

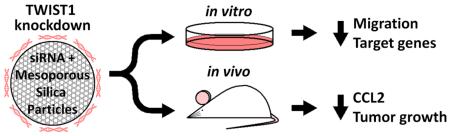
Abstract
Growth and progression of solid tumors depends on the integration of multiple pro-growth and survival signals, including the induction of angiogenesis. TWIST1 is a transcription factor whose reactivation in tumors leads to epithelial to mesenchymal transition (EMT), including increased cancer cell stemness, survival, and invasiveness. Additionally, TWIST1 drives angiogenesis via activation of IL-8 and CCL2, independent of VEGF signaling. In this work, results suggest that chemically modified siRNA against TWIST1 reverses EMT both in vitro and in vivo. siRNA delivery with a polyethyleneimine-coated mesoporous silica nanoparticle (MSN) led to reduction of TWIST1 target genes and migratory potential in vitro. In mice bearing xenograft tumors, weekly intravenous injections of the siRNA-nanoparticle complexes resulted in decreased tumor burden together with a loss of CCL2 suggesting a possible anti-angiogenic response. Therapeutic use of TWIST1 siRNA delivered via MSNs has the potential to inhibit tumor growth and progression in many solid tumor types.
Chemically modified siRNA against TWIST1 was complexed to cation-coated mesoporous silica nanoparticles and tested in vitro and in vivo. In cell culture experiments, siRNA reduced expression of TWIST1 and its target genes, and reduced cell migration. In mice, injections of the siRNA-nanoparticle complex led to reduced tumor weight. Data suggest that diminished tumor burden was the result of reduced CCL2 expression and angiogenesis following TWIST1 knockdown.
Keywords: TWIST1, angiogenesis, mesoporous silica, siRNA
Introduction
The majority of cancer associated deaths are attributable to metastatic disease.1 The processes by which cells from the primary tumor detach, invade, migrate, and colonize distant tissues are still not well understood. Designing therapeutic approaches to mitigate metastasis associated mortality is complicated by the fact that the majority of cancer research is based on cell lines from primary tumors.2 Nevertheless, there is a growing interest in better understanding metastasis and how to approach it therapeutically.
There are several epithelial-mesenchymal transition (EMT) characteristics that allow for metastatic lesions to develop, including cellular motility, breakdown of extracellular matrix, immune evasion, and invasiveness.3 EMT is implicated in many pro-metastatic events and upregulation of EMT-related genes are associated with poor clinical outcomes.4, 5
A prominent cancer related phenomenon driven by EMT is angiogenesis.6 Angiogenesis is the process by which endothelial cells differentiate and divide to form blood vessels branched from existing blood vessels.7, 8 While important during normal physiological processes, including wound healing and growth, angiogenesis has pathological implications in diseases such as cancer, rheumatoid arthritis, asthma, and diabetes.9, 10 Tumor growth and metastasis are limited by nutrients and oxygen supplied by the blood, thus angiogenesis is considered one of the “hallmarks” of cancer.11 Metastasis is the overwhelming problem in neoplastic diseases, responsible for 90% of cancer related deaths.12 Therefore, inhibiting the angiogenic properties of solid tumors can decrease tumor burden and metastatic disease, as shown with therapeutics such as bevacizumab, sorafenib, and sunitinib.13
Angiogenesis has a profound effect on the growth of solid tumors and contributes to their aggressive, metastatic nature.14 Vessels are recruited through various signaling pathways, including members of the vascular endothelial growth factor (VEGF) and angiopoietin (Ang) families, both of which are secreted by tumor cells.15, 16 Elevated levels of serum and tissue VEGF, Ang-1, and Ang-2 are associated with poor prognosis.17 Recently, it was shown that C–C motif chemokine ligand 2 (CCL2) is also associated with increased angiogenesis and metastasis, and its expression is at least partially driven by the transcription factor TWIST1.18
TWIST1 is a basic helix-loop-helix transcription factor that is involved in a variety of normal biological processes including embryogenesis, cellular motility, and tissue differentiation (Figure 1).19, 20 Mutations in TWIST1 can be embryonic lethal or result in severe craniofacial defects associated with Saethre-Chotzen Syndrome.21, 22 TWIST1 has also been shown to be a crucial contributor to cancer progression by enhancing EMT, promoting cancer stem cell phenotypes, and causing drug resistance (Figure 1).23, 24
Figure 1.

TWIST1 plays multiple roles in development and cancer progression. In utero, TWIST1 is required for mesoderm development . In adults, TWIST1 is involved in cell migration in wound healing. In cancer, TWIST1 lies at the hub of signaling pathways and transcriptional regulation of EMT/metastasis, cancer stem cell phenotype, acquired drug resistance, resistance to cell death, and angiogenesis.
TWIST1 is associated with angiogenesis, both in normal (development and wound healing) and pathologic neovascularization.25, 26 TWIST1 can induce angiogenesis through the recruitment of tumor macrophages with or without up-regulation of VEGF.18, 27 The chemokine responsible for macrophage recruitment is CCL2, and its secretion leads to an influx of macrophages into the tumor, resulting in immunosupression and angiogenesis. Because TWIST1 is known to enhance the expression of CCL2, TWIST1 inhibition is an attractive therapeutic strategy to reduce tumor burden through reduction of angiogenesis.18 IL-8 (another TWIST1 target gene) is also known to promote angiogenesis thus a reduction of TWIST1 expression would decrease the expression of IL-8 and further inhibit vessel formation.28
In the area of cancer therapeutics, nanomaterials have evolved to more efficiently accomplish therapeutic tasks. Mesoporous silica nanoparticles (MSNs, 50-200 nm in diameter) have gained favor because of their relatively large surface area, pore structure (allowing for drug delivery), and uniform size (which is easily controlled during synthesis).29, 30 MSNs are well suited for tumor therapeutic delivery because their small size allows them to escape the vasculature and enter into the tumor tissue. As a result of the enhanced permeability and retention (EPR) effect, nanoparticles become trapped in the tumor tissue, thereby having the effect of being “targeted” to the tumor itself to deliver their payload.31
In addition to chemotherapeutic payloads, MSNs are also capable of delivering small interfering RNA (siRNA), which results in posttranscriptional gene silencing in a sequence specific manner (Figure 2).32, 33 Delivery of siRNA molecules via MSNs helps overcome some of the inherent problems of siRNA therapeutics, by protecting siRNA from nucleotide degradation and targeting it to the tumor (via the EPR effect or surface modifications).34 The negatively charged siRNA molecules are complexed with the MSNs, which are produced with a cationic polyethyleneimine (PEI) coating. The low molecular weight PEI coating not only allows for the carrying of the siRNA, but it also facilitates the release of the siRNA from the endosome via the proton sponge effect.35 Furthermore, the low molecular weight PEI coating has been shown to be nontoxic and enhances the uptake of the MSN into the cell.36
Figure 2.

siRNA-MSN complexes for use in vitro and in vivo. A. Individual pores are visible in TEM micrographs of ~120 nm MSNs. B. Particle size within a batch is uniform. C. Schematic of siRNA silencing via RNA induced silencing complex (RISC). siRNA contains 2-OMe-U and inverted abasic ribose modifications, to decrease degradation and immune stimulation, and to guarantee the guide strand is loaded into RISC. TWIST1 siRNA binds to TWIST1 mRNA, leading to its cleavage by Ago2. The siRNA guide strand is reused to recognize additional TWIST1 mRNAs. D. Sequences of si419 showing positions of 2-OMe (green) and inverted abasic (red) modifications.
Despite being stable at physiologic pH, siRNA usually needs to be chemically modified to increase its nuclease resistance in an in vivo model.37 A common modification to the sugar moiety of the siRNA is a 2′-O-methyl substitution (Figure 2). This modification is known to stabilize siRNA in serum and minimize toll like receptor signaling, thereby decreasing immunogenicity.38 Inverted abasic ribose was added to the passenger strand of the siRNA to prevent its loading into the RNA-induced Silencing Complex (RISC) and promote loading of the guide strand (Figure 2).39
In this article we demonstrate siRNA delivery via a PEI-coated MSN. We show that the siRNA is able to knock down the expression of TWIST1 both in vitro and in vivo following MSN delivery. The knockdown of TWIST1 resulted in a functional change in the expression of TWIST1 targets and ultimately leads to decreased tumor burden in a xenograft mouse model.
Methods
MSN Production
The MSNs were produced using the sol-gel method as described previously.33 Briefly, the MSNs were created by dissolving 250 mg cetyltrimethylammonium bromide (CTAB, 95%) (Aldrich, St. Louis, MO) in 120 mL of water with 875 μL sodium hydroxide solution (2 M) and 48 mL water while heating the solution to 80°C. Next, 1.2 mL of tetraethylorthosilicate (Aldrich, 98%) was added to the solution. After 15 min, 300 μL of 3-(trihydroxysilyl) propyl methylphosphonate (Aldrich,42%) was added into the mixture. The solution was then stirred for 2 hours, during which MSNs began to form. Particles were collected following centrifugation and washed with methanol. Acidic methanol was then used to flush the pores in the MSNs to rid them of the CTAB surfactants. The resulting pore diameter was 2-3 nm. The addition of the cationic PEI coating (low molecular weight) to the MSNs has previously been described.40 In short, the PEI coating (1.8 kDa branched polymer) was added by suspending the MSNs in PEI ethanolic solution which was then sonicated and stirred (repeated twice). The MSNs were then washed in ethanol to remove unbound PEI.
Cell Culture and Transfection
MDA-MB-435S melanoma cells were obtained from ATCC (Manassas, VA). Cells were maintained at 37°C, 5% CO2, and 90% humidity in a tissue culture incubator. Media for the MDA-MB-435S cells was RPMI 1640 media (Genesee Scientific, San Diego, CA) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were passaged using 0.25% trypsin (Genesee Scientific, San Diego, CA) every 3-4 days.
To allow for imaging of cells in a xenograft model, we created a line of MDA-MB-435S that stably expressed GFP and firefly luciferase (ffluc). These cells were created with the aid of a CMV lentiviral construct that encodes a fusion protein of GFP and ffluc separated by a three glycine linker.41 This stable cell line (MDA-MB-435S-GFP+ffluc) was used for all experiments.
Previously published siRNA sequences against TWIST1 were used (si419-passenger, 5′-GGACAAGCUGAGCAAGAUU-3′; si419-guide, 5′-AAUCUUGCUCAGCUUGUCCUU-3′; si494-passenger, 5′-GCGACGAGCUGGACUCCAA-3′; si494-guide, 5′-UUGGAGUCCAGCUCGUCGCUU-3′).42 Two chemical modifications (addition of 2′-O-methyl and inverted abasic ribose, see Figure 2) were made to the si419 passenger/sense strand for all experiments except for those involving Lipofectamine® 2000 transfection. The chemically modified si419 duplex is referred to as si419Hybrid or si419H. No chemical modifications were made to the si494 sequences. siRNA duplexes were formed by mixing equal molar volumes of each strand together and heating them in a hot block (100°C) for 10 minutes, then removing the block from the heat source to cool to room temperature. Negative control siRNA (siQ, labeled with AlexaFluor® 647) was AllStars Negative Control siRNA from Qiagen (Valencia, CA).
Transient transfection of MDA-MB-435S-GFP+ffluc was carried out using Lipofectamine® 2000 (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Lipofectamine® 2000 transfection was used to confirm the functionality of the siRNA (si419 and si494) prior to testing their efficacy with MSNs. Transfection of siRNA with MSNs was carried out by incubating the MSNs with the siRNA overnight at 4°C while rotating the tube constantly. The mixture consisted of 7 parts MSN (diluted to 500 ng/ul in sterile PBS) to 1 part siRNA (diluted to 10 uM). The final concentrations for of MSNs and siRNA applied to cells were 17.5 ng/ul and 50 nM, respectively.
ELISA Assay
We used an ELISA assay to measure the amount of IL-8 secreted by MDA-MB-435S-GFP+ffluc cells following treatment with MSN+siRNA (si419H and si494). MSNs complexed with siRNA against GFP (siGFP) were used as a control. 250,000 MDA-MB-435S-GFP+ffluc cells were seeded in a 6-well tissue culture plate and allowed to adhere. After 24 hours, cells were incubated with the MSN+siRNA complexes for 72 hours at standard tissue culture conditions. After 72 hours, a sample of the conditioned media was collected for secreted IL-8 quantification. The IL-8 Human ELISA Kit (Thermo Fisher Scientific Inc., Waltham, MA) was used according to the manufacturer’s specifications.
Western Blot
Following siRNA treatment described above, cells were lifted from tissue culture wells with 0.25% trypsin, pelleted, and lysed in RIPA buffer. Protein concentration was determined using a BCA Assay (Thermo Fisher Scientific). A total of 30 ug of protein per lane was run on 4% stacking and 12% resolving polyacrylamide gels. Following gel electrophoresis, protein was transferred to Immobilon-P PVDF membrane (Millipore, Billerica, MA) using a Trans-Blot SD Semi-Dry Transfer Cell (Bio-Rad, Hercules, CA). Membranes were then blocked with 5% dry milk dissolved in 1X PBS with 0.1% Tween-20. Antibodies were diluted in blocking buffer (1:250 for anti-TWIST1 and 1:2,500 for anti-Actin). Antibodies used were: anti-TWIST, TWIST 2c1a (Santa Cruz Biotech, Dallas, TX); anti-β-Actin, A1978 (Sigma Aldrich, St. Louis, MO); and Horse Radish Peroxidase (HRP)-conjugated anti-mouse secondary antibody (Li-Cor, Lincoln, NE). ECL Plus chemiluminescent substrate (Pierce, Thermo Fisher Scientific, Waltham, MA) was used for the development of images.
Wound Healing Assay
In vitro wound healing assays were performed to examine directional cell migration.43 MDAMB-435S-GFP+ffluc cells were grown in the tissue culture conditions described previously in 6-well tissue culture plates. Cells were treated with MSN+siQ, MSN+si419H, or MSN+si494 for 24 hours (as described earlier in this section), then a sterile 200μl pipette tip was used to scratch a line in the monolayer of cells. Images were taken at several time points thereafter using a Nikon TE-2000S microscope (Nikon, Tokyo, Japan) and SPOT Advanced software (Diagnostic Instruments, Sterling Heights, MI). Cells were incubated with MSN+siRNA complexes at 37°C, 5% CO2, and 90% humidity in a tissue culture incubator at all times except for the imaging time points.
Quantitative PCR
Total cellular RNA was isolated using the RNeasy Plus kit (Qiagen, Valencia, CA). Synthesis of cDNA from total RNA was carried out using the iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA) with an equal amount of RNA used for all samples. Quantitative RT-PCR was performed using Maxima SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA) in 25μl reactions. Thermocycling was conducted in a Bio-Rad iQ5 thermal cycler for 40 cycles (95°C, 15s; 57°C, 60s; 79°C, 30s) followed by melt curve analysis. Data were analyzed using Bio-Rad iQ5 software. Primers used were: TWIST1-forward, 5′-CTATGTGGCTCACGAGCGGCTC-3′; TWIST1-reverse, 5′-CCAGCTCCAGAGTCTCTAGACTGTCC-3′; Vimentin-forward, 5′-TCGTCACCTTCGTGAATACCAAGA-3′, Vimentin-reverse, 5′-CCTCAGGTTCAGGGAGGAAAAGTT-3′; CCL2-forward, 5′-CAGCCAGATGCAATCAATGCC-3′; CCL2-reverse, 5′-TGGAATCCTGAACCCACTTCT-3′.18
Confocal Microscopy
MDA-MB-435S-GFP+ffluc cells were seeded into a 3.5 cm glass bottom tissue culture dish. Following attachment (24 hours), 2 ml of fresh media was added following the removal of old media. Next, MSN+siQ complexes (labeled with AlexaFluor® 647) were added to the dish for 24 hours at final concentrations of 17.5 ng/μl (MSN) and 50 μM (siQ). Following fixation with 4% paraformaldehyde, cells were counterstained with DAPI (300 nM for 2 min) and then mounted using ProLong® Gold (Thermo Fisher Scientific, Waltham, MA). Confocal images were obtained using a Zeiss LSM 700 Confocal Microscope and ZEN 2012 microscopy software (Zeiss AG, Oberkochen, Germany).
Tumor Engraftment and In vivo Imaging
All animal work was done following protocol approval by the Institutional Animal Care and Use Committee at the City of Hope Beckman Research Institute. A total of 18 female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory, Bar Harbor, ME) were used. Mice were approximately 10 weeks old at the time of the inoculation of tumor cells. Mice were randomly divided into four groups: control mice (no xenografts, 2 mice); negative control (MSN+siQ, 4 mice); si419H treatment group (MSN+si419H, 6 mice); and si494 treatment group (MSN+si494, 6 mice). An siRNA-only group was not included because previous in vitro experiments revealed no cellular uptake without MSNs (Supplemental Figure 7). Mice (other than the no xenograft controls) received bilateral inoculations into the 4th mammary fat pad set immediately adjacent to the nipple. Inoculation was carried out while mice were fully anesthetized using isoflurane (2-5%) delivered via a vaporizer. Inoculum for each mammary fat pad consisted of 3.2×106 MDA-MB-435S-GFP+ffluc cells suspended in 75 μl PBS. Following injections, mice were allowed to fully recover in a clean cage.
Bioluminescent imaging of mice began 12 days after initial inoculation of tumor cells and occurred every week for six weeks. Images were captured using the Xenogen IVIS 100 biophotonic imaging system (STTARR, Toronto, Ontario, Canada). Prior to being fully anesthetized with isoflurane (2-4%), mice were given a 200 μl intraperitoneal injection of 25 mg/ml D-Luciferin (PerkinElmer, Waltham, MA). Ten minutes after the D-luciferin injection, mice were placed in the biophotonic imager. Images were captured over a period of one minute.
Intravenous (IV) injections of MSN+siRNA (siQ, si419H, or si494; siQ fluorescently labeled with AlexaFluor® 647, si419H and si494 labeled with Cy5) were started two weeks after the inoculation of MDA-MB-435S-GFP+ffluc cells and done weekly for six weeks. Tumor size was evaluated manually and with bioluminescent imaging prior to MSN+siRNA administration and revealed that all mice had bilateral tumors of approximately the same size (Supplemental Figure 6). Prior to IV injections mice were randomly assigned to a treatment group. A 120 μl volume of MSN+siRNA was given in the lateral tail vein (excluding no-tumor controls). The injection consisted of 105 μl of 500 ng/μl MSN complexed with 15 μl of 10 μM siRNA (complexing took place overnight at 4°C). Ten minutes after the IV injection animals were anesthetized (2-4% isoflurane) and underwent infrared imaging using the Xenogen IVIS 100 biophotonic imaging system (STTARR) for 15 seconds.
At the end of the experiments, all animals were euthanized via CO2 asphyxiation followed by necropsy. Tumors were carefully dissected away from any adherent tissue and weighed and then placed in 10% formalin along with the heart, lungs, spleen, kidney, and liver for histological evaluation. Histopathological examination of tissues was interpreted by a board certified veterinary pathologist who was blinded to the treatment groups.
Statistical Analysis
Data were analyzed using Prism 6 (GraphPad Software, La Jolla, CA). All qPCR data were analyzed by a one-tailed unpaired t-test with Welch’s correction, separately comparing si419H to siQ and si494 to siQ. No correction for multiple comparisions was used. ELISA data were analyzed by Kruskal-Wallis non-parametric test and Dunn’s test for multiple comparisons. Data represents normalized data from two runs, each in duplicate, for a total n of 4. Tumor growth data were analyzed by Kruskal-Wallis and Dunn’s test as described above, and represent all individual tumors in each group (grown as 2 tumors per mouse). * = p<.05 and ** = p<.01 throughout.
Results
siRNA Design and Efficacy
We have designed and synthesized siRNA to inhibit TWIST expression, incorporating various chemical modifications to increase resistance to nuclease activity, decrease immunogenicity, and promote efficient loading of the guide strand into RISC (Figure 2). Our previous studies with a breast cancer cell line (SUM 1315) demonstrated the efficacy of the si419H and si494.42 To confirm efficacy of si419 and si494 (both not chemically modified) in MDA-MB-435S cells, Lipofectamine® 2000 transfection was performed. We observed a time dependent TWIST1 knockdown, with greater than 90% TWIST1 protein reduction at 72 hours following transfection (Figure 3A). Chemical modifications (2′-O-methyl and inverted abasic reoxyribose on passenger/sense strand) did not impact the efficacy of TWIST1 knock down (Supplemental Figure 2).
Figure 3.

siRNA enters cells and knocks down TWIST1 in vitro. A. TWIST1 siRNA delivered using Lipofectamine® 2000 decreases levels of TWIST1 protein up to 72 hours following transfection. B. i. Merged confocal image of GFP (green), DAPI (blue) and siRNA (pink). ii. Magnification of cell from center right of i. siRNA shows expected perinuclear localization. iii-v. Single color images. C. Delivery of TWIST1 siRNA using MSNs produces significant knockdown at 72 hours post transfection, but TWIST1 protein levels stabilize after one week. D. qRT-PCR data demonstrate that TWIST1 mRNA levels are also reduced 72 hours following MSN+siRNA treatment.
MSN Synthesis and Loading
MSNs produced had an average particle size of 127 nm (TEM image measurement, Figure 2) with a size distribution of +/− 7 nm. The zeta potential at 50 μg/mL was 43.75 mV. The above siRNAs were loaded onto MSNs that have a PEI coated cationic surface. Mixing of the two enables tight binding of siRNA to positively charged MSNs. Thus, PEI provides protection of siRNA and efficient delivery of siRNA to cancer cells.
MSN+siRNA Delivery and TWIST1 Silencing
A stable line of MDA-MB-435S that expresses GFP and ffluc was successfully produced via lentiviral transduction. Both GFP and ffluc were shown to be fully functional in vitro and in vivo (Supplemental Figure 3). Confocal microscopy of MDA-MB-435S-GFP+ffluc cells following incubation with fluorescently labeled MSN+siQ demonstrated perinuclear localization of the siRNA (Figure 3B). MTT assays confirmed that significant cell death did not occur until MSN+siRNA treatment was 2-5 times the concentration used for experiments (Supplemental Figure 1).
TWIST1 knock down in MDA-MB-435S-GFP+ffluc cells was observed at 72 hours post MSN+siRNA treatment. A significant decrease of TWIST1 was seen in both the RNA and protein measurements (Figure 3C). Basal levels of TWIST1 expression returned by one week.
TWIST Knockdown Resulted in Decreased Migration and Decreased IL8 Secretion
Two functional assays confirmed that TWIST1 knock down following MSN+siRNA treatment had downstream effects. A wound healing assay showed an appreciable difference in the migration capabilities of MDA MB 435S GFP+ffluc cells following treatment with MSN+si419H and MSN+si494 when compared to the MSN+siQ control (Figure 4A). IL-8 ELISA assays demonstrated a significant reduction in human IL-8 secretion from the MDA-MB-435S-GFP+ffluc cells following 72 hours of treatment with MSN+si419H or MSN+si494 when compared to the negative control (Figure 4B). Diminished IL-8 secretion was also observed at 48 and 96 hours post transfection, but not at 24 hours (data not shown).
Figure 4.

TWIST1 knockdown reduces downstream pathways in vitro. A. MDA-MB-435S cells with TWIST1 knocked down are slower to migrate and seal a scratch wound. B. ELISA reveals that MDA-MB-435S cells treated with TWIST1 siRNA secrete significantly less IL-8 than nontransfected cells or those transfected with irrelevant control (siGFP).
Tumor Burden Decreased Following Treatment with MSN+Chemically Modified siRNA
All mice that received an inoculation of tumor cells developed bilateral tumors in the area of the 4th mammary fat pad. All tumors were palpable and emitted a robust bioluminescent signal following IP injection of D-Luciferin. Following histopathological examination by an ACVP board certified pathologist, no changes were observed in the heart, lungs, liver, spleen, and kidneys, in mice receiving MSN+siRNA treatments when compared to control animals.
However, tumors collected from the MSN+si419H mice were significantly smaller when compared to the weights of the tumors from the MSN+siQ control mice (Figure 5A,B). Furthermore, by visual inspection, the blood vessels supplying the tumors were smaller in the MSN+si419H treated mice, and the tumors appeared less hemorrhagic than those of the other two groups (Figure 5A). However, the tumors from mice treated with MSN+si494 (without chemical modifications) were not significantly smaller than tumors of the control mice as would be expected from the in vitro TWIST1 knockdown studies for both si419H and si494 in Figure 3.
Figure 5.

MSN+siRNA therapy reduces TWIST expression and tumor growth in vivo. A. Gross tumor images reveal smaller, less vascularized tumors in mice treated with si419H than in control mice or those given non-chemically-modified si494. Representative images shown. B. si419H treated tumors show a significant drop in weight compared to untreated or si494-treated tumors. C-E. qPCR results from tumors collected at necropsy. Tumors exhibit loss of TWIST1 (C) and its target genes Vimentin (D) and CCL2 (E). In each case, more robust knockdown is observed for si419H than si494.
Tumor Characterization Demonstrated EMT Inhibition
Collected tumors were analyzed for the relative mRNA quantities of TWIST1, Vimentin (EMT marker), and CCL2 (chemokine involved in angiogenesis). There was a significant reduction in the amount of TWIST1, Vimentin, and CCL2 in the MSN+si419H and MSN+si494 treated mice when compared to the control mice (MSN+siQ) (Figure 5C, D, E). The average relative reduction of TWIST1 for MSN+si494 treated mice was less than that of those treated with MSN+si419H (p=0.0067), though no significant difference was observed for Vimentin or CCL2. Thus, these data suggest inhibition of EMT thourgh the delivery of TWIST1 siRNA.
No Evidence of Decreased Metastatic Lesions
Metastatic disease is the ultimate cause of mortality in most cancer related deaths. Hence, there are great efforts being made in understanding the mechanisms underlying metastasis as well as developing therapies to exploit those mechanisms to prevent the spread of cancer cells.44, 45 Contrary to what would be expected with significant TWIST1 knock down, there was no reduction in the number of metastatic lung lesions (Supplemental Figure 4). Metastatic lesions were categorized into 4 groups based on size, and no significant difference was seen among the MSN+siQ, MSN+si419H, and MSN+si494 treatment groups. The cause for this finding could be attributed to two possible elements of the experimental design. First, the initial MSN treatment occurred two weeks after the MDA-MB-435S-GFP+ffluc cells were inoculated. This period of time was designed to allow for the tumor cells to engraft unencumbered by any treatment. However, it is possible that this metastatic cell line spread to the lungs before any MSN treatment. Although less clinically relevant, future studies with this cell line might benefit from beginning MSN+siRNA treatments simultaneously with tumor cell inoculation. Second, in vitro results demonstrated that TWIST1 protein levels following MSN+siRNA treatment were reduced at 72 hours, but returned by 7 days (Figure 3C). It is possible that in order to reduce metastatic spread, it would have been necessary to provide MSN+siRNA treatments at more frequent intervals.
Discussion
Here, we demonstrate effective delivery of a chemically modified siRNA therapy via a silica nanoparticle carrier. Following delivery of the siRNA, there is significant knockdown of the transcription factor TWIST1, a known regulator of EMT and angiogenesis.46 TWIST1 knockdown was associated with decreased tumor burden in vivo as well as a reduction in the TWIST1-mediated targets Vimentin (EMT) and CCL2 (angiogenesis) (Figure 5).
Our results demonstrate that nanoparticle delivery of TWIST1 siRNA leads to a decrease in tumor burden, supporting the idea that TWIST1 is an important therapeutic target. TWIST1 was selected as a target for siRNA therapy because it is highly associated with metastasis, EMT, and a poor prognosis.46 TWIST1 is also an attractive therapeutic target because it is not expressed in most adult tissues, and therefore most normal tissues would not be negatively impacted by an siRNA silencing strategy.47 Further work is needed to assess the potential side effects of siRNA therapy such as immune stimulation, inflammation cascades, and off-target effects.48
The observed decrease of tumor burden appears to be due to the effect of reduced TWIST expression on EMT-mediated angiogenesis. Angiogenesis in cancer occurs following a variety of complex signaling pathways that result in increased blood supply to the tumor, thus allowing for continued growth and metastasis. Two key components of angiogenesis examined here are CCL2 and IL-8. CCL2 is a monocyte chemotactic protein secreted by tumor cells responsible for recruiting macrophages into the tumor to aid in establishing new blood vessels.18, 49 IL-8 is a pro-inflammatory cytokine that is known to work synergistically with VEGF to stimulate vessel growth in tumors.50 Furthermore, IL-8 is known to promote angiogenesis independent of VEGF and can be the cause of failure with anti-VEGF therapies.51 Reductions of both IL-8 (in vitro, secreted) and CCL2 (in vivo) were observed following treatment with MSN+si419H and MSN+si494 (Figure 4B and 5E). While reductions of these two promoters of angiogenesis were evident for both MSN+si419H and MSN+si494 treatment groups, reduced tumor burden was only observed with MSN+si419H.
In our experiments, we found that si419H exhibited excellent efficacy in vitro and in vivo. However, si494 exhibited efficacy in vitro but not in vivo. One of the possible explanations for these differences is that the si419H duplex had a chemically modified passenger strand (Figure 2) whereas si494 did not. The 2′-O-methyl modifications allow the siRNA to be protected from nucleases in the blood. In tissue culture conditions, there was no nuclease activity; therefore the chemical modifications present in si419H did not confer an inherent advantage over si494, thus explaining the similar knock down in vitro. A second chemical modification that was present only in si419H was the inverted abasic ribose at each end of the passenger strand. The abasic modification prevents the loading of the passenger strand into RISC, effectively increasing the number of available slots for the loading of the guide strand.38 Thus, the guide strand of si419H would have more efficient loading into RISC when compared to the guide strand of si494. Another possible reason for the tumor burden differences are the RNA sequences themselves. RNA-Seq analysis of a cell line stably expressing anti-TWIST1 shRNA demonstrated that sh419 knocked down TWIST1 and TWIST2 while sh494 only knocked down TWIST1 (Supplemental Figure 5). Given the high level of sequence similarity between TWIST1 and TWIST2, and because of the similar role each plays in EMT, it is possible that knocking down both would result in reduced tumor burden.52 Additionally, based on RNA sequence analysis described previously, it is expected that the si419H sequence could be less likely to illicit a TLR response compared to si494.53 However, this would need to be verified in future studies.
Our results show that MSNs provide efficient vehicle delivery of siRNA in vitro and in vivo. MSNs developed for this project were shown in vitro to successfully deliver their siRNA payload into melanoma cells (Figure 3B), and this delivery resulted in significant knockdown of TWIST1 (Figure 3C). These results further establish that MSNs are viable carriers for siRNA.36, 54 The PEI coated MSNs used in this project were shown to cause no cellular death in vitro (at normal concentrations, Supplemental Figure 1). Following weekly IV injection of MSN+siRNA there was no appreciable clinical or histopathologic evidence (heart, lung, spleen, liver, or kidney) of deleterious effects to the mice. Taken together, this would indicate that MSNs are efficacious and safe, as reported previously.55, 56 MSNs provide a number of advantages for future development as a siRNA vehicle. First, MSNs are highly customizable in both their size and shape, thus allowing for a finely tuned nanoparticle which can be optimized to specific delivery needs. Modifications to the size and structure of the MSNs allow for increased biocompatibility and safety.55 The porous nature of the MSNs used in this research is an untapped nanoparticle characteristic that should be explored in conjunction with the observed knockdown of TWIST1. Drug resistance is a major hindrance to the treatment of cancer and often results in a more aggressive phenotype. TWIST1 is a known contributor to chemoresistance in several types of cancer and its downregulation leads to cells that are more susceptible to traditional therapies.57, 58 Finally, MSNs can be modified to carry targeting moieties that allow them to home to specific tissues or tumors.59 Therefore, a MSN based co-delivery strategy of anti-TWIST1 siRNA together with chemotherapy could result in more pronounced tumor reduction. Increased efficacy and reduced dosage would also be possible with tumor targeting moieties as previously described.54
These data provide encouragement to continue development and optimization of MSNs as a delivery platform for the treatment of cancer. To our knowledge, this research represents the first example of silencing of an EMT-regulating transcription factor following siRNA delivery using an MSN. Further studies are warranted to build upon previous studies that examine the timing and biodistribution of the MSNs.59, 60
Supplementary Material
Acknowledgements
The authors wish to thank Dr. Paul Burke and Dr. John Rossi for their expertise in selecting and designing the siRNA sequences as well as the implemented chemical modifications. A special thanks to the Wold lab at CalTech for their assistance in analyzing the RNA-seq data. Special thanks to Ms. Brielle Finlay for her assistance with the confocal microscopy. The authors also wish to thank the staff of the Small Animal Imaging, Microscopy, Veterinary Pathology, Integrated Genomics, and Pathology Cores of the City of Hope-Beckman Research Institute.
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Funding for this project also includes an NCI Cancer Center Excellence Award and a generous gift from the Parvin Family Foundation. Further funding came from NIH grant CA133697. Funding sources did not have any role in study design, interpretation of data, or writing this manuscript.
Abbreviations
- MSN
Mesoporous Silica Nanoparticle
- CCL2
C–C motif chemokine ligand 2
- EMT
Epithelial-Mesenchymal Transition
- PEI
polyethyleneimine
- IL-8
Interleukin 8
- TEM
Transmission Electron Microscopy
- VEGF
Vascular Endothelial Growth Factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors do not have any conflict of interest to declare nor any competing interests.
References
- 1.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nature reviews Cancer. 2009;9:274–84. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 2.Coghlin C, Murray GI. Current and emerging concepts in tumour metastasis. J Pathol. 2010;222:1–15. doi: 10.1002/path.2727. [DOI] [PubMed] [Google Scholar]
- 3.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–81. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vernon AE, LaBonne C. Tumor metastasis: a new twist on epithelial-mesenchymal transitions. Curr Biol. 2004;14:R719–21. doi: 10.1016/j.cub.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 6.Niu RF, Zhang L, Xi GM, et al. Up-regulation of Twist induces angiogenesis and correlates with metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res. 2007;26:385–94. [PubMed] [Google Scholar]
- 7.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 8.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–4. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 9.Pralhad T, Madhusudan S, Rajendrakumar K. Concept, mechanisms and therapeutics of angiogenesis in cancer and other diseases. The Journal of pharmacy and pharmacology. 2003;55:1045–53. doi: 10.1211/0022357021819. [DOI] [PubMed] [Google Scholar]
- 10.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Spano D, Zollo M. Tumor microenvironment: a main actor in the metastasis process. Clin Exp Metastasis. 2012;29:381–95. doi: 10.1007/s10585-012-9457-5. [DOI] [PubMed] [Google Scholar]
- 13.Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13:1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi SH, Kwon OJ, Park JY, et al. Inhibition of tumour angiogenesis and growth by small hairpin HIF-1alpha and IL-8 in hepatocellular carcinoma. Liver international : official journal of the International Association for the Study of the Liver. 2014;34:632–42. doi: 10.1111/liv.12375. [DOI] [PubMed] [Google Scholar]
- 15.Semenza GL. Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. Journal of cellular biochemistry. 2007;102:840–7. doi: 10.1002/jcb.21523. [DOI] [PubMed] [Google Scholar]
- 16.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–8. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 17.Linardou H, Kalogeras KT, Kronenwett R, et al. The prognostic and predictive value of mRNA expression of vascular endothelial growth factor family members in breast cancer: a study in primary tumors of high-risk early breast cancer patients participating in a randomized Hellenic Cooperative Oncology Group trial. Breast Cancer Res. 2012;14:R145. doi: 10.1186/bcr3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Low-Marchelli JM, Ardi VC, Vizcarra EA, van Rooijen N, Quigley JP, Yang J. Twist1 induces CCL2 and recruits macrophages to promote angiogenesis. Cancer Res. 2013;73:662–71. doi: 10.1158/0008-5472.CAN-12-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bialek P, Kern B, Yang X, et al. A twist code determines the onset of osteoblast differentiation. Developmental cell. 2004;6:423–35. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- 20.Khan MA, Chen HC, Zhang D, Fu J. Twist: a molecular target in cancer therapeutics. Tumour biology : the journal of the International Society for Oncodevelopmental Biology and Medicine. 2013;34:2497–506. doi: 10.1007/s13277-013-1002-x. [DOI] [PubMed] [Google Scholar]
- 21.El Ghouzzi V, Lajeunie E, Le Merrer M, et al. Mutations within or upstream of the basic helix-loop-helix domain of the TWIST gene are specific to Saethre-Chotzen syndrome. European journal of human genetics : EJHG. 1999;7:27–33. doi: 10.1038/sj.ejhg.5200240. [DOI] [PubMed] [Google Scholar]
- 22.Xu Y, Xu Y, Liao L, et al. Inducible knockout of Twist1 in young and adult mice prolongs hair growth cycle and has mild effects on general health, supporting Twist1 as a preferential cancer target. Am J Pathol. 2013;183:1281–92. doi: 10.1016/j.ajpath.2013.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong D, Li Y, Wang Z, Sarkar FH. Cancer Stem Cells and Epithelial-to-Mesenchymal Transition (EMT)-Phenotypic Cells: Are They Cousins or Twins? Cancers (Basel) 2011;3:716–29. doi: 10.3390/cancers30100716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vesuna F, Lisok A, Kimble B, Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. 2009;11:1318–28. doi: 10.1593/neo.91084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Liu CH, Sun Y, et al. Endothelial TWIST1 promotes pathological ocular angiogenesis. Investigative ophthalmology & visual science. 2014;55:8267–77. doi: 10.1167/iovs.14-15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shalini Singh IWYM, Handa Divya, Ghert Michelle. The Role of TWIST in Angiogenesis and Cell Migration in Giant Cell Tumor of Bone. Advances in Biology. 2014;2014 Article ID 903259. [Google Scholar]
- 27.Mironchik Y, Winnard PT, Jr., Vesuna F, et al. Twist overexpression induces in vivo angiogenesis and correlates with chromosomal instability in breast cancer. Cancer Res. 2005;65:10801–9. doi: 10.1158/0008-5472.CAN-05-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S, Kendall SE, Raices R, et al. TWIST1 associates with NF-kappaB subunit RELA via carboxyl-terminal WR domain to promote cell autonomous invasion through IL8 production. BMC biology. 2012;10:73. doi: 10.1186/1741-7007-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu J, Liong M, Zink JI, Tamanoi F. Mesoporous silica nanoparticles as a delivery system for hydrophobic anticancer drugs. Small. 2007;3:1341–6. doi: 10.1002/smll.200700005. [DOI] [PubMed] [Google Scholar]
- 30.Liong M, Lu J, Kovochich M, et al. Multifunctional inorganic nanoparticles for imaging, targeting, and drug delivery. ACS nano. 2008;2:889–96. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greish K. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Methods Mol Biol. 2010;624:25–37. doi: 10.1007/978-1-60761-609-2_3. [DOI] [PubMed] [Google Scholar]
- 32.Castanotto D, Rossi JJ. The promises and pitfalls of RNA-interference-based therapeutics. Nature. 2009;457:426–33. doi: 10.1038/nature07758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hom C, Lu J, Liong M, et al. Mesoporous silica nanoparticles facilitate delivery of siRNA to shutdown signaling pathways in mammalian cells. Small. 2010;6:1185–90. doi: 10.1002/smll.200901966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.David S, Pitard B, Benoit JP, Passirani C. Non-viral nanosystems for systemic siRNA delivery. Pharmacol Res. 2010;62:100–14. doi: 10.1016/j.phrs.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 35.Yamazaki Y, Nango M, Matsuura M, Hasegawa Y, Hasegawa M, Oku N. Polycation liposomes, a novel nonviral gene transfer system, constructed from cetylated polyethylenimine. Gene therapy. 2000;7:1148–55. doi: 10.1038/sj.gt.3301217. [DOI] [PubMed] [Google Scholar]
- 36.Xia T, Kovochich M, Liong M, et al. Polyethyleneimine coating enhances the cellular uptake of mesoporous silica nanoparticles and allows safe delivery of siRNA and DNA constructs. ACS nano. 2009;3:3273–86. doi: 10.1021/nn900918w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo P, Coban O, Snead NM, et al. Engineering RNA for targeted siRNA delivery and medical application. Adv Drug Deliv Rev. 2010;62:650–66. doi: 10.1016/j.addr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Behlke MA. Chemical Modification of siRNAs for In Vivo Use. Oligonucleotides. 2008;18:305–19. doi: 10.1089/oli.2008.0164. [DOI] [PubMed] [Google Scholar]
- 39.Czauderna F, Fechtner M, Dames S, et al. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003;31:2705–16. doi: 10.1093/nar/gkg393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meng H, Xue M, Xia T, et al. Use of size and a copolymer design feature to improve the biodistribution and the enhanced permeability and retention effect of doxorubicin-loaded mesoporous silica nanoparticles in a murine xenograft tumor model. ACS nano. 2011;5:4131–44. doi: 10.1021/nn200809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brown CE, Starr R, Martinez C, et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res. 2009;69:8886–93. doi: 10.1158/0008-5472.CAN-09-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Finlay J, Roberts CM, Lowe G, Loeza J, Rossi JJ, Glackin CA. RNA-Based TWIST1 Inhibition via Dendrimer Complex to Reduce Breast Cancer Cell Metastasis. BioMed research international. 2015;2015:382745. doi: 10.1155/2015/382745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nature protocols. 2007;2:329–33. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 44.Vernon AE, Bakewell SJ, Chodosh LA. Deciphering the molecular basis of breast cancer metastasis with mouse models. Rev Endocr Metab Disord. 2007;8:199–213. doi: 10.1007/s11154-007-9041-5. [DOI] [PubMed] [Google Scholar]
- 45.Kuperwasser C, Dessain S, Bierbaum BE, et al. A mouse model of human breast cancer metastasis to human bone. Cancer Res. 2005;65:6130–8. doi: 10.1158/0008-5472.CAN-04-1408. [DOI] [PubMed] [Google Scholar]
- 46.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 47.Wang SM, Coljee VW, Pignolo RJ, Rotenberg MO, Cristofalo VJ, Sierra F. Cloning of the human twist gene: its expression is retained in adult mesodermally-derived tissues. Gene. 1997;187:83–92. [PubMed] [Google Scholar]
- 48.Dykxhoorn DM, Lieberman J. Knocking down disease with siRNAs. Cell. 2006;126:231–5. doi: 10.1016/j.cell.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonapace L, Coissieux MM, Wyckoff J, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515:130–3. doi: 10.1038/nature13862. [DOI] [PubMed] [Google Scholar]
- 50.Martin D, Galisteo R, Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1) complex. J Biol Chem. 2009;284:6038–42. doi: 10.1074/jbc.C800207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang D, Ding Y, Zhou M, et al. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70:1063–71. doi: 10.1158/0008-5472.CAN-09-3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Teng Y, Li X. The roles of HLH transcription factors in epithelial mesenchymal transition and multiple molecular mechanisms. Clin Exp Metastasis. 2014;31:367–77. doi: 10.1007/s10585-013-9621-6. [DOI] [PubMed] [Google Scholar]
- 53.Forsbach A, Nemorin JG, Montino C, et al. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. Journal of immunology. 2008;180:3729–38. doi: 10.4049/jimmunol.180.6.3729. [DOI] [PubMed] [Google Scholar]
- 54.Meng H, Mai WX, Zhang H, et al. Codelivery of an optimal drug/siRNA combination using mesoporous silica nanoparticles to overcome drug resistance in breast cancer in vitro and in vivo. ACS nano. 2013;7:994–1005. doi: 10.1021/nn3044066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu J, Liong M, Li Z, Zink JI, Tamanoi F. Biocompatibility, biodistribution, and drug-delivery efficiency of mesoporous silica nanoparticles for cancer therapy in animals. Small. 2010;6:1794–805. doi: 10.1002/smll.201000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferris DP, Lu J, Gothard C, et al. Synthesis of biomolecule-modified mesoporous silica nanoparticles for targeted hydrophobic drug delivery to cancer cells. Small. 2011;7:1816–26. doi: 10.1002/smll.201002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vesuna F, Lisok A, Kimble B, et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-alpha. Oncogene. 2011 doi: 10.1038/onc.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Ling MT, Guan XY, et al. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene. 2004;23:474–82. doi: 10.1038/sj.onc.1207128. [DOI] [PubMed] [Google Scholar]
- 59.Tarn D, Ashley CE, Xue M, Carnes EC, Zink JI, Brinker CJ. Mesoporous silica nanoparticle nanocarriers: biofunctionality and biocompatibility. Acc Chem Res. 2013;46:792–801. doi: 10.1021/ar3000986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yanes RE, Tamanoi F. Development of mesoporous silica nanomaterials as a vehicle for anticancer drug delivery. Therapeutic delivery. 2012;3:389–404. doi: 10.4155/tde.12.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.