

Abstract
Arsenic is a known carcinogen to humans, and chronic exposure to environmental arsenic is a worldwide health concern. As a dietary factor, ethanol carries a well-established risk for malignancies, but the effects of co-exposure to arsenic and ethanol on tumor development are not well understood. In the present study, we hypothesized that ethanol would enhance the function of an environmental carcinogen such as arsenic through increase in COX-2 expression. Our in vitro results show that ethanol enhanced arsenic-induced COX-2 expression. We also show that the increased COX-2 expression associates with intracellular ROS generation, up-regulated AKT signaling, with activation of both NFAT and NF-κB pathways. We demonstrate that antioxidant enzymes have an inhibitory effect on arsenic/ethanol-induced COX-2 expression, indicating that the responsive signaling pathways from co-exposure to arsenic and ethanol relate to ROS generation. In vivo results also show that co-exposure to arsenic and ethanol increased COX-2 expression in mice. We conclude that ethanol enhances arsenic-induced COX-2 expression in colorectal cancer cells via both the NFAT and NF-κB pathways. These results imply that, as a common dietary factor, ethanol ingestion may be a compounding risk factor for arsenic-induced carcinogenesis/cancer development.
Keywords: Arsenic, Ethanol, COX-2, NFAT, NF-κB
Graphical abstract
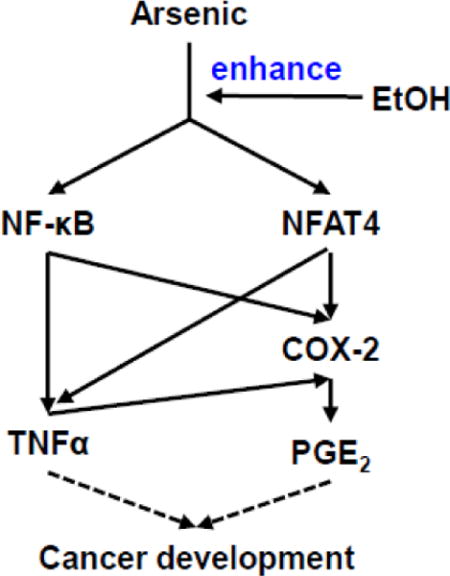
Introduction
Cyclooxygenase (COX) is the rate-limiting enzyme for the production of prostaglandins and thromboxanes from free arachidonic acid (Kitz et al., 2011). COX exists as two distinct isoforms, the constitutively expressed enzyme COX-1 and the inducible isoform COX-2 (Smith et al., 1996). COX-2 can be readily elevated in response to tumor promoters, inflammatory cytokines, and growth factors (Williams and DuBois, 1996). COX-2-produced PGs regulate tumor-associated angiogenesis, modulate the immune system, influence cell migration and invasion, and inhibit apoptosis, all of which are associated with cancer development (Cha and DuBois, 2007). Increasing evidence indicates that COX-2 plays an important role in carcinogenesis (Wang et al., 2013). A variety of human malignancies are associated with overexpression of COX-2 (Sung et al., 2011), which can lead to tumorigenesis (Liu et al., 2001). Mice deficient in COX-2 experience a 75% reduction in tumor formation in comparison with their wild type littermates (Tiano et al., 2002). High levels of prostaglandin E2 (PGE2), the main product of COX-2, are found in tumor cells (Mazhar et al., 2006). Importantly, arsenite exposure in human cells can induce COX-2 expression, which would contribute to arsenic-induced carcinogenesis (Chai et al., 2007; Ding et al., 2006).
Environmental exposure to arsenic is an ongoing worldwide health problem (Hughes, 2002). Arsenic has been classified as a class I human carcinogen by the International Agency of Research on Cancer (IARC). There are an increasing number of reports showing that human exposure to inorganic arsenic is related to lung cancer, skin cancer (Haque et al., 2003), gastrointestinal caner (Tchounwou et al., 2003), kidney cancer (Kitchin and Conolly, 2010), and liver cancer (Capra et al., 1944). Arsenic-induced reactive oxygen species (ROS) and its subsequent oxidative stress, cause genetic and/or epigenetic changes, uncontrolled cell growth, disordered cellular signaling, and eventual tumorigenesis, all of which are considered as key mediators for carcinogenesis (Valko et al., 2006). However, the exact molecular mechanism of arsenic-induced carcinogenesis and tumor progress are unclear and remain under investigation.
It is well recognized that carcinogenesis is affected to a great extent by the diet and nutritional status of the host (Sugimura, 2000). Ethanol or alcohol is a common dietary component and a well-established risk factor for a number of malignancies. Worldwide, approximately 3.6% of cancers are derived from chronic alcohol consumption (Baan et al., 2007). Animal studies support the concept that ethanol is not a pure carcinogen but a co-carcinogen and/or tumor promoter under certain experimental conditions. The role of ethanol as a risk factor for malignancies implicates ROS generation. Ethanol metabolizes to acetaldehyde with a concomitant generation of ROS, which consequently causes oxidative damage to proteins, nucleic acids, and lipids and induces signaling changes (Seitz and Meier, 2007). At present, the exact mechanisms by which chronic alcohol ingestion stimulates carcinogenesis are still unclear.
Previous studies have showed that arsenic has a stronger effect on inducing tumor development when combined with other factors (Qin et al., 2012). The high prevalence of arsenic exposure and the wide-ranging consumption of alcohol make it likely that the co-exposure exists and that this co-exposure may contribute to the environmental health issues (Bao and Shi, 2010). There are only limited epidemiological studies available on populations who consume both alcohol and arsenic-contaminated water. It has been reported that alcohol consumption resulted in increased arsenic accumulation in humans (Chiou et al., 1995; Tseng et al., 2005). Some reports also indicate that co-exposure to arsenic and alcohol caused cardiovascular and liver diseases (Engel et al., 1994). Exposure to arsenic and ethanol induced increases in VEGF and IGF-1 expressions in human microvascular endothelial cells and promoted angiogenesis by activating protein kinase C δ (Klei and Barchowsky, 2008). Animals co-exposed to ethanol and arsenic showed increase in arsenic uptake and retention in the liver and kidney (Flora et al., 1997).There are few reports on the influence of co-exposure to arsenic and ethanol on cancer development. Our previous study revealed that ethanol enhanced arsenic-induced tumor angiogenesis in colon cancer cells via the HIF-1α pathway (Wang et al., 2012). Thus, additional investigations on the effects of co-exposure of alcohol and arsenic were warranted to identify ways to alleviate harmful effects.
The present study tested the hypothesis that co-exposure to arsenic and ethanol promotes COX-2 expression in colorectal cancer cells. Our results indicate that ethanol enhances arsenic-induced COX-2 overexpression via both NFAT and NF-κB signalings. Our results also suggest that, as ethanol is a dietary element, consideration should be placed on the importance of this factor in the investigation of environmental arsenic-induced health effects.
Materials & methods
Materials
Sodium arsenite solution, catalase from bovine liver, and superoxide dismutase (SOD) from bovine liver were from Sigma-Aldrich (St. Louis, MO). GAPDH, p65, NFAT4, and lamin A/C antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA) with all other antibodies purchased from Cell Signaling Technology Inc. (Beverly, MA). Prostaglandin E2 EIA Kit was from Cayman Chemical Company (Ann Arbor, MI). The human TNF-alpha ELISA Kit was from RayBiotech, Inc. (Norcross, GA).
Cell lines and cell culture
Colorectal adenocarcinoma HT29, DLD-1, and colorectal carcinoma HCT116 were obtained from the American Type Culture Collection (ATCC; Rockville, MD). HCT116 cells were cultured in RPMI 1640 medium with penicillin (100 IU/mL), streptomycin (100 μg/mL) and 10% fetal bovine serum (FBS). HT29 and DLD-1 cells were maintained in DMEM medium supplemented with penicillin (100 IU/mL), streptomycin (100 μg/mL), and 10% FBS. Cells were maintained at 37°C in humidified incubator with 5% CO2.
Western blot analysis
Whole-cell extracts were prepared by adding RIPA buffer (Sigma-Aldrich) containing protease inhibitor cocktail. Protein concentrations were determined using coomassie (Bradford) protein assay reagent (Thermo, Rockford, IL). Proteins were separated on SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were probed with primary antibodies followed by incubation with horseradish peroxidase (HRP) conjugated secondary antibodies (Pierce, Rockford, IL). The proteins of interest were visualized using a Chemiluminescent Detection Kit (Pierce) and blots exposed to Hyperfilm (Amersham Pharmacia Biotech, Piscataway, NJ).
Luciferase reporter assay
The luciferase reporter assay was performed as described previously (Wang et al., 2010). Briefly, cells (1 × 106 cells/dish) seeded onto a 10 cm cell culture dish were allowed to reach 60% confluence. Reporter gene constructs were transfected with 8 μg luciferase vector/plate using lipofectamine 2000 (Invitrogen) as recommended by the manufacturer. After transfection, cells were re-seeded onto 24-well plates and subsequently pretreated with the indicated concentration of arsenic and ethanol for 24 hr. Rinsed cells were lysed with luciferase lysis buffer (Promega, Madison, WI). Renilla luciferase reporter was used as a transfection efficiency control. The luciferase activity of lysates were measured following the manufacturer’s protocol (Luciferase Assay System, Promega) using a GloMax® 96 Microplate Luminometer (Promega). The luciferase vectors, which include COX-2 luciferase vector, NFAT luciferase vector, and NF-κB luciferase vector, were maintained in our lab.
Measurement of Tumor necrosis factor-alpha and Prostaglandin E2
TNF-alpha (TNFα) secreted into culture medium was measured using RayBio Human TNF-alpha ELISA kit following the manufacture’s protocol. Prostaglandin E2 (PGE2) secreted into culture medium was measured using Prostaglandin E2 EIA Kit following the manufacture’s protocol. Concentrations were estimated using a generated standard curve.
Subcellular fractionations
Washed cells were lysed in Buffer A (10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA plus the protease inhibitor cocktail (Cell Signaling Technology Inc)) and incubated on ice for 15 min. After incubation, addition of NP40 (final concentration 0.3%) to the lysate was followed by a 10 second vortex and centrifugation for 1 min at 4°C. The supernatant containing the cytoplasmic extract was retained. The pellet was re-suspended in ice cold Buffer B (20 mM HEPES, 400 mM KCl, 1 mM EDTA, 1 mM EGTA plus the protease inhibitor cocktail) and incubated on ice for 30 min with occasional vortexing every 5 min at 4°C. Samples were then centrifuged at full speed for 5 min at 4°C. The supernatant containing the nuclear fraction was retained.
Immunofluoresence microscopy
Cells cultured on chambers slides were washed with PBS and fixed with 4% paraformaldehyde for 10 min. Cells were permeabilized with a 1% glycine/0.5 Triton X-100 solution for 15 min. After blocking with 5% bovine serum albumin for 1 hr, cells were incubated with a primary antibody overnight at 4°C. Washed cells (PBST; PBS containing 0.1% Tween-20) were incubated with a secondary antibody for 45 min. Final rinses included PBST and then PBS alone. The slides were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Labs, Burlingame, CA).
In vivo study and Immunohistochemistry
Twenty Big Blue Mice (about six-week-old), obtained from our collaborator Dr. Zhigang Wang, University of Kentucky, USA, were maintained at the University of Kentucky Animal Facility according to the Institutional Animal Care Guidelines and breeding by our collaborator. Mice were randomized into 4 groups: control, arsenic, ethanol, and arsenic plus ethanol. Arsenic containing drinking water was prepared by dissolving sodium arsenite in water at a concentration of 100 ppm. Ethanol concentration was 10 % in water. The treatment continued four weeks. All mice were sacrificed by CO2 asphyxiation 7 days after the last treatment and tissue samples immediately fixed in 10% formalin or frozen in liquid nitrogen.
Statistical analysis
All arrays were performed with at least three independent experiments. Presented data are the mean ± SE, and statistical comparisons among groups were performed using one-way ANOVA followed by Dunnet test. P value ≤0.05 was considered statistically significant.
Results
The present study was designed to investigate the effect of co-exposure of arsenic and ethanol on COX-2 expression and identify the underlying mechanism in colorectal cancer cells. The results showed that ethanol enhances arsenic-induced COX-2 overexpression via both NFAT and NF-κB signaling.
Ethanol enhances arsenic-induced COX-2 expression in colorectal cancer cells
Western blot analysis assessed the effect of arsenic and ethanol on COX-2 expression in colorectal cancer cells. After incubation with different concentrations of arsenic for 24 hours, COX-2 levels were dramatically increased in multiple colorectal cancer cell lines including HT29, DLD1, and HCT116. Co-treatment with ethanol further increased the COX-2 expression (Figures 1A and 1B). COX-2 expression increased in a time dependent manner with exposure to both arsenic and ethanol (Figure 1C). The COX-2 promoter of HT29 cells exposed to either arsenic or ethanol alone and combination showed significant transcriptional changes (Figure 1D). Taken together, the above results indicate that ethanol enhances arsenic-induced COX-2 expression in colorectal cancer cells and that the increased expression in different cell lines suggests the enhancement is not cell type specific.
Figure 1. Ethanol enhances arsenic-induced COX-2 expression in colorectal cancer cells.
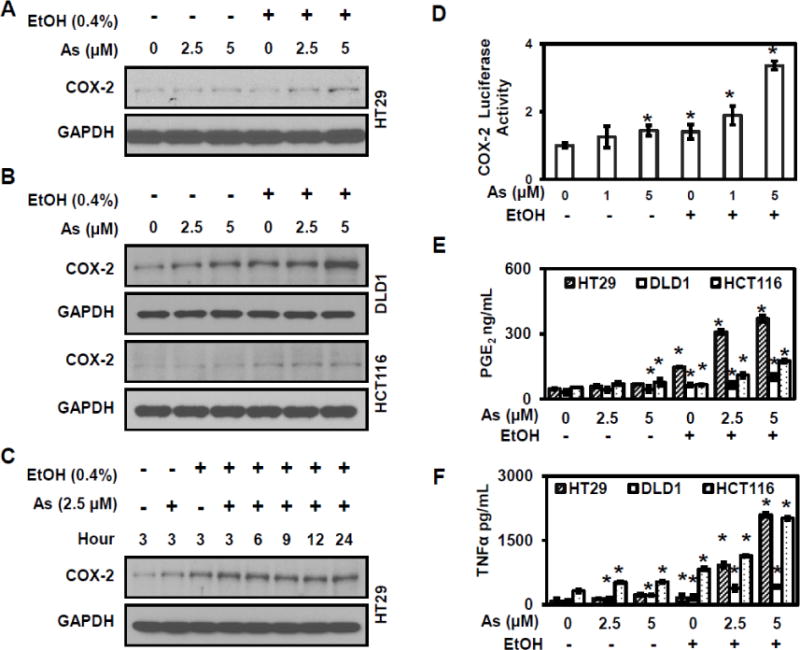
(A) Western blot analysis showing ethanol enhanced arsenic-induced COX-2 expression in HT29 cells occurs in dose dependent manner. (B) Ethanol enhances arsenic-induced COX-2 expression in DLD1 and HCT116 cells. (C) Ethanol enhances arsenic-induced COX-2 expression in HT29 cells in time dependent manner. (D) The luciferase-reported transcriptional activity of the COX-2 promoter in HT29 cells exposed to arsenic and ethanol, either alone or combination. (E) ELISA assay of PGE2 levels in culture medium from various colorectal cancer cell lines. (F) ELISA assay of TNFα levels in culture medium in different colorectal cancer cell lines. Columns are the mean from at least three replicates with the SE indicated (*, P<0.05 versus control).
To determine the effect of arsenic and ethanol on downstream targets of the COX-2 pathway, ELISA analyses were carried out to measure PGE2 levels. Ethanol markedly enhanced arsenic-induced secretion of PGE2 into culture medium in multiple colorectal cancer cell lines (Figure 1E). Among the three cell lines used, HT29 cells provided the most robust response to co-exposure. These results indicate that ethanol enhances arsenic-induced PGE2 production and secretion, suggesting that the enhanced COX-2 expression observed with co-exposure to arsenic and ethanol is functionally relevant. Additionally, ELISA results show that ethanol also enhanced arsenic-induced secretion of TNFα into culture medium in multiple colorectal cancer cell lines (Figure 1F), suggesting that the ethanol-related enhancement is associated with the nuclear factor of activated T-cells (NFAT) or the nuclear factor κB (NF-κB) signaling pathways.
Ethanol enhances arsenic-induced NFAT4 signaling in colorectal cancer cells
It is well known that COX-2 is regulated by different transcriptional factors including NFAT and NF-κB (Cai et al., 2011). Western blot analysis evaluated the combined effect of ethanol and arsenic on NFAT4 in HT29 cells. NFAT4 expression shows a robust increase in HT29 cells with exposure to both arsenic and ethanol (Figure 2A). Treatment with cyclosporin A (CsA), an NFAT inhibitor, resulted in significant decreases in COX-2 levels (Figure 2B). Next, western blot analysis evaluated NFAT4 nuclear translocation with the finding that cells exhibited more NFAT4 nuclear accumulation under co-exposure to arsenic and ethanol (Figure 2C). In addition, luciferase activity demonstrated a significant increase in transcriptional activity at the NFAT promoter following exposure to arsenic and ethanol in HT29 cells (Figure 2D). These results indicate that the enhancement of arsenic-induced COX-2 expression by ethanol is mediated through NFAT signaling.
Figure 2. Ethanol enhances arsenic-induced NFAT signaling.
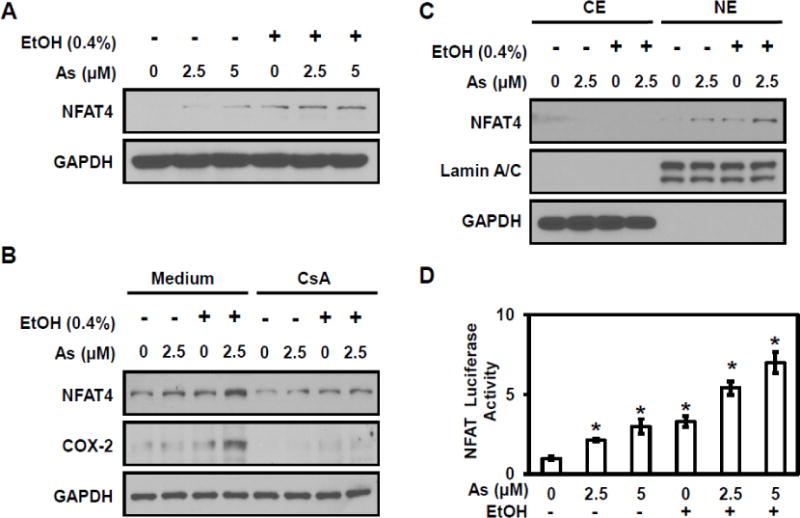
(A) Arsenic and ethanol induced NFAT4 accumulation in HT29 cells. (B) NFAT and COX-2 levels decreased when treated with the NFAT inhibitor Cyclosporin A (CsA) in HT29 cells. (C) NFAT nuclear translocation increased in HT29 cells when exposed to arsenic and ethanol, either alone or combination. (D) Luciferase-reported NFAT transcriptional activity in HT29 cells exposed to arsenic and ethanol, either alone or combination. Columns are the mean from at least three replicates with the SE indicated (*, P<0.05 versus control).
Ethanol enhances arsenic-induced NF-κB signaling in colorectal cancer cells
NF-κB signaling also plays an important role in COX-2 expression (Shishodia and Aggarwal, 2004). Western blot analysis evaluated NF-κB signaling (Figure 3A); the protein level of IκB decreased in cells exposed to arsenic combined with ethanol. Cells treated with an NF-κB inhibitor showed an increase in IκB levels and a dramatic decrease in COX-2 levels (Figure 3B), confirming the association between NF-κB signaling and COX-2 expression. The translocation of p65, a NF-κB subunit, was also investigated. As show in Figure 3C, exposure to either arsenic or ethanol increased p65 nuclear translocation; as expected, co-exposure to arsenic and ethanol resulted in an enhanced p65 translocation into nucleus. Significant increases in NF-κB luciferase activity were observed in HT29 cells exposed to either arsenic or ethanol alone or combination (Figure 3D). The results indicate that ethanol enhanced arsenic-induced NF-κB activity in colorectal cancer cells, but since p65 protein remained in cytoplasmic, the NF-κB signaling pathway appears to be only partially activated.
Figure 3. Ethanol enhances arsenic-induced NF-κB signaling.
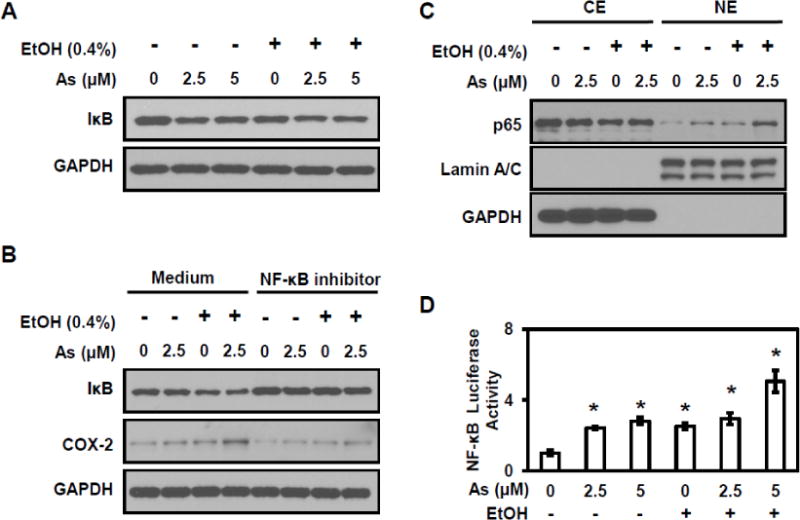
(A) Arsenic and ethanol induce IkBα protein level decreases in HT29 cells. (B) Mitigation of the COX-2 and IκB levels after treatment with an NF-κB inhibitor in HT29 cells. (C) Nuclear translocation of the NF-κB subunit p65 increased in HT 29 cells with exposure to arsenic and ethanol, either alone or combination. (D) Luciferase-reported NF-κB transcriptional activity in HT29 cells exposed to arsenic and ethanol, either alone or combination. Columns are the mean from at least three replicates with the SE indicated (*, P<0.05 versus control).
Immunofluoresence staining shows that arsenic, ethanol, and combined exposures stabilized NFAT4 and p65 proteins, with increased nuclear translocation of these two proteins (Figure 4A). These results suggest that ethanol enhanced arsenic-induced COX-2 expression is mediated by both NF-κB and NFAT4 signaling pathways. The luciferase activity at the COX-2 promoter confirmed that both the NF-κB and NFAT signaling pathways contributed to increased COX-2 expression (Figure 4B). The increased COX-2 expression results from activities related to both of NF-κB and NFAT signaling.
Figure 4. Both NFAT4 and NF-κB signaling pathways contribute to COX-2.
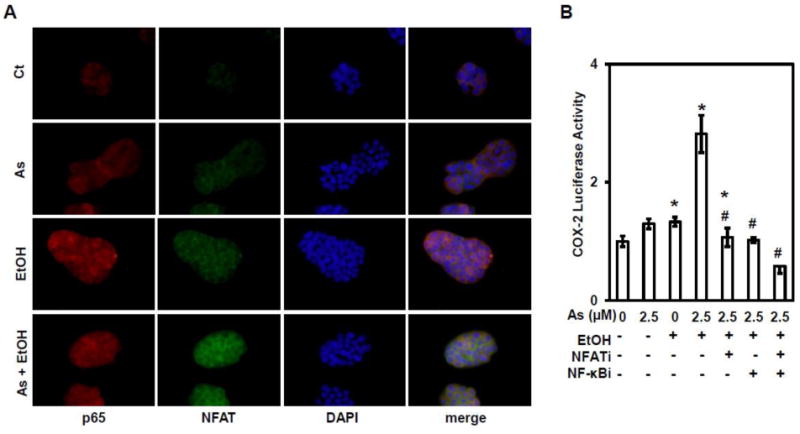
(A) Immunofluoresence staining of p65 (red) and NFAT4 (green) in HT29 cells exposed to arsenic and ethanol, either alone or combination. (B) Luciferase-reported COX-2 transcriptional activity in HT29 cells treated with an NF-κB inhibitor (NF-κBi) and NFAT inhibitor (NFATi). Columns are the mean from at least three replicates with the SE indicated (*, P<0.05 versus control; #, P<0.05 versus combine exposure of As2.5μM and 0.4% EtOH).
ROS/AKT signaling is upstream of ethanol/arsenic-induced NF-κB and NFAT4 activation
ROS plays an important role in metal-mediated carcinogenesis (Lee et al., 2012). ROS signaling is upstream to both NF-κB and NFAT4. ROS activity was detected by following of hydrogen peroxide (H2O2) metabolism and monitoring the response with fluorescence microsopy; oxidation of the fluorescent dye 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate ethyl ester (H2DCFDA) to the fluorescent product, 2′,7′-dichlorofluorescein (DCF) occurs in the presence of H2O2. Cells treated with 5 μM arsenic or 0.4% ethanol alone exhibited visible fluorescence, which represents the generation of H2O2, while cells co-exposed to arsenic and ethanol showed markedly increased fluorescence (Figure 5A). Similar results were observed using dihydroethidium (DHE, a specific fluorescent dye for superoxide anion, O2 •−) staining (Figure 5A). To confirm that the ROS activity was related to arsenic and/or ethanol exposure, the antioxidant enzyme catalase (500 U/mL) or SOD (500 U/mL) was applied to cells. The results demonstrate that ROS generation in arsenic and/or ethanol treated cells was reduced after treatment with either catalase or SOD (Figure 5A). Taken together, these results suggest that ethanol enhances arsenic-induced ROS generation, which could be rescued by antioxidant enzymes.
Figure 5. ROS/AKT signaling is activation.
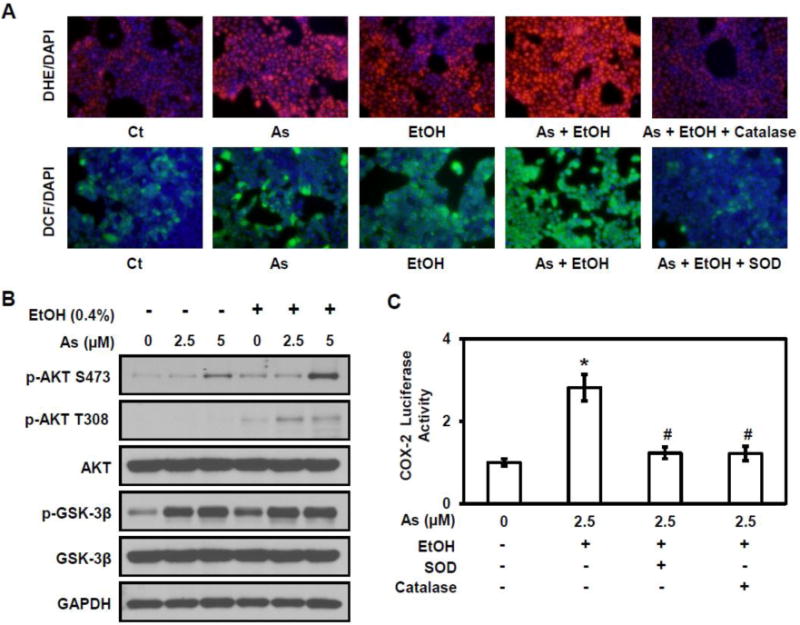
(A) Detection of hydrogen peroxide (H2O2) and superoxide anion (O2•−) using the fluorescent dye 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate ethyl ester (DCF, green) and dihydroethidium (DHE, red) respectively in HT29 cells. (B) Western blot analysis of the Akt signaling pathway. (C) Luciferase-reported COX-2 transcriptional activity in HT29 cells treated with catalase and SOD. Columns are the mean from at least three replicates with SE indicated (*, P<0.05 versus control; #, P<0.05 versus combine exposure of 2.5μM As and 0.4% EtOH).
Akt signaling, activated by intracellular ROS, is important during cancer development. Downstream targets can activate both NF-κB and NFAT4 signaling (Chatterjee et al., 2012). Western blot analysis evaluated the effect of ethanol and/or arsenic on Akt signaling. Increased expression of the phosphorylated forms of Akt and GSK-3β was observed in cells exposed to ethanol or arsenic. Cells co-exposed to arsenic and ethanol also show a further increase in the expression of these phosphorylated proteins; the increase was more pronounced for Akt (Figure 5B). These results indicate greater activation of the Akt pathway with co-exposure to arsenic and ethanol. The effect of ROS on arsenic and ethanol-induced COX-2 expression was investigated, using a COX-2 promoter-driven luciferase assay. Results show that there was a significant decrease in COX-2 luciferase activity with catalase (500 U/mL) or SOD (500 U/mL) treatment to arsenic and ethanol exposed cells (Figure 5C). These results further support the contention that the ethanol enhancement of arsenic-induced COX-2 expression is associated with ROS/Akt signaling pathway.
Ethanol enhances arsenic-induced COX-2 expression in vivo
Since the in vitro results show the ethanol enhances arsenic-induced COX-2 expression, we wanted to investigate the effects in vivo. Big Blue Mice were supplied water containing arsenic, ethanol or both for up to four weeks, sacrificed and colon tissues obtained for western blot analysis. As shown in Figure 6A, both NFAT4 and COX-2 protein levels were robustly increased with co-exposure to arsenic and ethanol. Immunohistochemical staining results confirmed overexpression of NFAT4 and COX-2 in the colon of mice co-treated with arsenic and ethanol (Figure 6B). These results indicate that ethanol enhances arsenic-induced COX-2 expression in vivo, which is consistent with our in vitro results.
Figure 6. Ethanol enhances arsenic-induced COX-2 expression in vivo.
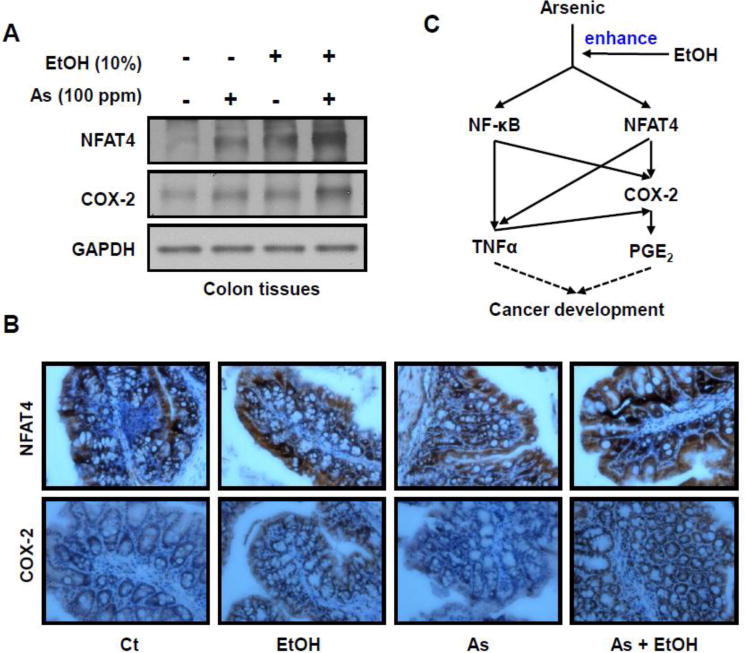
Big Blue Mice were provided with arsenic and/or ethanol containing water for four weeks and colon tissues collected. (A) Western blot analysis of NFAT4 and COX-2 expression levels. (B) Immunohistochemical staining for COX-2 and NFAT4 expression in colonic tissue of mice co-treated with arsenic and ethanol. (C) Diagram of ethanol enhanced arsenic-induced COX-2 expression via both NFAT and NF-κB signaling.
Discussion
Arsenic exposure is a major environmental issue because of its world-wide distribution and its ability to cause serious health problems. Ethanol, a recreational beverage, is consumed globally and has the potential to influence human health. In this study, we initially investigated the effects of co-exposure to arsenic and ethanol on colorectal cancer cells. We found that arsenic-induced COX-2 expression was enhanced by co-exposure to ethanol. We also found that ethanol potentiated arsenic-induced ROS/Akt signaling. In addition to this, the co-exposure activated transcriptional factors NFAT and NF-κB, which may result in an accelerated colorectal cancer development.
In this study, we investigated the effects of co-exposure to arsenic and ethanol in colon cancer cells to provide mechanistic evidence of pathogenic interactions. The arsenic and ethanol concentration used in this study was selected based on previous studies (Klei and Barchowsky, 2008; Wang et al., 2012).
COX-2 is an enzyme responsible for the biogenesis of prostanoids, signaling molecules with multiple functions including inflammation, cell transformation, and cancer metastasis (Vane et al., 1998). COX-2 is known to promote cancer development. Using COX-2 knockout mice, a strong correlation between COX-2 expression and cancer development was revealed, indicating that COX-2 contributes to carcinogenesis and oncogenesis (Muller-Decker et al., 2006). In this study, we report that co-exposure to arsenic and ethanol induces high COX-2 expressions in a variety of colorectal cancer cells. COX-2 associated functions such as PGE2 and TNFα secretion are also enhanced with co-treatment. These results reveal that the combination of a dietary factor (i.e. ethanol) with an environmental factor (i.e. arsenic) may have a dramatic effect on cancer development, and serious consideration for the prevention and treatment of arsenic-induced carcinogenesis and cancer progression should be given.
It is well-known that excessive ROS cause oxidative stress, which consequently leads to various diseases. ROS are believed to act as major mediators in carcinogenesis and cancer development (Valko et al., 2006). In arsenic-induced carcinogenesis, although the molecular mechanism remains unknown ROS generation is considered an important factor. Experimental results show that H2O2 and O2•− are produced in various cellular systems when exposed to arsenic (Zhang et al., 2011). Arsenic-induced ROS causes DNA damage, lipid peroxidation, and chronic inflammation, which broadly lead to carcinogenic responses. It is also well known that ROS play an important role in alcohol-induced cell injury (Wu et al., 2006). The metabolism of ethanol promotes ROS generation and accumulation, followed by oxidative damage. Therefore, both arsenic and ethanol can generate ROS and could have additive effects on ROS in cells. It has been reported that glutathione levels were reduced in rats co-exposed to arsenic and ethanol, indicating that this co-exposure has an additive effect on ROS generation (Flora et al., 1997). Our results show that co-exposure to arsenic and ethanol increased the generation of H2O2 and O2•−. It has been reported that combined treatment with arsenic and ethanol activates different signaling pathways, such as PKCδ in human microvascular endothelial cells (Klei and Barchowsky, 2008), and PI3K/AKT in colorectal cancer cells (Wang et al., 2012). In this study, we have demonstrated that combined treatment with arsenic and ethanol induced ROS generation and subsequently activated downstream AKT signaling. In addition, both catalase and SOD were found to significantly decrease COX-2 luciferase activity. These results suggest that the ethanol-enhanced arsenic-induced COX-2 expression is associated with the ROS/Akt signaling pathway.
The COX-2 promoter region contains binding sites for both NFAT (Iniguez et al., 2000) and NF-κB (Crofford et al., 1997). NFAT is a master transcription factor that controls expressions of a variety of genes involved in inflammation, such as iNOS, COX-2 and TNFα (Ke et al., 2006). NF-κB has been reported to be a major mediator for the regulation of cell proliferation, differentiation, and transformation (Huang et al., 1999). Previous studies have demonstrated that metal exposure could activate both NF-κB and NFAT pathways (Cai et al., 2011). Many metals, such as arsenic, nickel, and chromium (IV), share similar biological roles in COX-2 induction (Zuo et al., 2012). It has also been reported that arsenite exposure induces COX-2 expression concurrent with significant increases in PGE2 generation, leading to an inflammatory effect in rat liver epithelial cells (Lee et al., 2010). Our results showed that either arsenic or ethanol alone could activate Akt and inhibit GSK3β, while co-exposure to arsenic and ethanol exhibit a stronger effect compared to single exposure. The luciferase reporter assay showed enhancement for both NF-κB and NFAT transcriptional activity after co-exposure to arsenic and ethanol. The in vivo results also show that COX-2 and NFAT levels in mice colon were increased after supplying mice with water containing arsenic and ethanol for one month. Since the mice we used for above experiment were of normal health, the results indicate that COX-2 induced chronic inflammation has the potential to increase the risk of carcinogenesis and accelerate cancer development. This finding may extend our understanding of ethanol that could be a compounding risk factor for arsenic-induced carcinogenesis.
In summary, the results obtained from the present study show that ethanol enhanced arsenic-induced COX-2 expression via both NF-κB and NFAT signaling in colorectal cancer cells. These results indicated that as a dietary factor, alcohol or ethanol should be given serious consideration when investigating environmental arsenic-induced health issues.
Supplementary Material
Highlights.
-
►
Arsenic is able to induce Cox-2 expression in colorectal cancer cells.
-
►
Ethanol, a diet nutritional factor, could enhance arsenic-induced Cox-2.
-
►
The up-regulation of Cox-2 via both NFAT and NF-κB activities.
Acknowledgments
We would like to thank Hong Lin for her technical help. This work was supported by the National Institutes of Health [R01ES021771, R01ES025515, R01ES020870, and R01ES017244].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
References
- Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, Altieri A, Cogliano V. Carcinogenicity of alcoholic beverages. Lancet Oncol. 2007;8:292–293. doi: 10.1016/s1470-2045(07)70099-2. [DOI] [PubMed] [Google Scholar]
- Bao L, Shi H. Potential molecular mechanisms for combined toxicity of arsenic and alcohol. J Inorg Biochem. 2010;104:1229–1233. doi: 10.1016/j.jinorgbio.2010.08.005. [DOI] [PubMed] [Google Scholar]
- Cai T, Li X, Ding J, Luo W, Li J, Huang C. A cross-talk between NFAT and NF-kappaB pathways is crucial for nickel-induced COX-2 expression in Beas-2B cells. Current cancer drug targets. 2011;11:548–559. doi: 10.2174/156800911795656001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra F, Grant C, Massey R, Lorre P, Warner Bros Pictures (1923–1967), LC Purchase Collection(Library of Congress), and Copyright Collection (Library of Congress) Criterion collection 136 (pp 1 videodisc of 1 (laser) (ca 118 min) Warner Brothers The Voyager Company; United States: 1944. Arsenic and old lace. [Google Scholar]
- Cha YI, DuBois RN. NSAIDs and cancer prevention: targets downstream of COX-2. Annu Rev Med. 2007;58:239–252. doi: 10.1146/annurev.med.57.121304.131253. [DOI] [PubMed] [Google Scholar]
- Chai CY, Huang YC, Hung WC, Kang WY, Chen WT. Arsenic salt-induced DNA damage and expression of mutant p53 and COX-2 proteins in SV-40 immortalized human uroepithelial cells. Mutagenesis. 2007;22:403–408. doi: 10.1093/mutage/gem035. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Browning EA, Hong N, DeBolt K, Sorokina EM, Liu W, Birnbaum MJ, Fisher AB. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am J Physiol Heart Circ Physiol. 2012;302:H105–114. doi: 10.1152/ajpheart.00298.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiou HY, Hsueh YM, Liaw KF, Horng SF, Chiang MH, Pu YS, Lin JS, Huang CH, Chen CJ. Incidence of internal cancers and ingested inorganic arsenic: a seven-year follow-up study in Taiwan. Cancer Res. 1995;55:1296–1300. [PubMed] [Google Scholar]
- Crofford LJ, Tan B, McCarthy CJ, Hla T. Involvement of nuclear factor kappa B in the regulation of cyclooxygenase-2 expression by interleukin-1 in rheumatoid synoviocytes. Arthritis Rheum. 1997;40:226–236. doi: 10.1002/art.1780400207. [DOI] [PubMed] [Google Scholar]
- Ding J, Li J, Xue C, Wu K, Ouyang W, Zhang D, Yan Y, Huang C. Cyclooxygenase-2 induction by arsenite is through a nuclear factor of activated T-cell-dependent pathway and plays an antiapoptotic role in Beas-2B cells. J Biol Chem. 2006;281:24405–24413. doi: 10.1074/jbc.M600751200. [DOI] [PubMed] [Google Scholar]
- Engel RR, Hopenhayn-Rich C, Receveur O, Smith AH. Vascular effects of chronic arsenic exposure: a review. Epidemiol Rev. 1994;16:184–209. doi: 10.1093/oxfordjournals.epirev.a036150. [DOI] [PubMed] [Google Scholar]
- Flora SJ, Pant SC, Malhotra PR, Kannan GM. Biochemical and histopathological changes in arsenic-intoxicated rats coexposed to ethanol. Alcohol. 1997;14:563–568. doi: 10.1016/s0741-8329(97)00048-7. [DOI] [PubMed] [Google Scholar]
- Haque R, Mazumder DN, Samanta S, Ghosh N, Kalman D, Smith MM, Mitra S, Santra A, Lahiri S, Das S, De BK, Smith AH. Arsenic in drinking water and skin lesions: dose-response data from West Bengal, India. Epidemiology. 2003;14:174–182. doi: 10.1097/01.EDE.0000040361.55051.54. [DOI] [PubMed] [Google Scholar]
- Huang C, Ma WY, Li J, Goranson A, Dong Z. Requirement of Erk, but not JNK, for arsenite-induced cell transformation. J Biol Chem. 1999;274:14595–14601. doi: 10.1074/jbc.274.21.14595. [DOI] [PubMed] [Google Scholar]
- Hughes MF. Arsenic toxicity and potential mechanisms of action. Toxicol Lett. 2002;133:1–16. doi: 10.1016/s0378-4274(02)00084-x. [DOI] [PubMed] [Google Scholar]
- Iniguez MA, Martinez-Martinez S, Punzon C, Redondo JM, Fresno M. An essential role of the nuclear factor of activated T cells in the regulation of the expression of the cyclooxygenase-2 gene in human T lymphocytes. J Biol Chem. 2000;275:23627–23635. doi: 10.1074/jbc.M001381200. [DOI] [PubMed] [Google Scholar]
- Ke Q, Li J, Ding J, Ding M, Wang L, Liu B, Costa M, Huang C. Essential role of ROS-mediated NFAT activation in TNF-alpha induction by crystalline silica exposure. Am J Physiol Lung Cell Mol Physiol. 2006;291:L257–264. doi: 10.1152/ajplung.00007.2006. [DOI] [PubMed] [Google Scholar]
- Kitchin KT, Conolly R. Arsenic-induced carcinogenesis–oxidative stress as a possible mode of action and future research needs for more biologically based risk assessment. Chem Res Toxicol. 2010;23:327–335. doi: 10.1021/tx900343d. [DOI] [PubMed] [Google Scholar]
- Kitz K, Windischhofer W, Leis HJ, Huber E, Kollroser M, Malle E. 15-Deoxy-Delta12,14-prostaglandin J2 induces Cox-2 expression in human osteosarcoma cells through MAPK and EGFR activation involving reactive oxygen species. Free Radic Biol Med. 2011;50:854–865. doi: 10.1016/j.freeradbiomed.2010.12.039. [DOI] [PubMed] [Google Scholar]
- Klei LR, Barchowsky A. Positive signaling interactions between arsenic and ethanol for angiogenic gene induction in human microvascular endothelial cells. Toxicol Sci. 2008;102:319–327. doi: 10.1093/toxsci/kfn003. [DOI] [PubMed] [Google Scholar]
- Lee KM, Hwang MK, Lee DE, Lee KW, Lee HJ. Protective effect of quercetin against arsenite-induced COX-2 expression by targeting PI3K in rat liver epithelial cells. J Agric Food Chem. 2010;58:5815–5820. doi: 10.1021/jf903698s. [DOI] [PubMed] [Google Scholar]
- Lee JC, Son YO, Pratheeshkumar P, Shi X. Oxidative stress and metal carcinogenesis. Free Radic Biol Med. 2012;53:742–757. doi: 10.1016/j.freeradbiomed.2012.06.002. [DOI] [PubMed] [Google Scholar]
- Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, Haudenschild C, Lane TF, Hla T. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J Biol Chem. 2001;276:18563–18569. doi: 10.1074/jbc.M010787200. [DOI] [PubMed] [Google Scholar]
- Mazhar D, Ang R, Waxman J. COX inhibitors and breast cancer. Br J Cancer. 2006;94:346–350. doi: 10.1038/sj.bjc.6602942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Decker K, Furstenberger G, Neumann M, Schnolzer M. Differential protein expression in the epidermis of wild-type and COX-2 transgenic mice. Skin Pharmacol Physiol. 2006;19:89–94. doi: 10.1159/000091975. [DOI] [PubMed] [Google Scholar]
- Qin XJ, Liu W, Li YN, Sun X, Hai CX, Hudson LG, Liu KJ. Poly(ADP-ribose) polymerase-1 inhibition by arsenite promotes the survival of cells with unrepaired DNA lesions induced by UV exposure. Toxicol Sci. 2012;127:120–129. doi: 10.1093/toxsci/kfs099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, Meier P. The role of acetaldehyde in upper digestive tract cancer in alcoholics. Transl Res. 2007;149:293–297. doi: 10.1016/j.trsl.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Shishodia S, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates activation of cigarette smoke-induced nuclear factor (NF)-kappaB by suppressing activation of IkappaBalpha kinase in human non-small cell lung carcinoma: correlation with suppression of cyclin D1, COX-2, and matrix metalloproteinase-9. Cancer Res. 2004;64:5004–5012. doi: 10.1158/0008-5472.CAN-04-0206. [DOI] [PubMed] [Google Scholar]
- Smith WL, Garavito RM, DeWitt DL. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J Biol Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- Sung S, Park Y, Jo JR, Jung NK, Song DK, Bae J, Keum DY, Kim JB, Park GY, Jang BC, Park JW. Overexpression of cyclooxygenase-2 in NCI-H292 human alveolar epithelial carcinoma cells: roles of p38 MAPK, ERK-1/2, and PI3K/PKB signaling proteins. J Cell Biochem. 2011;112:3015–3024. doi: 10.1002/jcb.23226. [DOI] [PubMed] [Google Scholar]
- Sugimura T. Nutrition and dietary carcinogens. Carcinogenesis. 2000;21:387–395. doi: 10.1093/carcin/21.3.387. [DOI] [PubMed] [Google Scholar]
- Tchounwou PB, Patlolla AK, Centeno JA. Carcinogenic and systemic health effects associated with arsenic exposure–a critical review. Toxicol Pathol. 2003;31:575–588. doi: 10.1080/01926230390242007. [DOI] [PubMed] [Google Scholar]
- Tiano HF, Loftin CD, Akunda J, Lee CA, Spalding J, Sessoms A, Dunson DB, Rogan EG, Morham SG, Smart RC, Langenbach R. Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 2002;62:3395–3401. [PubMed] [Google Scholar]
- Tseng CH, Huang YK, Huang YL, Chung CJ, Yang MH, Chen CJ, Hsueh YM. Arsenic exposure, urinary arsenic speciation, and peripheral vascular disease in blackfoot disease-hyperendemic villages in Taiwan. Toxicol Appl Pharmacol. 2005;206:299–308. doi: 10.1016/j.taap.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Vane JR, Bakhle YS, Botting RM. Cyclooxygenases 1 and 2. Annu Rev Pharmacol Toxicol. 1998;38:97–120. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- Wang F, Liu S, Xi S, Yan L, Wang H, Song Y, Sun G. Arsenic induces the expressions of angiogenesis-related factors through PI3K and MAPK pathways in SV-HUC-1 human uroepithelial cells. Toxicol Lett. 2013;222:303–311. doi: 10.1016/j.toxlet.2013.08.008. [DOI] [PubMed] [Google Scholar]
- Wang L, Kuang L, Pan X, Liu J, Wang Q, Du B, Li D, Luo J, Liu M, Hou A, Qian M. Isoalvaxanthone inhibits colon cancer cell proliferation, migration and invasion through inactivating Rac1 and AP-1. Int J Cancer. 2010;127:1220–1229. doi: 10.1002/ijc.25119. [DOI] [PubMed] [Google Scholar]
- Wang L, Son YO, Ding S, Wang X, Hitron JA, Budhraja A, Lee JC, Lin Q, Poyil P, Zhang Z, Luo J, Shi X. Ethanol enhances tumor angiogenesis in vitro induced by low-dose arsenic in colon cancer cells through hypoxia-inducible factor 1 alpha pathway. Toxicol Sci. 2012;130:269–280. doi: 10.1093/toxsci/kfs242. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Williams CS, DuBois RN. Prostaglandin endoperoxide synthase: why two isoforms? Am J Physiol. 1996;270:G393–400. doi: 10.1152/ajpgi.1996.270.3.G393. [DOI] [PubMed] [Google Scholar]
- Wu D, Zhai Q, Shi X. Alcohol-induced oxidative stress and cell responses. J Gastroenterol Hepatol. 2006;21(Suppl 3):S26–29. doi: 10.1111/j.1440-1746.2006.04589.x. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang X, Cheng S, Sun L, Son YO, Yao H, Li W, Budhraja A, Li L, Shelton BJ, Tucker T, Arnold SM, Shi X. Reactive oxygen species mediate arsenic induced cell transformation and tumorigenesis through Wnt/beta-catenin pathway in human colorectal adenocarcinoma DLD1 cells. Toxicol Appl Pharmacol. 2011;256:114–121. doi: 10.1016/j.taap.2011.07.016. [DOI] [PubMed] [Google Scholar]
- Zuo Z, Cai T, Li J, Zhang D, Yu Y, Huang C. Hexavalent chromium Cr(VI) up-regulates COX-2 expression through an NFkappaB/c-Jun/AP-1-dependent pathway. Environ Health Perspect. 2012;120:547–553. doi: 10.1289/ehp.1104179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.