of SIH analogs at 50 μM, with or without preincubation for 1 h with anti- or pro-oxidants (DIP or BSO, respectively). Nuclear Nrf2 binding was assessed by binding of nuclear Nrf2 to an ARE oligonucleotide by ELISA. Data are expressed as mean ± standard error of the mean of three independent experiments.
* p<0.05, ANOVA, with respect to solvent-control cells.
Figure 5. Effect of SIH and analogs on Gclc expression in Nrf2 knock-down HepG2 cells.

(A) Flow cytometry histogram of HepG2 cells transfected with control lentiviral particles (left) or copGFP lentiviral particles (right), followed by puromycin selection. M1 represents the percentage of copGFP-transfected cells expressing high green (Fl1) fluorescence. Insert: Nrf2 protein levels in HepG2 cells transfected with control or Nrf2 shRNA lentiviral particles, followed by puromycin selection, evaluated by western blot of whole cell lysates. One representative plot out of two independent samples is shown. (B) Gclc mRNA was determined in RNA extracts by reverse transcriptase-RT PCR in HepG2 cells transfected with control or Nrf2 shRNA lentiviral particles, followed by puromycin selection, and incubated for 24h in the presence or absence of HAPI at 50 μM. Data are expressed as mean ± standard error of the mean of three independent samples. Solvent control refers to cells incubated in 0.2% DMSO. (C) GCLC protein levels were determined by western blot in whole cell lysates from HepG2 cells transfected with control or Nrf2 shRNA lentiviral particles, followed by puromycin selection, and incubated for 24h in the presence or absence of HAPI at 50 μM. Numbers below the representative blots are the relative mean GCLC band intensity normalized by its loading control (± standard error of the mean of three independent samples). Solvent control refers to cells incubated in 0.2% DMSO.
* p<0.05, ANOVA, with respect to solvent-control cells.
4. Discussion
The redox cycling of iron in the presence of oxygen generates ROS and induces oxidative stress [20]. SIH is usually classified as a preventive antioxidant, because SIH typically binds Fe3+ with high affinity, a reaction that prevents the reduction of iron by cellular reductants, inhibiting the generation of ROS [5,15,16]. However, SIH and its stable analog HAPI actually increased ROS levels and produced a more oxidized redox state in HepG2 and FaO cells, as determined by increased oxidation of DCFH, a redox indicator probe that responds to oxidants generated by redox-active iron [33]. We propose that SIH (and its stable analog HAPI) promote the generation of ROS by chelating Fe3+ and increasing the autoxidation rate of Fe2+. The experimental evidence to support this conclusion is the following: 1) SIH and HAPI promote the autoxidation of ferrous iron, as evidenced by the rapid disappearance of Fe2+ and O2 in solutions containing these iron chelators; 2) The Fe2+:O2 stoichiometric ratios at completion of the autoxidation reaction was 2:1 in the presence of HAPI and 2.8:1 in the presence of SIH; any ratio lower than 4:1 suggests the generation of partially reduced oxygen species [29]; 3) The Fe2+:O2 stoichiometric ratios at completion of the autoxidation reaction was 4:1 in the presence of HAPI and catalase. This conversion suggests the generation of H2O2 as a product of iron autoxidation [29]; 4) SIH and HAPI promoted the generation of H2O2 as assessed by the oxidation of the redox probe amplex red (in the presence of peroxidase). This oxidation was inhibited by catalase, further demonstrating the generation of H2O2; 5) in vitro, DIP blocked the autoxidation of Fe2+ in the presence of HAPI, and in HepG2 cells, DIP blocked ROS generation in the presence of HAPI, suggesting that O2 consumption and ROS generation by HAPI is an Fe2+-dependent redox reaction; 6) an SIH analog devoid of iron chelating properties did not increase Fe2+ autoxidation or H2O2 generation in vitro, and did not increase cellular ROS in HepG2 cells, suggesting that Fe3+ chelation is essential for SIH and HAPI prooxidant activity.
SIH and HAPI in HepG2 cells increased the concentration of the main cellular water-soluble antioxidant, glutathione. Our results suggest that SIH iron chelators increase the concentration of cellular glutathione by transcriptional activation of the Gclc gene via ROS-dependent activation of the transcription factor Nrf2. Experimental results that support this conclusion are the following: 1) Increased glutathione occurred together with oxidative stress, Nrf2 activation, and induction of the expression of Gclc, suggesting a correlation between these events; 2) An antioxidant such as DIP inhibited DCFH oxidation and Nrf2 activation in the presence of HAPI, suggesting that ROS induces Nrf2 activation in HAPI-treated cells; 3) Nrf2 knock-down with shRNAs inhibited the induction of Gclc in the presence of HAPI, suggesting that Nrf2 induces Gclc in HAPI-treated cells; 4) inhibition of GCL enzymatic activity with BSO inhibited the increase in glutathione caused by HAPI, suggesting that HAPI increases GSH content by increasing GCL activity. On the contrary, HAPI did not affect glutathione metabolism in FaO cells, suggesting that activation of glutathione synthesis is not a general response of hepatoma cells to SIH iron chelators. The differential effect of HAPI on glutathione metabolism in HepG2 and FaO cells might depend on different cellular redox status, LIP levels, or degree of activation of signaling pathways.
What is the biological effect of the increased GSH caused by HAPI? Our results suggest that increased GSH counteracted the increased generation of ROS by HAPI, providing a partial antioxidant protection. The main experimental evidence to support this conclusion is the fact that conditions that depleted GSH (i.e. BSO) increased ROS in the presence of HAPI. In spite of increased ROS in the presence of HAPI and BSO, nuclear Nrf2 binding activity in cells incubated with HAPI in the presence or absence of BSO was comparable, probably because HAPI by itself induced maximal nuclear Nrf2 binding.
Oxidative hormesis is defined as a process in which exposure to a low dose of an oxidant induces an adaptive antioxidant protective response [34]. While SIH is usually classified as a preventive antioxidant, our results suggest that SIH iron chelators can also display antioxidant effects via an oxidative hormesis effect mediated by Nrf2 activation. Reports in the literature show that other molecules besides SIH iron chelators can induce oxidative hormesis via Nrf2 in mammalian cells, suggesting a general antioxidant mechanism. For example, oxidative hormesis effects induced by Nrf2 were observed in murine fibrosarcoma L929 cells exposed to H2O2 [35], in human endothelial EA.hy926 cells exposed to docosahexaenoic acid [36], and in human lung fibroblast HELF cells exposed to polychlorinated biphenyls [37].
Antioxidants are broadly defined as agents that decrease steady-state ROS levels [38]. Steady state ROS levels are determined by the balance between the rates of ROS generation and ROS elimination. Our results show that SIH iron chelators can affect both sides of this balance in HepG2 cells, both increasing ROS production and inducing antioxidant protection. Previous reports informed net anti- or pro-oxidant effects of SIH iron chelators in different cellular models [3,4,9,10]. Our results suggest that whether SIH iron chelators display net anti- or pro-oxidant effects depends on the effect of SIH iron chelators on the prooxidant/antioxidant balance, which in turn depends on variable cellular factors including the degree of activation of antioxidant pathways, cellular redox state and LIP levels.
The cells used in our study are hepatoma cells derived from human (HepG2) or rat (FaO) liver carcinoma. The cancer phenotype is associated with high iron requirements resulting in high oxidative stress. Because of this, it has been proposed that the cancer phenotype can be selectively targeted by decreasing the LIP through iron depletion and/or by increasing ROS levels [39]. Other iron chelators such as 3-aminopyridine-2- carboxaldehyde (Triapine®) and deferoxamine have anti-cancer effects associated with LIP binding and ROS induction [40, 41]. Our results indicate that SIH iron chelators might constitute an effective therapy against liver cancer, because SIH iron chelators both bind iron in the LIP and induce oxidative stress in hepatoma cells. Nrf2 activation and Gclc induction in HepG2 human hepatoma cells (but not FAO rat hepatoma cells) exposed to SIH iron chelators might represent a resistance mechanism developed specifically in HepG2 cells to overcome the increased oxidative stress and promote survival. In this case, the combination of SIH iron chelators and inhibition of Nrf2 pathways might prove an effective therapy against human liver cancer.
5. Conclusion
In conclusion, we propose that SIH iron chelators display both prooxidant and antioxidant properties at low concentrations in HepG2 cells. Prooxidant properties are determined by their ability to increase ferrous iron autoxidation rate, and antioxidant properties are determined by their ability to prevent ferric iron reduction and increase the concentration of cellular glutathione via the ROS-dependent activation of Nrf2. Activation by SIH iron chelators of a hormetic antioxidant response therefore contributes to its antioxidant properties. Net anti- or pro- oxidant effects of SIH iron chelators depend on its overall effects on the anti- and pro- oxidant balance.
Highlights.
SIH increased ferrous iron autoxidation and ROS generation in HepG2 cells.
SIH increased total glutathione and induced GCLC in HepG2 cells.
GCLC induction by SIH in HepG2 cells depended on Nrf2 activation.
Activation by SIH of antioxidant signaling contributes to its antioxidant properties.
SIH modulates the anti- / pro-oxidant balance by both anti- and pro-oxidant effects.
Acknowledgments
The study was supported by grants from the National Center for Research Resources (5P20RR16460-11) and the National Institute of General Medical Sciences (8 P20 GM103429-11).
Abbreviations
- BIH
benzaldehyde isonicotinoyl hydrazone
- Gclc
catalaytic subunit of γ-glutamate cysteine ligase
- DCFH-DA
dichlorodihydrofluorescein diacetate
- DIP
2,2′-dipyridyl
- Gcl
γ-glutamate cysteine ligase
- HAPI
N′-[1-(2-Hydroxyphenyl)ethyliden]isonicotinoyl hydrazide
- LIP
labile iron pool
- MEM
minimal essential medium
- PG SK-DA
Phen Green SK diacetate
- ROS
reactive oxygen species
- SIH
salicylaldehyde isonicotinoyl hydrazone
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hašková P, Kovaříková P, Koubková L, Vávrová A, Macková E, Šimůnek T. Iron chelation with salicylaldehyde isonicotinoyl hydrazone protects against catecholamine autoxidation and cardiotoxicity. Free Radical Biology and Medicine. 2011;50(4):537–549. doi: 10.1016/j.freeradbiomed.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Šimůnek T, Boer C, Bouwman RA, Vlasblom R, Versteilen AM, Štěrba M, Musters RJ. SIH—a novel lipophilic iron chelator—protects H9c2 cardiomyoblasts from oxidative stress-induced mitochondrial injury and cell death. Journal of molecular and cellular cardiology. 2005;39(2):345–354. doi: 10.1016/j.yjmcc.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Hruskova K, Kovarikova P, Bendova P, Haskova P, Mackova E, Stariat J, Simunek T. Synthesis and initial in vitro evaluations of novel antioxidant aroylhydrazone iron chelators with increased stability against plasma hydrolysis. Chemical research in toxicology. 2011;24(3):290–302. doi: 10.1021/tx100359t. [DOI] [PubMed] [Google Scholar]
- 4.Kaiserova H, Hartog GJM, Šimůnek T, Schröterová L, Kvasničková E, Bast A. Iron is not involved in oxidative stress-mediated cytotoxicity of doxorubicin and bleomycin. British journal of pharmacology. 2006;149(7):920–930. doi: 10.1038/sj.bjp.0706930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Charkoudian LK, Dentchev T, Lukinova N, Wolkow N, Dunaief JL, Franz KJ. Iron prochelator BSIH protects retinal pigment epithelial cells against cell death induced by hydrogen peroxide. Journal of inorganic biochemistry. 2008;102(12):2130–2135. doi: 10.1016/j.jinorgbio.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kakhlon O, Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radical Biology and Medicine. 2002;33(8):1037–1046. doi: 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 7.Šimůnek T, Štĕrba M, Popelova O, Kaiserova H, Adamcova M, Hroch M, Geršl V. Anthracycline toxicity to cardiomyocytes or cancer cells is differently affected by iron chelation with salicylaldehyde isonicotinoyl hydrazone. British journal of pharmacology. 2008;155(1):138–148. doi: 10.1038/bjp.2008.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caro AA, Cederbaum AI. Antioxidant properties of S-adenosyl-l-methionine in Fe 2+-initiated oxidations. Free Radical Biology and Medicine. 2004;36(10):1303–1316. doi: 10.1016/j.freeradbiomed.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Bendova P, Mackova E, Haskova P, Vavrova A, Jirkovsky E, Sterba M, Simunek T. Comparison of clinically used and experimental iron chelators for protection against oxidative stress-induced cellular injury. Chemical research in toxicology. 2010;23(6):1105–1114. doi: 10.1021/tx100125t. [DOI] [PubMed] [Google Scholar]
- 10.Hofer T, Jørgensen TØ, Olsen RL. Comparison of food antioxidants and iron chelators in two cellular free radical assays: strong protection by luteolin. Journal of agricultural and food chemistry. 2014;62(33):8402–8410. doi: 10.1021/jf5022779. [DOI] [PubMed] [Google Scholar]
- 11.Buss JL, Neuzil J, Ponka P. Oxidative stress mediates toxicity of pyridoxal isonicotinoyl hydrazone analogs. Archives of biochemistry and biophysics. 2004;421(1):1–9. doi: 10.1016/j.abb.2003.09.044. [DOI] [PubMed] [Google Scholar]
- 12.Glickstein H, El RB, Shvartsman M, Cabantchik ZI. Intracellular labile iron pools as direct targets of iron chelators: a fluorescence study of chelator action in living cells. Blood. 2005;106(9):3242–3250. doi: 10.1182/blood-2005-02-0460. [DOI] [PubMed] [Google Scholar]
- 13.González PM, Piloni NE, Puntarulo S. Iron overload and lipid peroxidation in biological systems. INTECH Open Access Publisher; 2012. [Google Scholar]
- 14.Fernandez AM, Van derpoorten K, Dasnois L, Lebtahi K, Dubois V, Lobl TJ, Trouet A. N-Succinyl-(β-alanyl-l-leucyl-l-alanyl-l-leucyl) doxorubicin: an extracellularly tumor-activated prodrug devoid of intravenous acute toxicity. Journal of medicinal chemistry. 2001;44(22):3750–3753. doi: 10.1021/jm0108754. [DOI] [PubMed] [Google Scholar]
- 15.Charkoudian LK, Pham DM, Franz KJ. A pro-chelator triggered by hydrogen peroxide inhibits iron-promoted hydroxyl radical formation. Journal of the American Chemical Society. 2006;128(38):12424–12425. doi: 10.1021/ja064806w. [DOI] [PubMed] [Google Scholar]
- 16.Charkoudian LK, Pham DM, Kwon AM, Vangeloff AD, Franz KJ. Modifications of boronic ester pro-chelators triggered by hydrogen peroxide tune reactivity to inhibit metal-promoted oxidative stress. Dalton Transactions. 2007;(43):5031–5042. doi: 10.1039/b705199a. [DOI] [PubMed] [Google Scholar]
- 17.Galey JB, Destrée O, Dumats J, Pichaud P, Marché J, Génard S, Cantoni O. Protection of U937 cells against oxidative injury by a novel series of iron chelators. Free Radical Biology and Medicine. 1998;25(8):881–890. doi: 10.1016/s0891-5849(98)00113-0. [DOI] [PubMed] [Google Scholar]
- 18.Bou-Abdallah F, Lewin AC, Le Brun NE, Moore GR, Chasteen ND. Iron Detoxification Properties of Escherichia coliBacterioferritin ATTENUATION OF OXYRADICAL CHEMISTRY. Journal of Biological Chemistry. 2002;277(40):37064–37069. doi: 10.1074/jbc.M205712200. [DOI] [PubMed] [Google Scholar]
- 19.Caro AA, Cederbaum AI. Ca 2+-dependent and independent mitochondrial damage in HepG2 cells that overexpress CYP2E1. Archives of biochemistry and biophysics. 2002;408(2):162–170. doi: 10.1016/s0003-9861(02)00544-1. [DOI] [PubMed] [Google Scholar]
- 20.Caro AA, Cederbaum AI. Role of calcium and calcium-activated proteases in CYP2E1-dependent toxicity in HEPG2 cells. Journal of Biological Chemistry. 2002;277(1):104–113. doi: 10.1074/jbc.M107864200. [DOI] [PubMed] [Google Scholar]
- 21.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Analytical biochemistry. 1969;27(3):502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 22.Marí M, Cederbaum AI. CYP2E1 overexpression in HepG2 cells induces glutathione synthesis by transcriptional activation of γ-glutamylcysteine synthetase. Journal of Biological Chemistry. 2000;275(20):15563–15571. doi: 10.1074/jbc.M907022199. [DOI] [PubMed] [Google Scholar]
- 23.Arab K, Rossary A, Flourié F, Tourneur Y, Steghens JP. Docosahexaenoic acid enhances the antioxidant response of human fibroblasts by upregulating γ-glutamyl-cysteinyl ligase and glutathione reductase. British journal of nutrition. 2006;95(01):18–26. doi: 10.1079/bjn20051626. [DOI] [PubMed] [Google Scholar]
- 24.Petrat F, Rauen U, de Groot H. Determination of the chelatable iron pool of isolated rat hepatocytes by digital fluorescence microscopy using the fluorescent probe, phen green SK. Hepatology. 1999;29(4):1171–1179. doi: 10.1002/hep.510290435. [DOI] [PubMed] [Google Scholar]
- 25.Du J, Wagner BA, Buettner GR, Cullen JJ. Role of labile iron in the toxicity of pharmacological ascorbate. Free Radical Biology and Medicine. 2015;84:289–295. doi: 10.1016/j.freeradbiomed.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian SY, Buettner GR. Iron and dioxygen chemistry is an important route to initiation of biological free radical oxidations: an electron paramagnetic resonance spin trapping study. Free radical biology and medicine. 1999;26(11):1447–1456. doi: 10.1016/s0891-5849(99)00002-7. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Dai J, Fournier J, Ali AM, Zhang Q, Frenkel K. Ferrous ion autoxidation and its chelation in iron-loaded human liver HepG2 cells. Free Radical Biology and Medicine. 2002;32(1):84–92. doi: 10.1016/s0891-5849(01)00770-5. [DOI] [PubMed] [Google Scholar]
- 28.Stäubli A, Boelsterli UA. The labile iron pool in hepatocytes: prooxidant-induced increase in free iron precedes oxidative cell injury. American Journal of Physiology-Gastrointestinal and Liver Physiology. 1998;274(6):G1031–G1037. doi: 10.1152/ajpgi.1998.274.6.G1031. [DOI] [PubMed] [Google Scholar]
- 29.Welch KD, Davis TZ, Aust SD. Iron autoxidation and free radical generation: effects of buffers, ligands, and chelators. Archives of biochemistry and biophysics. 2002;397(2):360–369. doi: 10.1006/abbi.2001.2694. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP. Glutamate Cysteine ligase catalysis dependence on ATP and modifier subunit for regulation of tissue glutathione levels. Journal of Biological Chemistry. 2005;280(40):33766–33774. doi: 10.1074/jbc.M504604200. [DOI] [PubMed] [Google Scholar]
- 31.Levy S, Jaiswal AK, Forman HJ. The role of c-Jun phosphorylation in EpRE activation of phase II genes. Free Radical Biology and Medicine. 2009;47(8):1172–1179. doi: 10.1016/j.freeradbiomed.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lau A, Tian W, Whitman SA, Zhang DD. The predicted molecular weight of Nrf2: it is what it is not. Antioxidants & redox signaling. 2013;18(1):91–93. doi: 10.1089/ars.2012.4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radical Biology and Medicine. 2012;52(1):1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luna–López A, González-Puertos VY, López-Diazguerrero NE, Königsberg M. New considerations on hormetic response against oxidative stress. Journal of cell communication and signaling. 2014;8(4):323–331. doi: 10.1007/s12079-014-0248-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luna-López A, Triana-Martínez F, López-Diazguerrero NE, Ventura-Gallegos JL, Gutiérrez-Ruiz MC, Damián-Matsumura P, Königsberg M. Bcl-2 sustains hormetic response by inducing Nrf-2 nuclear translocation in L929 mouse fibroblasts. Free Radical Biology and Medicine. 2010;49(7):1192–1204. doi: 10.1016/j.freeradbiomed.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 36.Yang YC, Lii CK, Wei YL, Li CC, Lu CY, Liu KL, Chen HW. Docosahexaenoic acid inhibition of inflammation is partially via cross-talk between Nrf2/heme oxygenase 1 and IKK/NF-κB pathways. The Journal of nutritional biochemistry. 2013;24(1):204–212. doi: 10.1016/j.jnutbio.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 37.Hashmi MZ, Khan KY, Hu J, Su X, Abbas G, Yu C, Shen C. Hormetic effects of noncoplanar PCB exposed to human lung fibroblast cells (HELF) and possible role of oxidative stress. Environmental toxicology. 2014 doi: 10.1002/tox.22008. [DOI] [PubMed] [Google Scholar]
- 38.Day BJ. Antioxidants as potential therapeutics for lung fibrosis. Antioxidants & redox signaling. 2008;10(2):355–370. doi: 10.1089/ars.2007.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bystrom LM, Rivella S. Cancer cells with irons in the fire. Free Radical Biology and Medicine. 2015;79:337–342. doi: 10.1016/j.freeradbiomed.2014.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Callens C, Coulon S, Naudin J, Radford-Weiss I, Boissel N, Raffoux E, Hermine O. Targeting iron homeostasis induces cellular differentiation and synergizes with differentiating agents in acute myeloid leukemia. The Journal of experimental medicine. 2010;207(4):731–750. doi: 10.1084/jem.20091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Myers JM, Antholine WE, Zielonka J, Myers CR. The iron-chelating drug triapine causes pronounced mitochondrial thiol redox stress. Toxicology letters. 2011;201(2):130–136. doi: 10.1016/j.toxlet.2010.12.017. [DOI] [PubMed] [Google Scholar]