

Abstract
Niemann-Pick type C (NPC) disease is a fatal neurodegenerative disorder characterized by the accumulation of unesterified cholesterol in the late endosomal/lysosomal compartments. Mutations in the NPC1 protein are implicated in 95% of patients with NPC disease. The most prevalent mutation is the missense mutation I1061T that occurs in ∼15–20% of the disease alleles. In our study, an isobaric labeling-based quantitative analysis of proteome of NPC1I1061T primary fibroblasts when compared with wild-type cells identified 281 differentially expressed proteins based on stringent data analysis criteria. Gene ontology enrichment analysis revealed that these proteins play important roles in diverse cellular processes such as protein maturation, energy metabolism, metabolism of reactive oxygen species, antioxidant activity, steroid metabolism, lipid localization, and apoptosis. The relative expression level of a subset of differentially expressed proteins (TOR4A, DHCR24, CLGN, SOD2, CHORDC1, HSPB7, and GAA) was independently and successfully substantiated by Western blotting. We observed that treating NPC1I1061T cells with four classes of seven different compounds that are potential NPC drugs increased the expression level of SOD2 and DHCR24. We have also shown an abnormal accumulation of glycogen in NPC1I1061T fibroblasts possibly triggered by defective processing of lysosomal alpha-glucosidase. Our study provides a starting point for future more focused investigations to better understand the mechanisms by which the reported dysregulated proteins triggers the pathological cascade in NPC, and furthermore, their effect upon therapeutic interventions.
Niemann-Pick type C (NPC)1 disease is a rare autosomal recessive neurodegenerative disorder in which the transport of cholesterol and glycosphingolipids from late endosomal/lysosomal (LE/Ly) compartments to plasma membrane or endoplasmic reticulum (ER) is impaired. The trafficking defect leads to an excessive accumulation of these lipids in the LE/Ly compartments (1). The disease is often diagnosed in early childhood, and as it progresses there is a gradual loss of Purkinje cells in the cerebellum leading to ataxia, dysarthria, vertical supranuclear gaze palsy, and decline of neurological functions (2). NPC disease occurs with an estimated frequency of 1 in 120,000 to 150,000 live births (1). Currently, there is no cure for NPC disease, and available therapeutic efforts are focused on symptom treatment.
Approximately 95% of NPC cases are caused by mutations in the NPC1 gene, whereas the remaining 5% are because of mutations in the NPC2 gene (3). NPC1 is a large glycoprotein of 140–170 kDa with 13 transmembrane domains that resides primarily on the limiting membrane of LE/Ly compartments. At steady state, NPC1 is synthesized in the ER and targeted to the LE/Ly compartments where it mediates cholesterol transport via unknown mechanisms. To date over 254 disease-causing mutations, including both missense and nonsense mutations, have been reported on the various domains of NPC1 (4). Among these mutations, I1061T occurs in the luminal side of NPC1 protein and accounts for ∼15–20% of the disease-causing alleles in NPC patients (5). NPC1I1061T protein is synthesized but fails to advance in the secretory pathway because of its recognition as a misfolded protein by the ER quality control machinery and is consequently targeted for proteasomal degradation (5). Interestingly, if the NPC1I1061T mutant protein escapes from the ER quality control, it can properly localize to the late endosome and is functional in maintaining cellular cholesterol homeostasis (5). Because NPC1 containing the I1061T mutation is the most common mutation, detailed exploration of the proteome of NPC1I1061T cells and its comparison to wild-type will further enhance our insight into its molecular mechanisms. Moreover a better understanding of the pathophysiology of the NPC disease from such studies will facilitate implementation of effective therapeutic strategies.
Mass spectrometry-based proteomics has emerged as a preferred method for in-depth characterization and quantification of the protein components of biological systems (6). Furthermore, isobaric labeling is a powerful tool for quantitative proteomics studies, which enables concurrent identification and multiplexed quantification of proteins in different samples using tandem mass spectrometry (MS/MS) (7). To identify proteins with relevance to NPC pathogenesis because of I1061T mutation, we have used an amine-reactive six-plex tandem mass tags (TMT) isobaric reagent to differentially label and perform a proteomics comparison of primary fibroblasts derived from healthy and I1061T-mutant individuals. Three biological replicates of NPC1I1061T and NPC1WT cells were labeled with different isotopic variant of the TMT 6-plex tag, combined, and analyzed by the multidimensional protein identification technology (MudPIT) technique (8). After filtering MS/MS spectra with low reporter ion intensities from 4308 nonredundant identified proteins, a total of 3553 distinct proteins were quantified. Further data analysis enabled characterization of 281 differentially expressed proteins (DEPs) that were statistically significant (False discovery rate (FDR) = 5%). We assessed our TMT results by validating the expression level of seven proteins by Western blotting. From a therapeutic perspective, we monitored the expression of two DEPs, SOD2 and DHCR24, in the NPC1I1061T fibroblasts upon treatment with potential NPC drugs: β-cycodextrins (MβCD and HPCD) (9), histone deacetylase inhibitors (HDACIs, such as CI-994, SAHA, and VPA) (10), antioxidant N-acetyl cysteine (NAC), and an oxysterol derivative pharmacological chaperone, mo56HC (11). We have also examined the cellular glycogen levels in NPC1WT and NPC1I1061T fibroblasts by staining with periodic acid-Schiff reagents.
EXPERIMENTAL PROCEDURES
Materials
Tetraethylammonium bicarbonate (TEAB), tris(2-carboxyethyl)phosphine (TCEP), iodoacetamide, and SDS were obtained from Sigma-Aldrich (St. Louis, MO). Sequencing grade trypsin was purchased from Promega (Madison, WI). Tandem mass tags (TMT 6-plex) kit was obtained from Thermo Fisher Scientific (Rockford, IL). 2-Hydroxypropyl-β-cyclodextrin (HPCD), Methyl-β-cyclodextrin (MβCD), Valproic acid (VPA), and N-acetyl cysteine (NAC) were purchased from Sigma-Aldrich. Histone deacetylase inhibitor (HDACI) SAHA (Chemical item number 10009929) was purchased from Cayman Chemicals (Ann Arbor, MI), whereas CI-994 was obtained from Dr. Edward Holson's laboratory (Broad Institute, Cambridge). Oxysterol-derived pharmacological chaperone, mo56HC, was a gift from Dr. Kenji Ohgane's laboratory (Institute of Molecular and Cellular Biosciences, The University of Tokyo, Japan). Periodic acid-Schiff (PAS) Kit was obtained from Sigma-Aldrich.
Antibodies and Reagents
Rat anti-NPC1 monoclonal antibody was developed against the C-terminal peptide (amino acids 1261–1278) of the NPC1 protein. Mouse anti-actin (Millipore, Billerica, MA), mouse anti-tubulin (Sigma-Aldrich), and mouse anti-DHCR24/Seladin (Sigma-Aldrich) antibodies were obtained from the respective vendors. Rabbit anti-calmegin and rabbit anti-TOR4A antibodies were purchased from Abgent (San Diego, CA). Rabbit anti-SOD2 antibody was obtained from Genetex (Irvine, CA). Rabbit anti-HSPB7, rabbit anti-CHORDC1, and rabbit anti-GAA antibodies were purchased from Bioss antibodies (Boston, MA).
Cell Lines and Cell Culture
Human wild-type fibroblasts (GM05659) and NPC1-mutant fibroblasts, I1061T/I1061T (GM18453) were purchased from Coriell Cell Repositories (Coriell Institute for Medical Research). The cells were grown in DMEM medium supplemented with 2 mm l-Glutamine, 10% FBS, 50 units/ml penicillin, and 50 μg/ml streptomycin antibiotics.
Filipin Staining
Filipin staining was performed to visualize the intracellular cholesterol accumulation in NPC1 fibroblasts. Briefly, human NPC1I1061T and NPC1WT fibroblasts were grown on a 22-mm coverslip in a 6-well plate until they were 60–80% confluent. The cells were washed twice with 1× PBS (phosphate-buffer saline), fixed with 4% formaldehyde in PBS for 10 min and subsequently washed three times with PBS, followed by incubation with 1.5 mg/ml glycine for 10 min to quench the formaldehyde effect. The cells were then stained with 25 μg/ml filipin for 30 min at room temperature and rinsed three times with PBS. The coverslip was mounted on a glass slide using a Fluoromount G solution (Electron Microscopy Sciences, Hatfield, PA) and visualized by fluorescence microscopy using a UV filter set at 340–380 nm excitation.
Cell Lysis for TMT and Western Blot Analyses
Cell lysates were obtained from three biological replicates of NPC1WT and NPC1I1061T fibroblasts grown at early passages. Cells were grown to nearly 100% confluence and washed twice with ice-cold 1X PBS. Cells were then incubated with TNI lysis buffer (250 mm NaCl, 50 mm Tris pH 7.5, 1 mm EDTA, 0.5% IGEPAL CA-630, 1X protease inhibitor mixture, and 1 mm PMSF) on ice for 30 min. Clear cell lysates were obtained by centrifugation (16,000 × g) for 20 min at 4 °C. Protein concentration was measured using standard BCA assay. Fifty micrograms of cell lysates from each NPC1WT and NPC1I1061T were further processed for isobaric labeling with TMT 6-plex reagent as described in the section below.
TMT Labeling
TMT labeling was performed according to the manufacturer's instructions with some modifications. Fifty micrograms of protein from each sample was aliquoted and subjected to acetone precipitation. The protein pellets were resolubilized in 100 mm TEAB supplemented with 0.1% SDS, reduced with TCEP for 1 h at 55 °C, and then alkylated with iodoacetamide for 30 min in the dark. The denatured proteins were digested with trypsin by overnight incubation at 37 °C. Each sample digest was labeled with a specific TMT tag reconstituted in acetonitrile (ACN) and incubated for 1 h at room temperature. The NPC1WT digests were derivatized with isobaric labels 126, 128, and 130 whereas the labels 127, 129, and 131 were added to the NPC1I1061T tryptic digests. After incubation, 15 μl of 5% hydroxylamine solution was added to quench the labeling reaction. The tagged samples were combined and the ACN in the mixture was evaporated in a SpeedVac. The combined TMT-labeled peptides were further diluted in 1 ml buffer A (95% ddH2O/5% ACN/0.1% formic acid (FA)) and centrifuged at 14,000 rpm for 30 min to remove particulates. The labeled sample pool was further split into aliquots of 75 μg of peptide digest. Two of the aliquots were loaded into a biphasic trapping column (see next section) for duplicate runs of LC-MS/MS data acquisition by MudPIT method.
Mass Spectrometry (MS) Analysis
MS analysis of the samples was performed using MudPIT technology. An analytical RPLC column was generated by pulling a 100 μm ID/360 μm OD capillary (Polymicro Technologies, Inc., Phoenix, AZ) to 3 μm ID tip. The pulled column was packed with reverse phase particles (Aqua C18, 3 μm dia., 90 Å pores, Phenomenex, Torrance, CA) until 15 cm long. A biphasic trapping column was prepared by creating a Kasil frit at one end of an undeactivated 250 μm ID/360 μm OD capillary (Agilent Technologies, Inc., Santa Clara, CA), which was then successively packed with 2.5 cm strong cation exchange (SCX) particles (Partisphere SCX, 5 μm dia., 100 Å pores, Phenomenex) and 2.5 cm reverse phase particles (Aqua C18, 5 μm dia., 90 Å pores, Phenomenex). The trapping column was equilibrated using buffer A prior to sample loading. After sample loading and prior to MS analysis, the resin-bound peptides were desalted with buffer A by letting it flow through the biphasic trap column. The trap and analytical columns were assembled using a zero-dead-volume union (Upchurch Scientific, Oak Harbor, WA).
LC-MS/MS analysis was performed on LTQ Orbitrap Velos (Thermo Scientific, San Jose, CA) interfaced at the front end with a quaternary HP 1100 series HPLC pump (Agilent Technology) using an in-house built electrospray stage. Electrospray was performed directly from the analytical column by applying the ESI voltage at a tee (150 μm ID, Upchurch Scientific) downstream of a 1:1000 split flow used to reduce the flow rate to 250 nL/min through the columns. A fully automated 12-step MudPIT run was performed on each sample using a three mobile phase system consisting of buffer A (5% ACN; 0.1% FA), buffer B (80% ACN, 0.1% FA), and buffer C (500 mm ammonium acetate, 5% ACN, 0.1% FA). The first step was a 60 min reverse-phase run, whereas subsequent steps were of 120 min duration and included steps with 10, 20, 30, 40, 50, 60, 70, 80, 90, and 100% buffer C run for 4 min at the beginning of the gradient and a last step that includes salt bump of 90% buffer C with 10% buffer B for 4 min.
As peptides eluted from the microcapillary column, they were electrosprayed directly into the mass spectrometer with the application of a distal 2.4 kV spray voltage. Peptides were analyzed using a Top-10 data-dependent acquisition method in which fragmentation spectra are acquired for the top 10 peptide ions above a predetermined signal threshold. For each cycle, full-scan MS spectra (m/z range 300–1600) were acquired in the Orbitrap with the resolution set to a value of 30,000 at m/z 400 and an automatic gain control (AGC) target of 1 × 106 ions and a maximal injection time of 250 ms. Each full scan was followed by the selection of the most intense ions, up to 10, for higher-energy collisional dissociation (HCD)-MS/MS analysis with a mass resolution of 7500 in the Orbitrap. For MS/MS scans, the target value was 30,000 ions with an injection time of 150 ms. Once analyzed, the selected peptide ions were dynamically excluded from further analyses for 120 s to allow for the selection of lower-abundance peptide ions for subsequent fragmentation and detection. The settings for repeat count = 1, repeat duration = 30 ms, and exclusion list size = 500. Charge state filtering was enabled to reject fragmenting of ions with singly or unassigned charge states. The minimum MS signal for triggering MS/MS was set to 5000 and an activation time of 0.1 ms was used. All tandem mass spectra were collected using an isolation window of 2 Th and a normalized collision energy of 45%.
Data Analysis
Tandem mass spectra were extracted from the Xcalibur data system format (.raw) into MS2 format using RawXtract1.9.9.2. The MS/MS spectra were searched with the ProLuCID algorithm against the human SwissProt database (downloaded in July 2013; number of protein entries = 20,210), which was concatenated to a decoy database in which the sequence for each entry in the original database was reversed. The search parameters include 10 ppm peptide precursor mass tolerance and 25 ppm for the fragment mass tolerance acquired in the Orbitrap; carbamidomethylation (57.02146) on cysteine and TMT modification (229.1629) on N terminus and lysine were defined as fixed modification in the search criteria. The search space only included fully tryptic peptide candidates of length of at least six amino acids. The maximum number of internal miscleavages was kept unlimited, thereby allowing all cleavage points for consideration. ProLuCID outputs were assembled and filtered using the DTASelect2.0 (12) program that groups related spectra by protein and removes those that do not pass basic data-quality criteria. DTASelect2.0 combines the XCorr and ΔCn measurements and computes confidence scores to achieve a user-specified protein false discovery rate (1% for the current study).
Census (13), a software tool for quantitative proteomics analysis, was used to extract the TMT reporter ion intensities from the identified tandem mass spectra, correction of isotope contamination, and normalization. Mass tolerance and intensity thresholds (sum of six reporter ion intensities in each spectrum) of the reporter ions in Census were set at 0.05 Da and ≥ 2.5 × 104, respectively. Because equal amounts of proteins were loaded across all six channels, reporter ion intensities in each reporter ion channel were normalized by the sum of all reported ion intensities of the corresponding channel. A box plot of the log10-transformed normalized reporter ion intensity illustrating well-centered median intensity of each isobaric tags of a TMT 6-plex experiment is shown in supplemental Fig. S1A in Supporting Information. Normalized reporter ion intensities from Census were used to compute protein expression ratios (mutant/wild-type) and assess the statistical significance of protein differential expression. Ratios of the normalized reporter ion intensities (NPC1I1061T/NPC1WT = 127/126, 129/128 and 131/130) were calculated from each MS/MS spectrum and log2-transformed. Protein ratios are computed as the median of the log2-transformed reporter ion intensity ratios of all the spectra of peptides assigned to a given protein. The MS/MS spectra with redundant peptide sequences were included when computing the protein expression ratio, as each spectrum provides unique TMT quantitation measurements. To assess the variation in ratio measurements, median absolute deviation (MAD) of the reporter ion intensity log2 ratios for each quantified protein was calculated from the equation:
where Xi are log2-transformed individual reporter ion ratios. MAD values can be used as an indicator of the quantification quality for each protein. Lower MAD values represent a greater precision in protein expression ratios. Histograms displaying the distribution of MAD in the two TMT-MudPIT analyses are shown in supplemental Fig. S1D in Supporting Information.
Assessment of Statistical Significance of Protein Differential Expression
To maximize specificity and exclusively report high confidence results, a candidate protein must meet two criteria in order to qualify as a statistically significantly differentially expressed protein (DEP). Protein expression ratios for channel pairings 127/126, 129/128, and 131/130 were obtained by averaging the normalized reporter ion intensity ratios of all spectra of the peptides assigned to a given protein. One sample t-tests were performed on these ratios to assess the statistical significance of protein differential expression between NPC1I1061T and NPC1WT fibroblasts and assign a p value to each protein in the data set. Because 3553 proteins were quantified, we used the Benjamini-Hochberg procedure to estimate a false discovery rate (FDR) at a given p value threshold to control for multiple hypothesis testing. A given protein to be classified as a DEP must obtain a p value that corresponds to a 5% FDR in each replicate in which it was quantified (p value < 0.013 for experimental replicate-01 and p value < 0.008 for experimental replicate-02). For the second criterion, we transformed the previously described protein median expression ratios (NPC1I1061T/NPC1WT fold-change) into Z-scores. A candidate protein to be considered as a DEP must therefore also be associated in all replicates in which it was quantified with Z-scores smaller than −1 or greater than 1 (i.e. a protein ratio, which is at least one standard deviation away from the mean protein expression ratio in the entire data set). Supplemental Tables S2 in Supporting Information includes the list of the 281 DEPs identified in the proteome comparison of NPC1I1061T versus NPC1WT fibroblasts.
Gene Ontology Enrichment Analysis
To obtain an overview of the biological significance of the DEP data set, we performed a Gene Ontology (GO) (14) enrichment analysis using the Ontologizer (15) software tool. The Gene Ontology human database was downloaded on December 19, 2013. The set of 281 DEPs was interrogated for GO term enrichment, whereas the total number of quantified proteins in the replicate TMT-MudPIT analyses (3553) was used as background. The reported p values of significantly enriched GO terms are corrected for multiple hypothesis testing.
Western Blot Validation
Western blot analysis of selective proteins was performed to validate TMT-MudPIT results. Human NPC1I1061T and NPC1WT fibroblasts were lysed with TNI lysis buffer and centrifuged at 16,000 × g for 20 min at 4 °C, as described in the Cell lysis section above. Twenty-five μg of cell lysates were subjected to SDS-PAGE and Western blotting against the specific primary antibodies to analyze the expression level of respective proteins in NPC1I1061T and NPC1WT fibroblasts. Actin, tubulin, GAPDH or HSP90 was used as a loading control in the immunoblotting. The dilutions of antibody used are as follows: rat anti-NPC1 antibody (1:1000), mouse anti-DHCR24 (1:1000), rabbit anti-TOR4A (1:1000), rabbit anti-CLGN antibody (1:1000), rabbit anti-SOD2 (1:2000), rabbit anti-CHORDC1 (1:1000), rabbit anti-HSPB7 (1:1000), rabbit anti-GAA (1:2000), mouse anti-actin (1:10,000), and rat anti-tubulin (1:10,000). To study the effect of potential NPC drugs on the expression of SOD2 and DHCR24, we have treated NPC1WT fibroblasts with DMSO and NPC1I1061T fibroblasts with either DMSO or the drugs (concentration indicated in the legend of Fig. 6) for 72 h followed by Western blotting with respective antibodies. The blots were quantified with Image J software.
Fig. 6.

Effect of drugs on expression level of SOD2 and DHCR24. A, Human NPC1WT and NPC1I1061T fibroblasts were treated with DMSO or 10 μm of SAHA for 72 h. Cell lysates were subjected to SDS-PAGE, followed by Western blotting with primary antibodies against NPC1 and tubulin proteins. B, Human NPC1WT fibroblast was treated with DMSO and NPC1I1061T fibroblast with DMSO or seven drugs of four different categories—cyclodextrins: MβCD (0.5 mm) and HPCD (0.5 mm); HDACIs: CI-994 (10 μm), SAHA (10 μm), and VPA (4 mm); antioxidant: NAC (5 mm); and an oxysterol derivative pharmacological chaperone: mo56HC (10 μm) for 72 h. Cell lysates were immunoblotted against rabbit anti-SOD2 antibody. C, Human NPC1WT and NPC1I1061T fibroblasts were treated with the drugs and for the duration as mentioned above and immunoblotted against mouse anti-DHCR24 antibody. Tubulin was used as a loading control. The blots were quantified with the Image J software. In the bar graph panels, * indicates statistically significant (p < 0.05) difference in drug-treated NPC1I1061T cells compared with DMSO-treated NPC1I1061T cells.
Periodic Acid-Schiff (PAS) Staining
PAS staining was performed in NPC1WT and NPC1I1061T fibroblasts according to the manufacturer's instructions (Sigma-Aldrich). Briefly, human NPC1I1061T and NPC1WT fibroblasts were grown on a 22-mm coverslip in a 6-well plate until they were 60–70% confluent. The cells were washed twice with 1X PBS, fixed with 4% formaldehyde for 30 min at room temperature and subsequently washed three times with PBS. The cells were then incubated with periodic acid solution for 5 min at room temperature and rinsed with four changes of distilled water. The cells were further incubated with Schiff's reagent for 15 min at room temperature and rinsed with several changes of distilled water. The cells were counterstained in Hematoxylin solution for 90 s and washed four to five times with distilled water. The coverslips were mounted on a glass slide using a Fluoromount G solution (Electron Microscopy Sciences) and examined under a bright-field microscopy (Carl Zeiss). Images were taken with 20× objective magnification.
RESULTS
NPC1 protein with I1061T mutation is unable to fold correctly and consequently targeted for proteasomal degradation (5). Fig. 1A shows the lower expression level of NPC1I1061T compared with NPC1WT in primary fibroblasts. Cells harboring NPC1I1061T exhibit abnormal accumulation of unesterified cholesterol within LE/Ly compartments (5). In these cells, LDL receptor expression is not down-regulated, and consequently, LDL uptake continues to occur despite the increased cellular content of free cholesterol (16). Staining the cells with cholesterol specific dye filipin reveals massive accumulation of unesterified cholesterol in NPC1I1061T cells, whereas NPCWT cells exhibited normal cholesterol homeostasis (Fig. 1B).
Fig. 1.
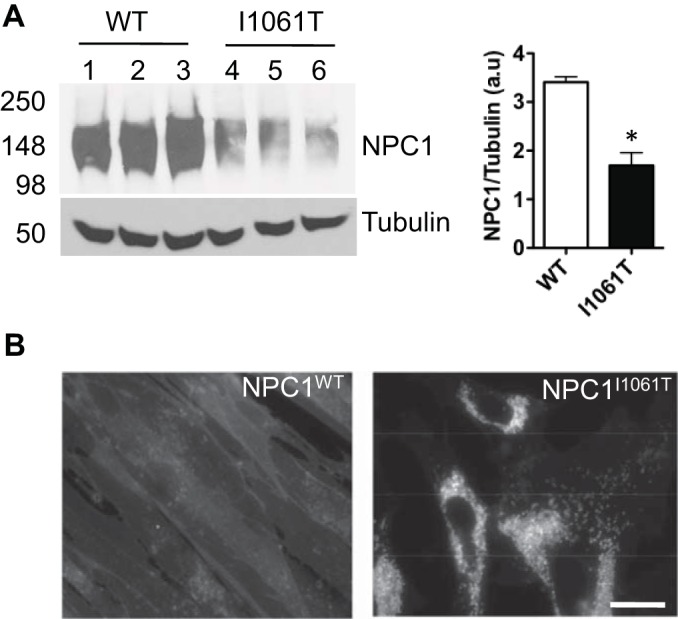
A, Western blot showing the down-regulation of NPC1 protein in NPC1I1061T fibroblasts when compared with NPC1WT. Twenty-five μg of cell lysates were resolved in 8% SDS-PAGE and immunoblotted with rat anti-NPC1 antibody. For equivalent loading control, tubulin was probed with mouse anti-tubulin antibody. Band intensities were quantified using the Image J software. * indicates statistically significant (p < 0.05) difference in NPC1I1061T expression level relative to NPC1WT. B, Filipin staining visualization of massive accumulation of unesterified cholesterol in late endosomal/lysosomal compartments in NPC1I1061T cells relative to wild-type skin fibroblasts. Cells were visualized by fluorescence microscopy using a UV filter set at 340–380 nm excitation. Scale bar is 10 μm.
Isobaric Labeling-based Quantitative Proteome Profiling of NPC1I1061T and NPC1WT Cells
Fibroblasts derived from skin of NPC1 patients exhibit profound and reproducible cholesterol accumulation in LE/Ly compartments, and therefore, provide a robust in vitro cellular model to study NPC1 disease (17). In an effort to investigate the relative expression level of proteins in NPC1I1061T compared with NPC1WT fibroblasts, we have used TMT-based isobaric labeling followed by data acquisition using MudPIT with a LTQ Orbitrap Velos instrument. Isobaric labeling allows quantitative comparison of protein abundances by measuring peak intensities of reporter ions released from TMT-tagged peptides by fragmentation during MS/MS. The MudPIT technology is an unbiased discovery-based method for rapid yet nearly comprehensive proteome analysis where increasing levels of salt are used for stepwise elution of peptides from the SCX resin onto the reversed-phase resin (8). An overview of the experimental approach used in this study is depicted diagrammatically in Fig. 2A.
Fig. 2.

A, Diagrammatic representation of the experimental workflow. B, Venn diagram of the proteins identified in the two TMT-MudPIT experimental replicates. From a total of 4308 nonredundant proteins (3282 proteins in replicate-01 and 3849 proteins in replicate-02), 2823 were detected in both data sets. C, Venn diagram of the proteins quantified in the two TMT-MudPIT experimental replicates. Out of 3553 total quantified proteins from two replicates, an overlap of 2283 quantified proteins were observed in both. D, Scatter plot showing the number (in log base 2) of peptides and spectra match per protein in TMT-MudPIT experimental replicate-01. The color intensity of the hexagon indicates the number of binned proteins. For example, in the TMT-MudPIT experimental replicate-01, around 800 proteins (blue dot) were quantified with a single peptide and one MS/MS spectrum each and so on. The actual number of proteins and their respective spectral counts are tabulated in supplemental Fig. S1C. Results from replicate-02 are shown in the supplemental Fig. S1B.
Six isobaric tags from the TMT 6-plex reagent were utilized to label three biological replicates of each NPC1WT and NPC1I1061T samples. The analyses of the TMT-labeled samples in two MudPIT experimental replicates resulted in an identification of 3282 nonredundant proteins in one replicate and 3849 in the other, with a total of 4308 distinct identified proteins (Fig. 2B). Of the total identified proteins, 66% (2823) identifications were observed in both TMT-MudPIT replicate runs. For further determination of protein quantification ratios, only the identified peptides yielding MS/MS spectra with high reporter ion intensities (i.e. sum of six reporter ion intensities ≥ 2.5 × 104) were used. Intensity filtering is performed because low reporter ion signals tend to produce important quantification inaccuracies (18). Protein expression ratios were calculated as the median of the log2 ratios of reporter ion intensities of all MS/MS spectra of their corresponding peptides. A total of 3553 proteins were quantified from the two replicates of TMT-MudPIT runs with 2283 quantified proteins present in both data sets whereas an additional 629 and 641 proteins were unique to the two replicates (Fig. 2C). The number of peptides and the number of spectra used in the estimation of each protein ratio is illustrated in Fig. 2D and supplemental Fig. S1B and tabulated in supplemental Fig. S1C. The log2-transformed reporter ion intensity ratios were also used to calculate the median absolute deviation (MAD) for each protein in order to assess the variability in measured protein expression ratios (supplemental Fig. S1D). Approximately 95% of the proteins have a MAD value below 0.25. The complete list of all the quantified proteins and their abundance ratios are shown in supplemental Table S1 in the Supporting Information section.
Inference of Differentially Expressed Proteins (DEPs) in NPC1I1061T Relative to NPC1WT Fibroblasts
We used one-sample t-test with multiple hypothesis testing adjustment to identify proteins in NPC1I1061T cells that show statistically significant changes in expression when compared with wild-type fibroblasts. Proteins were considered as differentially expressed if their adjusted p values correspond to an FDR smaller or equal to 5% and their measured ratio (NPC1I1061T/NPC1WT) Z-score is ≤ −1 or ≥ 1. In addition, if observed in both replicates, these proteins should be quantified with consistent expression ratios i.e. up- or down-regulated in both the experimental replicates. Based on these parameters, a total of 281 proteins were qualified as significantly differentially expressed in NPC1I1061T fibroblasts relative to NPC1WT, among which 168 proteins appeared in both replicates. One hundred thirteen (40%) of the 281 DEPs were found to be significantly up-regulated, whereas the other 168 (60%) were down-regulated. The plots in Fig. 3(A, B) and supplemental Fig. S2 show the log2 protein expression ratios and corresponding peptide reporter ion intensity ratios of the DEPs in NPC1I1061T versus NPC1WT comparison. The validity of our TMT-based experimental setup and data analysis strategy is supported by the fact that the relative reporter ion intensity of NPC1 in NPC1I1061T cells, which is expected to be lower when compared with NPC1WT, was indeed decreased (Fig. 3C). The list of 281 DEPs along with their measured ratios, number of observed MS/MS spectra, p values, and MAD values are provided in supplemental Table S2 in Supporting Information section.
Fig. 3.

Differentially expressed proteins (DEPs) that are observed in both TMT-MudPIT experimental replicates: A, down-regulated B, up-regulated. The protein expression ratios are computed as the median of the log2 ratios of the reporter ion intensities of all the MS/MS spectra of peptides determined for each protein. Protein expression ratios in NPC1I1061T/NPC1WT are depicted by larger squares whereas the smaller squares represent the peptide reporter ion intensity ratios. TMT-MudPIT experimental replicates are color-coded (blue = replicate-01 and red = replicate-02). DEPs observed in only one of the replicates are shown in supplemental Fig. 2. C, Box plot showing reporter ion intensity values of a peptide mapped to the NPC1 protein in NPC1WT and NPC1I1061T fibroblasts.
Gene Ontology Enrichment Analysis
To understand the biological significance of the 281 DEPs and gain insight of their roles in the pathophysiological processes of NPC disease, a primary question to ask is whether these differentially expressed proteins are enriched for in certain Gene Ontology (GO) terms. Hence, we assessed the statistical significance of the DEPs identified by our proteomics analysis on enriched processes with Ontologizer (15). Of the total input of 281 proteins, 158 were mapped to at least one GO term (supplemental Table S3) that was statistically significantly enriched (adjusted p value < 0.05). Fig. 4A shows the observed profile of enriched and highly diverse GO terms such as lipid localization, lipid binding, reactive oxygen species (ROS) metabolic process, antioxidant activity, steroid metabolic process, apoptosis, and others that are of particular interest in the context of NPC disease. The overrepresentation of proteins with function in ROS metabolic process and antioxidant activity (19–21), apoptosis, and energy metabolism (22) corroborates previous observations on NPC and thus adds an independent validation to our data set. The bar chart in Fig. 4B displays selective DEPs and their log2 protein expression ratios that are involved in biological functions such as energy metabolism, apoptosis and anti-apoptosis, lipid metabolism and localization, and ROS metabolism and antioxidant activity.
Fig. 4.

A, Gene Ontology (GO) terms that are significantly enriched among differentially expressed proteins (DEPs). The number in the bubbles corresponds to the number of proteins present in our DEP data set that are annotated to the respective GO term. Out of 281 DEPs, 158 distinct proteins are represented in the depicted 27 GO terms. B, Bar graph of selective DEPs with log2 ratios that are involved in energy metabolism, lipid metabolism and localization, ROS metabolism and antioxidant activity, and apoptosis and anti-apoptosis. Observations from TMT-MudPIT experimental replicates are color-coded (blue = replicate-01 and red = replicate-02). Note: Unpaired bars represent proteins that are observed in only one of the experimental replicates. C, Subcellular localizations of 107 DEPs. Blue indicates up-regulated and green indicates down-regulated proteins in NPC1I1061T relative to NPC1WT cells.
In addition, 107 DEPs were linked to key subcellular organelles such as mitochondria, ER, Golgi apparatus, lysosome, and endosome (Fig. 4C). Mitochondrial dysfunction plays an important role in NPC pathogenesis (19). The perturbation of mitochondrial proteins can be associated with the observation that endosomal cholesterol is transported to the mitochondria in the absence of functional NPC1 leading to a buildup of cholesterol (19, 23). Mitochondria are important for sterol metabolism and they harbor the cytochrome P450 side chain cleavage enzyme, which converts cholesterol into pregnenolone, the precursor of steroids (24). Additionally, mitochondria could be a significant source of oxidative stress in NPC disease (19). Proteins that are localized in the lysosomal and endosomal compartments function in maintaining normal cholesterol homeostasis. Fifteen out of 20 DEPs that are annotated as lysosomal in the GO database (Fig. 4C) were also present in The Human Lysosome Gene Database (hLGDB) (25) (http://lysosome.unipg.it/) (supplemental Fig. S3A) thus substantiating the correct cellular localization of these proteins in the GO database. The hLGDB database is a first resource that provides a comprehensive and accessible census of the human genes belonging to the lysosomal system (25). The database was developed by collecting and annotating gene lists from many different sources. The overlap and unique sets of proteins mapped to the three different sub-endosomal compartments, early endosomes, late endosomes, and recycling endosomes, are shown in Venn diagram in supplemental Fig. S3B.
Validation of TMT Results of Selective Proteins from DEPs Data Set
The use of specific antibodies provides a method for protein identification and quantification that is independent of mass spectrometry. We used Western blotting to validate the expression of seven differentially expressed proteins with roles in biological processes such as protein folding and maturation, cholesterol metabolism, glycogen metabolism, and antioxidant activity. These proteins are localized at various cellular compartments, for example, CLGN (Calmegin), DHCR24 (3 β-hydroxysteroid-Δ 24 reductase), and TOR4A (Torsin Family 4, Member A) are ER proteins whereas chaperones HSPB7 and CHORDC 1 (cochaperone of HSP90) are cytosolic proteins. SOD2 (Superoxide dismutase 2, mitochondrial) is localized in a mitochondrion matrix and GAA (lysosomal α-glucosidase) is a lysosomal protein.
In corroboration with the TMT data, TOR4A was up-regulated in NPC1I1061T cells compared with NPC1WT fibroblasts (Fig. 5A). TOR4A is a member of AAA+ ATPase superfamily of proteins. It is localized in the lumen of the ER and the nuclear envelope and actively involved in the degradation of misfolded proteins through the ERAD pathway (26). Up-regulation of TOR4A protein may promote the degradation of NPC1I1061T protein through the ERAD pathway. An increased abundance of DHCR24 was also observed in the TMT data set and was further confirmed by Western blotting (Fig. 5B). DHCR24 protein is of particular interest and potentially relevant in the context of NPC disease. It is an ER-localized multifunctional enzyme and has shown to protect neuronal cells from apoptosis induced by ER stress (27). Interestingly, apoptosis is one of the enriched GO terms in the analysis of DEPs data set with Ontologizer (Fig. 4A and 4B). DHCR24 is also one of the essential enzymes involved in the final step of cholesterol synthesis (28).
Fig. 5.

Western blot analyses confirming the identity and differential expression of selected candidates identified by our TMT quantitative study. Twenty-five μg of cell lysates, obtained from an independent set of NPC1I1061T and NPC1WT fibroblasts, were subjected to 8%, 10% or 12% SDS-PAGE and Western blotting. (A) TOR4A and (B) DHCR24 proteins were up-regulated in NPCII1061T cells, whereas (C) CLGN, (D) SOD2, (E) CHORDC1, (F) HSPB7, and (G) GAA proteins were down-regulated. The expression of actin, tubulin, GAPDH, and HSP90 proteins were unchanged and used as a loading control. In all the bar graph images, * indicates statistically significant (p < 0.05) difference in I1061T cells relative to wild-type cells. H, Bright-Field microscopy images of NPC1WT and NPC1I1061T fibroblasts stained with Periodic Acid-Schiff (PAS) reagent and showing cellular glycogen content.
CLGN, SOD2, CHORDC1, HSPB7, and GAA are among the proteins that were found to be significantly down-regulated in the TMT results and were further confirmed by Western blotting (Fig. 5C–G). CLGN is an ER-localized molecular chaperone involved in spermatogenesis and fertility; however, the molecular mechanism of CLGN in protein folding is poorly understood (29). SOD2 localizes in the inner membrane of mitochondria and defends against oxidative stress by transforming toxic superoxide into hydrogen peroxide (30). CHORDC1 is an ADP-dependent HSP90-interacting protein and is proposed to act as its cochaperone and assist in protein folding (31). Chaperone HSPB7 belongs to a family of small heat shock proteins that have a role in preventing aggregation of unfolded or misfolded proteins (32). GAA is synthesized as a precursor protein of 110 kDa, which undergoes different stages of proteolytic processing and N-glycosylation events before proceeding to the lysosomal compartments. GAA is essential for the degradation of glycogen to glucose in lysosomes. Interestingly, GAA showed the processing defect in NPC1I1061T fibroblasts compared with NPC1WT and is mostly retained in its precursor form (Fig. 5G). This observation also prompted us to compare the level of glycogen in NPC1I1061T fibroblasts to that of NPC1WT fibroblasts because deficiency of GAA causes the accumulation of glycogen in the lysosome resulting in a glycogen storage disease (Pompe disease) (33). Periodic acid-Schiff (PAS) staining result shows the abnormal accumulation of glycogen in NPC1I1061T fibroblasts compared with NPC1WT (Fig. 5H).
Drug Treatment Increases the Expression Level of SOD2 and DHCR24 in NPC1I1061T Fibroblasts
Various classes of potential drugs have been investigated in the treatment of NPC disease in both cellular and animal models (9, 10, 34) and some of them are in clinical trials. We have examined the effect of seven types of drugs, belonging to four different classes of compounds—cyclodextrins, histone deacetylase inhibitors (HDACIs), N-acetylcysteine (NAC), and oxysterol-derived pharmacological chaperone (mo56HC)—on the expression level of SOD2 and DHCR24 in NPC1I1061T fibroblasts. These two proteins were chosen as representatives because they are involved in ROS or cholesterol metabolism, or a combination of both.
Treatment with cyclodextrins, methyl-β-cyclodextrin (MβCD) and 2-hydroxylpropyl-β-cyclodextrin (HPCD) marginally increased the expression of SOD2 protein in NPC1I1061T fibroblasts compared with DMSO-treated cells (Fig. 6B). The expression of DHCR24 was also slightly increased when treated with MβCD and HPCD cyclodextrins (Fig. 6C).
HDACIs are a class of structurally diverse compounds that interfere with the function of different classes of histone deacetylases (HDACs) (10). Our study confirms that treatment with Vorinostat (Suberanilohydroxamic acid, SAHA), an inhibitor of class I and II HDACs, increases the expression and stabilization of NPC1I1061T in mutant fibroblasts compared with DMSO-treated cells (Fig. 6A). No effect was observed on the expression of NPC1 in SAHA- or DMSO-treated wild-type fibroblasts. Next, we analyzed the effect of three different HDACIs—CI-994 (benzamide derivative inhibitor of class I HDAC), SAHA, and valproic acid (VPA, short-chain aliphatic acid inhibitor of class I and II HDACs)—on the expression level of SOD2 and DHCR24 proteins. Treatment with these compounds increased the level of SOD2 in NPC1I1061T fibroblasts to the level nearly equivalent to DMSO-treated wild-type fibroblasts (Fig. 6B). Interestingly, treatment with the HDACIs (CI-994, SAHA, and VPA) also increased the level of DHCR24 in NPC1I1061T fibroblasts but beyond its level in DMSO-treated wild-type fibroblasts (Fig. 6C).
Finally, we tested the effect of drugs NAC and mo56HC on SOD2 and DHCR24 levels. Our Western blot results show that treatment with NAC and mo56HC increased the level of SOD2 and DHCR24 in NPC1I1061T fibroblasts when compared with DMSO-treated cells (Fig. 6B, 6C).
DISCUSSION
An unbiased proteomics profiling experiment holds the potential for discovery of perturbed molecular pathways underlying a complex disease process. Only a few profiling studies for NPC disease have been reported to date. In one study, Cluzeau et al. characterized changes in liver gene expression in NPC mouse model at six ages spanning the pathological progression of the disease in order to identify mechanisms underlying NPC and uncover potential biomarkers (35). In another study, the liver and cerebellum gene expression patterns in NPC mice were analyzed by qPCR and oligonucleotide microarray techniques (36). Their study shows a differential expression of hepatic as well as cerebellar genes associated with oxidative stress, fibrosis, and inflammation. Using cDNA microarrays, Reddy et al. analyzed the genome-wide expression patterns of human fibroblasts homozygous for the NPC1I1061T mutation (37). The study identified a distinct gene expression profile in NPC fibroblasts when compared with fibroblasts isolated from normal subjects revealing many intriguing similarities with classic neurodegenerative diseases. Gene expression analysis by Windt et al. identified 66 differentially expressed genes in the NPC cells when compared with healthy controls (37 up- and 29 down-regulated) (38). Although these studies are useful, direct investigation at the protein level is important because gene or mRNA expression changes do not necessarily act as accurate predictors for changes in the proteome (39). Protein levels are influenced by the amount of mRNA and its rate of translation as well as the rate of protein turnover based on their half-life. For example, despite similar NPC1 mRNA levels between wild-type and NPC1I1061T fibroblasts, NPC1 protein level decreases by 85% in NPC1I1061T cells (5). Hence, only comparative proteome analysis provides a functional understanding of the biochemical processes that are deranged in NPC1I1061T cells. The outcome of such studies will further enable us to explore avenues for potential drugs based on observed dysfunctional molecular manifestations. In a quantitative proteomics study using two-dimensional gel electrophoresis and mass spectrometry, 77 DEPs were identified in NPC1-mutant mice cerebella compared with controls (40). Two-dimensional gel electrophoresis and MALDI-TOF mass spectrometry were used to resolve the hippocampal protein expression profiles of 4- and 8-week old wild-type and NPC-deficient mice (41).
In this study, we have used a high throughput isobaric labeling-based protein quantification approach in conjunction with MudPIT to characterize differentially regulated proteins in NPC1I1061T in relation to NPC1WT fibroblasts.
Isobaric Labeling-based Relative Quantification
Isobaric quantification is an effective tool for global proteome profiling and characterization of proteins that are differentially expressed in NPC fibroblasts. The TMT isobaric reagent consists of three groups: a mass reporter group, a mass-balancing group, and an amine-reactive functional group that labels primary amines (NH2-terminal and ε-amino group of the lysine side chain) of peptides. The isobaric mass design of the TMT six-plex reagent results in the differentially labeled peptides from six samples appearing as a single peak in MS scan, but when subjected to MS/MS analysis, the mass-balancing carbonyl moiety is released as a neutral loss and liberates isotope-encoded reporter ions (7). The peak intensity of the reporter ions provides relative quantitative information on the proteins from which the peptides originate. The relatively nonspecific nature of TMT labeling permits quantification of the identified proteins by multiple peptides, thereby increasing the confidence in the reported protein ratios. Additionally, the additive effect on precursor intensities when samples are multiplexed results in increased sensitivity. Also, because six samples can be combined and analyzed together, the MS analysis time is considerably reduced. With this strategy more than 4000 proteins were identified from two experimental replicates with quantitative information of a total of 3553 distinct proteins. We identified 281 proteins that were statistically significant and differentially regulated in NPC1I1061T fibroblasts relative to NPC1WT.
Corroboration of DEPs in our Data Set with Previous Studies on NPC Disease
Literature searches showed that a subset of DEPs observed in our data set have previously been found to be associated with NPC pathogenesis. For example, we observed an up-regulation in the expression level of Galectin-3 (LGALS3) protein in NPC1I1061T cells compared with NPC1WT. LGALS3, a pro-inflammatory molecule, showed an increased expression in the liver of NPC1 mouse model as well as elevated levels in serum of NPC1 patients (35). Their serum concentration is found to be correlated with neurological disease severity and appears to be specific for NPC1, suggesting that it could be an indicator of disease progression and might serve as a biomarker to monitor efficacy in therapeutic trials (35). In accordance with our results in fibroblasts, FABP5, a member of the fatty acid binding protein family, was found to be increased, whereas the level of IDH2 (isocitrate dehydrogenase), an enzyme involved in citric acid cycle, was decreased in the NPC1-mutant cerebellum relative to control (40). Our proteomic observation also substantiated the gene expression analyses results for NPC and healthy fibroblasts by Windt et al. (38) for proteins EPB41L3, COL12A1, and POSTN that were up-regulated and MYH11, PPL, NT5E, RABGAP1, and CRABP2 that were down-regulated in their study. Other proteins, FDPS, LSS, WNT5A, SPARC, SLC1A5, PTGIS, HMOX1, FBLN1, and ITGA11, were also detected to be differentially expressed (corroborating Windt et al. observations); however, these proteins did not surpass the stringent p value and/or Z-score thresholds of our study and were therefore not listed in the DEPs data set. Furthermore, our DEPs data set includes proteins such as STUB1, UCHL1, SUMO1, and many others that were found to be perturbed in NPC1I1061T fibroblasts. The roles of these proteins are reported in various neurodegenerative diseases (42–44).
The 281 differentially regulated proteins observed in the NPC1I1061T cells belong to diverse functional categories (Fig. 4A) and are localized at various cellular subcompartments (Fig. 4C). A subset of these proteins and enriched GO categories have been shown previously in their association with NPC; however, others have not been noted prior to this work. The functional roles of some of these additional proteins that are of potential relevance to NPC pathogenesis are discussed below.
Protein Folding and Degradation
Protein maturation in the ER is subjected to stringent quality control. The role of ER quality control machinery is to keep the polypeptides from being delivered to their sites of function until they are properly folded, thereby limiting cytotoxicity of accumulated misfolded proteins (45). Proteins recognized as misfolded by ER quality control are degraded through the ubiquitin-proteasome pathway. The molecular mechanism of loss of function for NPC1I1061T has been proposed to involve a folding defect that fails to undergo normal post-translational glycosylation (5). Nearly all of the NPC1I1061T mutant proteins are retained in the ER and targeted for proteasomal degradation (5). We have identified a repertoire of proteins in our DEP data set that are implicated in protein folding and degradation. For example, TOR4A, a cofactor required for chaperone-mediated protein folding (26) increased in its abundance in NPC1I1061T fibroblasts. CLGN, a chaperone protein localized in the ER membrane and belonging to Calreticulin protein family was detected as being down-regulated (29). Wolframin (WFS1) is an ER membrane glycoprotein that participates in the regulation of cellular calcium homeostasis. The expression of a number of calcium-regulated and regulating proteins has been shown to be perturbed in NPC fibroblasts by microarray studies (37). NPC1-mutant cells are known to have defective lysosomal Ca2+ homeostasis when compared with wild-type cells (46). Targeting ER calcium levels using ryanodine receptor (RyR) antagonists increases the steady-state levels and promotes trafficking of the NPC1I1061T protein to the LE/Ly compartments (47). STUB1 (E3 ubiquitin-protein ligase CHIP) acts as a link between the chaperone and proteasome systems (48). It interacts with the molecular chaperones and results in client substrate ubiquitylation and degradation by the proteasome. The relative expression level of STUB1 was up-regulated in NPC1I1061T compared to NPCWT cells. However, the relative expression level of another E3 ubiquitin ligase, RBCK1 (RanBP-type and C3HC4-type zinc finger-containing protein 1), decreased. We observed a slight up-regulation of protein AUP1 (ancient ubiquitous protein 1). AUP1 is involved in the degradation of misfolded proteins and localizes to both the ER and to lipid droplets (49). Its expression level affects the abundance of cellular lipid droplets and as such represents the first protein with lipid droplet regulatory activity to be linked to ER quality control (49).
HSPB7 (heat shock 27 kDa protein family, member 7), which is down-regulated in our study, has been identified as the most active member in the HSPB family for preventing toxicity of polyglutamine-containing proteins in cells and Drosophila (32). HSPB7 lacks several characteristics of the more classical small heat-shock proteins and instead acts in a noncanonical mechanism by assisting the loading of misfolded proteins or small protein aggregates into autophagosomes and facilitates their degradation (32). Autophagy regulates the metabolism of lipids including cholesterol; impaired maturation of autophagosomes and defective autophagy is observed in NPC disease (50). CRYAB (Alpha-crystallin B chain or Heat Shock Protein Beta-5, HSPB5), which is down-regulated in NPC1I1061T fibroblasts, belongs to a superfamily of small heat-shock proteins. CHORDC1 (Cysteine and histidine-rich domain-containing protein 1) appeared to be down-regulated in NPC1I1061T cells and is an ADP-dependent HSP90-interacting protein. It is proposed to act as its co-chaperone and assist in protein folding (31). CHORDC1 is stimulated to bind HSP90 when the ATP/ADP ratio falls below 1, raising the possibility that the HSP90-CHORDC1 interaction may be involved in the response to altered energy balance in vivo (31). Reduced ATP/ADP ratio is observed in conditions of oxidative stress combined with dysfunction in the calcium homeostasis and impaired energy status (51).
Cholesterol Homeostasis
Cholesterol is an essential component of cellular membranes. It is central in maintaining membrane integrity and fluidity as well as regulating membrane protein function, cell signaling, and ion permeability (52). In addition, cholesterol is the precursor molecule for synthesis of cellular components such as bile acids, oxysterols, and steroid hormones. Cholesterol homeostasis is tightly regulated at both the cellular and whole-body levels (3). The primary defect of NPC1 causes a secondary disruption in cholesterol biosynthesis. The reduction of NPC1 level in NPC disease leads to a massive accumulation of cholesterol in the LE/Ly compartments and cell death. Several cholesterol oxidation products were markedly elevated in the tissues of NPC mice and in plasma of NPC subjects (53).
We have identified several dysregulated proteins that are involved in cholesterol homeostasis or the sterol biosynthetic process in NPC1I1061T cells. The differential expression of few of them is shown as bar graphs in Fig. 4B. OSBPL1A (Oxysterol-binding protein-related protein 1), which was significantly up-regulated in NPC1I1061T cells, has been reported to stabilize GTP-bound RAB7A on LE/Ly and alter functional properties of late endocytic compartments via its interaction with RAB7A (54). Other proteins include DHCR24, FDPS (Farnesyl pyrophosphate synthase), APOB, and AKR1C1 (Aldo-keto reductase family 1 member C1). FDPS and APOB have been shown to be associated with Alzheimer's disease, a neurodegenerative disorder (55, 56). During cholesterol biosynthesis, DHCR24 catalyzes the reduction of the Δ24 double bond of sterol intermediates; deficiency of DHCR24 causes desmosterolosis (28). Patients with desmosterolosis have elevated levels of the cholesterol precursor desmosterol in plasma and tissues (28). AKR1C1 is mainly involved in the conversion of progesterone into its biologically inactive metabolite 20α-hydroxyprogesterone (57). Together with AKR1C3 these enzymes likely control the estradiol/progesterone level formed locally in the endometrium and the mammary gland. Both AKR1C1 and AKR1C3 expression levels were down-regulated in NPC1I1061T cells in our study. Expression of prosaposin (PSAP), a precursor of small heat-stable glycoproteins, saposins A, B, C, and D, was also detected to be decreased in NPC1I1061T cells in our study. The mature saposins, as well as PSAP, activate several lysosomal hydrolases involved in the metabolism of various sphingolipids (58). We also observed a decrease in abundance of glucosylceramidase (GBA), a lysosomal membrane protein that cleaves the β-glucosidic linkage of glycosylceramide to free glucose and ceramide, in NPC1I1061T cells. GBA interacts with and requires saposin-C for activity (59). Mutations in GBA cause Gaucher disease, a lysosomal storage disease characterized by an accumulation of glucocerebrosides (59).
Energy Metabolism
Mitochondrial cholesterol accumulation can increase oxidative stress and in turn cause metabolic alterations in NPC1-deficient cells (21). Alterations in mitochondrial abundance, quality and electron transport chain have been suggested to affect energy homeostasis in the cerebellum of NPC1-deficient mice (22). An impaired glucose uptake and utilization was reported to be a novel mechanism contributing to the pathology of NPC (40). The glycolytic enzymes were shown to be perturbed in NPC1-deficient mice cerebellum (22). The GO enrichment analysis of DEPs data set in our study revealed that the dysregulated proteins comprised a range of proteins involved in energy metabolism (Fig. 4A, 4B). We observed down-regulation of lactate dehydrogenase (LDH-A) in NPC1I1061T cells. Lactate dehydrogenase catalyzes the interconversion of pyruvate and lactate. NPC1-deficient cells show increased lactate secretion, decreased glutamine-dependent mitochondrial respiration, and decreased ATP transport across mitochondrial membranes (21). Studies from NPC1-deficient mice cerebellum and cerebral cortex revealed an increase in lactate and a decrease in acetate/acetyl-CoA levels with disease progression as compared with wild-type (22). Other enzymes with a role in metabolism and energy production that we found to be dysregulated in NPC1I1061T include PGK1 (phosphoglycerate kinase 1), PGM2 (phosphoglucomutase 2), IDH2 (isocitrate dehydrogenase 2), ALDOC (aldolase C), and SUCLA2 (succinyl-CoA ligase subunit beta) (Fig. 4B). The processing defect and down-regulation of GAA (acid α-glucosidase) protein that we observed in NPC1I1061T fibroblasts indicated dysfunction in degradation of glycogen to glucose in lysosomes. PAS staining shows an abnormal accumulation of glycogen in NPC1I1061T cells compared with NPC1WT (Fig. 5H). However, further study would be necessary to determine the reason for defective processing of GAA and whether mobilization of glycogen alleviates the disease symptoms in NPC patients.
Oxidative Stress
Several lines of evidence suggest that lysosomal lipid accumulation in NPC disease is accompanied by cellular oxidative stress. For example, the oxidative stress pathway is activated in liver and brain of NPC mice, NPC cellular models (37, 60), and in serum of NPC patients (20). In the NPC mouse model, oxidative stress is accompanied by elevated ROS and nonenzymatic oxidation of cholesterol in multiple tissues (53, 61, 62). In NPC1-deficient cells, cellular oxidative stress is evident by increased production of ROS, oxidative damage, and a gene expression profile indicative of oxidative stress (37, 60). Concentrations of ROS and lipid peroxidation were higher in fibroblasts from NPC patients than in fibroblasts from normal subjects (63). Oxidative stress is the main upstream stimulus activating apoptosis in NPC neurons (64, 65). Studies from NPC patients showed significant reduction in both antioxidant capacity (expressed as Trolox equivalents) and reduced coenzyme Q10 in serum compared with controls (20). This finding confirms the potential role of increased oxidative stress in the pathology of NPC.
We observed significant changes in proteins that are known to play an important role in maintaining redox regulation and homeostasis against ROS (Fig. 4B). Antioxidant enzyme SOD2 is well known to play a role in combating oxidative stress and detoxifying superoxide to less reactive hydrogen peroxide (66). The level of SOD2 was significantly down-regulated in NPC1I1061T fibroblasts. Other proteins with decreased abundance levels include glutathione transferases, such as GSTT1 and GSTO1, and mitochondrial thioredoxin reductase, TXNRD2. GSTT1 and GSTO1 are members of a superfamily of proteins that catalyze the conjugation of reduced glutathione to a variety of electrophilic and hydrophobic compounds. TXNRD2 is a selenocysteine-containing enzyme that is essential for the removal of toxic ROS species and the maintenance of mitochondrial integrity (67). TXNRD2-deficient fibroblasts have an increased sensitivity against oxidative stress (67). Interestingly, peroxiredoxin 6 (PRDX6), a member of a thiol-specific antioxidant protein family was found to be significantly up-regulated NPC1I1061T fibroblasts. PRDX6 is a bifunctional enzyme with both GSH peroxidase and phospholipase A2 activities and may play a role in the regulation of phospholipid turnover as well as in cellular protection against oxidative injury (68). The increased level of PRDX6 may represent a compensatory response to the decline in the levels of other ROS metabolic enzymes in an attempt to reestablish homeostasis.
Effect of Potential NPC Drugs on Expression of SOD2 and DHCR24 in NPC1I1061T Fibroblasts
SOD2 converts superoxide to the less reactive hydrogen peroxide that can freely diffuse across the mitochondrial membrane for further detoxification. Loss of SOD2 is involved in the progression of neurodegenerative diseases such as strokes and Alzheimer's, and Parkinson's diseases, as well as normal age-related cognitive decline (66). SOD2 is known to be a significant player in counteracting neurodegeneration (66). DHCR24 (also known as Seladin-1) is involved in the final step of cholesterol biosynthesis, catalyzing the conversion of desmosterol to cholesterol by saturating the C-24,25 double bond in its side-chain (28). DHCR24 suppresses the excess generation of intracellular ROS from ER stress and functions as an anti-apoptotic and neuroprotective protein (27). Therefore, we examined the effect of various classes of drugs that are currently being tested for efficacy in NPC treatment, such as cyclodextrins (HPCD/MβCD), HDACIs (Cl-994, SAHA, VPA), antioxidant NAC, and an oxysterol derivative pharmacological chaperone (mo56HC) on the expression level of SOD2 and DHCR24 in NPC1I1061T cells.
Cyclodextrins are nonreducing cyclic glucose oligosaccharides that are good chelators and have been shown to have a very high affinity for sterols. They bypass the requirement of NPC1- and NPC2-dependent pathway and facilitate the efflux of sequestered unesterified cholesterol from LE/Ly compartment to the metabolically active pool of cholesterol in the cytosolic compartment of cells (9, 69). Cyclodextrin treatment significantly increased Purkinje cell survival and markedly prolonged the life of the mutant mice (34). Inhibition of HDACs reduces the accumulation of cholesterol and other lipids in the LE/Ly compartments in NPC1I1061T fibroblasts (70). This correction of phenotype may be achieved by increasing the expression of NPC1I1061T, facilitating egress of cholesterol from LE/Ly compartments (70). However, the exact mechanism of action of the HDACIs in correcting cholesterol-storage defect requires further research. As reported previously (70), we also observed that treatment with SAHA corrects the folding and trafficking defect of I1061T-mutant NPC1 and restores the cholesterol homeostasis at the LE/Ly compartments (data not shown). NAC, an acetylated form of the amino acid cysteine, is an antioxidant molecule. It undergoes deacetylation in vivo to yield cysteine, which is the rate-limiting precursor in the synthesis of the tripeptide glutathione, a molecule essential for regulation of intracellular oxidative stress. The effect of NAC in alleviating oxidative stress and disease symptoms in two distinct NPC disease mouse models and individuals with NPC disease has been studied (71). mo56HC is an oxysterol derivative that acts as a pharmacological chaperone, which enhances the folding/maturation and corrects the trafficking defect of NPC1I1061T mutant protein (11).
The substantial decrease of SOD2 level in NPC1I1061T fibroblasts observed in our study suggests increased vulnerability against ROS-induced oxidative damage. The restoration of the cellular SOD2 level to that of wild-type following treatment with the aforementioned drugs (Fig. 6B) indicates the potential recovery in the capability of mutant cells to fight against oxidative stress. Also, the dramatic increase in the level of DHCR24 in drug-treated NPC1I1061T cells, beyond its expression level in DMSO-treated wild-type cells (Fig. 6C), may help cells defend against oxidative stress and/or attain cholesterol homeostasis. However, further studies will be necessary to address the exact role and molecular mechanism of the DHCR24 protein in the context of NPC disease. Although a treatment of oxidative stress would not correct the primary underlying defect in NPC1, it could provide some benefit, especially for treating other aspects of the downstream pathological cascade. Hence, monitoring the proteins representative of a certain class of biological processes (such as oxidative stress), although not specific for NPC, is of great potential utility to track disease progression and therapeutic intervention.
CONCLUSIONS
Mutations in NPC1 gene are responsible for an estimated 95% of human NPC disease. The most prevalent mutation I1061T, representing 15–20% of disease-causing alleles in NPC disease, is recognized as misfolded by the ER quality control machinery and subsequently targeted for proteasomal degradation. The consequence of reduced NPC1 levels is a massive intracellular accumulation of unesterified cholesterol in the LE/Ly compartments. To successfully devise a therapeutic strategy to rescue the mutated NPC1 protein, it will be necessary to understand the global proteome alteration in mutant cells when compared with controls. In this study we have used a high throughput isobaric labeling approach to identify differentially regulated proteins in NPC1I1061T relative to NPC1WT primary fibroblasts. With stringent data analysis criteria, a total of 4308 proteins were identified from two TMT-MudPIT experimental replicates, out of which 3553 distinct proteins were quantified. We observed 281 proteins that were statistically significantly differentially expressed. Of these, 113 proteins showed an up-regulation, whereas 168 were down-regulated in NPC1I1061T cells. The detected dysregulated proteins are associated with a diverse array of biological functional annotations including ROS metabolic process, antioxidant activity, steroid metabolic process, lipid localization, apoptosis, and energy metabolism among many others. They were also mapped to key cellular organelles such as, ER, mitochondria, Golgi apparatus, lysosome, and endosome, which are of relevance in the context of NPC disease pathogenesis.
To demonstrate the reliability of our data set, the expression level of a subset of significantly altered proteins (TOR4A, DHCR24, CLGN, SOD2, CHORDC1, HSPB7, and GAA) was substantiated by Western blotting. Furthermore, our study showed that treatment of NPC1I1061T cells with potential NPC drugs increased the expression level of SOD2 and DHCR24. We also show an abnormal accumulation of glycogen in NPC1I1061T fibroblasts compared with NPC1WT. Thus, the differentially expressed proteins observed in this study provide a starting point for future investigations and may serve as a useful resource to the NPC research community. More focused experiments will be needed to better understand how these proteins interact and how they are linked in the pathological cascade because of mutation of NPC1. This could potentially provide insights on effective therapeutic interventions for clinically reported NPC patients.
Supplementary Material
Acknowledgments
We thank Drs. Daniel B. McClatchy and Claire Delahunty for their helpful suggestions and critical reading of the manuscript. We thank Sung Kyu Robin Park for bioinformatics support.
Footnotes
Author contributions: N.R., K.S., W.E.B., and J.R.Y. designed research; N.R. and K.S. performed research; N.R., K.S., M.L., S.M., W.E.B., and J.R.Y. contributed new reagents or analytic tools; N.R., K.S., M.L., and S.M. analyzed data; N.R., K.S., M.L., and S.M. wrote the paper; W.E.B. and J.R.Y. project supervisor.
* This work was supported by National Institutes of Health grants P41 GM103533 and R01 HL079442-09 (to J.R.Y) and GM33301 (to W.E.B). K.S. is supported by Ara Parseghian Medical Research Foundation Fellowship and NPC-SOAR (Support Of Accelerated Research for NPC Disease) and M.L.-A. holds a postdoctoral fellowship from FRQNT.
 This article contains supplemental Figs. S1 to S3 and Table S1 to S3.
This article contains supplemental Figs. S1 to S3 and Table S1 to S3.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (72) via the PRIDE partner repository with the data set identifier PXD001938.
1 The abbreviations used are:
- NPC
- Niemann-Pick type C
- LE/Ly
- Late endosomal/lysosomal compartments
- TMT
- Tandem mass tags
- MudPIT
- Multi-dimensional protein identification technology
- HCD
- Higher-energy collisional dissociation
- FDR
- False discovery rate
- DEP
- Differentially expressed protein
- ROS
- Reactive oxygen species
- HPCD
- 2-hydroxypropyl-β-cyclodextrin
- MβCD
- Methyl-β-cyclodextrin
- CI-994
- N-acetyldinaline
- SAHA
- Suberoylanilide hydroxamic acid
- VPA
- Valproic acid
- NAC
- N-acetyl cysteine.
REFERENCES
- 1. Vanier M. T. (2010) Niemann-Pick disease type C. Orphanet J. Rare Dis. 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Patterson M. C., Hendriksz C. J., Walterfang M., Sedel F., Vanier M. T., Wijburg F. (2012) Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol. Genet. Metab. 106, 330–344 [DOI] [PubMed] [Google Scholar]
- 3. Ory D. S. (2004) The niemann-pick disease genes; regulators of cellular cholesterol homeostasis. Trends Cardiovasc. Med. 14, 66–72 [DOI] [PubMed] [Google Scholar]
- 4. Runz H., Dolle D., Schlitter A. M., Zschocke J. (2008) NPC-db, a Niemann-Pick type C disease gene variation database. Hum. Mutat. 29, 345–350 [DOI] [PubMed] [Google Scholar]
- 5. Gelsthorpe M. E., Baumann N., Millard E., Gale S. E., Langmade S. J., Schaffer J. E., Ory D. S. (2008) Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 283, 8229–8236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cravatt B. F., Simon G. M., Yates J. R., 3rd (2007) The biological impact of mass-spectrometry-based proteomics. Nature 450, 991–1000 [DOI] [PubMed] [Google Scholar]
- 7. Dayon L., Hainard A., Licker V., Turck N., Kuhn K., Hochstrasser D. F., Burkhard P. R., Sanchez J. C. (2008) Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6-plex isobaric tags. Anal. Chem. 80, 2921–2931 [DOI] [PubMed] [Google Scholar]
- 8. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 9. Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., Walkley S. U. (2009) Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One 4, e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Helquist P., Maxfield F. R., Wiech N. L., Wiest O. (2013) Treatment of Niemann–pick type C disease by histone deacetylase inhibitors. Neurotherapeutics 10, 688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohgane K., Karaki F., Dodo K., Hashimoto Y. (2013) Discovery of oxysterol-derived pharmacological chaperones for NPC1: implication for the existence of second sterol-binding site. Chem. Biol. 20, 391–402 [DOI] [PubMed] [Google Scholar]
- 12. Tabb D. L., McDonald W. H., Yates J. R., 3rd (2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park S. K., Aslanian A., McClatchy D. B., Han X., Shah H., Singh M., Rauniyar N., Moresco J. J., Pinto A. F., Diedrich J. K., Delahunty C., Yates J. R., 3rd (2014) Census 2: isobaric labeling data analysis. Bioinformatics 30, 2208–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bauer S., Grossmann S., Vingron M., Robinson P. N. (2008) Ontologizer 2.0–a multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 24, 1650–1651 [DOI] [PubMed] [Google Scholar]
- 16. Liscum L., Faust J. R. (1987) Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 262, 17002–17008 [PubMed] [Google Scholar]
- 17. Tangemo C., Weber D., Theiss S., Mengel E., Runz H. (2011) Niemann-Pick Type C disease: characterizing lipid levels in patients with variant lysosomal cholesterol storage. J. Lipid Res. 52, 813–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Karp N. A., Huber W., Sadowski P. G., Charles P. D., Hester S. V., Lilley K. S. (2010) Addressing accuracy and precision issues in iTRAQ quantitation. Mol. Cell. Proteomics 9, 1885–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vazquez M. C., Balboa E., Alvarez A. R., Zanlungo S. Oxidative stress: a pathogenic mechanism for Niemann-Pick type C disease. Oxid. Med. Cell Longev. 2012:205713, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fu R., Yanjanin N. M., Bianconi S., Pavan W. J., Porter F. D. (2010) Oxidative stress in Niemann-Pick disease, type C. Mol. Genet. Metab. 101, 214–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kennedy B. E., Madreiter C. T., Vishnu N., Malli R., Graier W. F., Karten B. (2014) Adaptations of Energy Metabolism Associated with Increased Levels of Mitochondrial Cholesterol in Niemann-Pick Type C1-deficient Cells. J. Biol. Chem. 289, 16278–16289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kennedy B. E., LeBlanc V. G., Mailman T. M., Fice D., Burton I., Karakach T. K., Karten B. (2013) Pre-symptomatic activation of antioxidant responses and alterations in glucose and pyruvate metabolism in Niemann-Pick Type C1-deficient murine brain. PLoS One 8, e82685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Charman M., Kennedy B. E., Osborne N., Karten B. (2010) MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res. 51, 1023–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller W. L. (1995) Mitochondrial specificity of the early steps in steroidogenesis. J. Steroid Biochem. Mol. Biol. 55, 607–616 [DOI] [PubMed] [Google Scholar]
- 25. Brozzi A., Urbanelli L., Germain P. L., Magini A., Emiliani C. (2013) hLGDB: a database of human lysosomal genes and their regulation. Database 2013, bat024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nery F. C., Armata I. A., Farley J. E., Cho J. A., Yaqub U., Chen P., da Hora C. C., Wang Q., Tagaya M., Klein C., Tannous B., Caldwell K. A., Caldwell G. A., Lencer W. I., Ye Y., Breakefield X. O. (2011) TorsinA participates in endoplasmic reticulum-associated degradation. Nat. Commun. 2, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu X., Li Y., Wang W., Chen S., Liu T., Jia D., Quan X., Sun D., Chang A. K., Gao B. (2014) 3 beta-hydroxysteroid-Delta 24 reductase (DHCR24) protects neuronal cells from apoptotic cell death induced by endoplasmic reticulum (ER) stress. PLoS One 9, e86753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waterham H. R., Koster J., Romeijn G. J., Hennekam R. C., Vreken P., Andersson H. C., FitzPatrick D. R., Kelley R. I., Wanders R. J. (2001) Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am. J. Hum. Genet. 69, 685–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikawa M., Wada I., Kominami K., Watanabe D., Toshimori K., Nishimune Y., Okabe M. (1997) The putative chaperone calmegin is required for sperm fertility. Nature 387, 607–611 [DOI] [PubMed] [Google Scholar]
- 30. Schieber M., Chandel N. S. (2014) ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gano J. J., Simon J. A. (2010) A proteomic investigation of ligand-dependent HSP90 complexes reveals CHORDC1 as a novel ADP-dependent HSP90-interacting protein. Mol. Cell. Proteomics 9, 255–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vos M. J., Zijlstra M. P., Kanon B., van Waarde-Verhagen M. A., Brunt E. R., Oosterveld-Hut H. M., Carra S., Sibon O. C., Kampinga H. H. (2010) HSPB7 is the most potent polyQ aggregation suppressor within the HSPB family of molecular chaperones. Hum. Mol. Genet. 19, 4677–4693 [DOI] [PubMed] [Google Scholar]
- 33. Moreland R. J., Jin X., Zhang X. K., Decker R. W., Albee K. L., Lee K. L., Cauthron R. D., Brewer K., Edmunds T., Canfield W. M. (2005) Lysosomal acid alpha-glucosidase consists of four different peptides processed from a single chain precursor. J. Biol. Chem. 280, 6780–6791 [DOI] [PubMed] [Google Scholar]
- 34. Liu B., Li H., Repa J. J., Turley S. D., Dietschy J. M. (2008) Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 49, 663–669 [DOI] [PubMed] [Google Scholar]
- 35. Cluzeau C. V., Watkins-Chow D. E., Fu R., Borate B., Yanjanin N., Dail M. K., Davidson C. D., Walkley S. U., Ory D. S., Wassif C. A., Pavan W. J., Porter F. D. (2012) Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 21, 3632–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vazquez M. C., del Pozo T., Robledo F. A., Carrasco G., Pavez L., Olivares F., Gonzalez M., Zanlungo S. (2011) Alteration of gene expression profile in Niemann-Pick type C mice correlates with tissue damage and oxidative stress. PLoS One 6, e28777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Reddy J. V., Ganley I. G., Pfeffer S. R. (2006) Clues to neuro-degeneration in Niemann-Pick type C disease from global gene expression profiling. PLoS One 1, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Windt A., Rai M., Kytomaki L., Thelen K. M., Lutjohann D., Bernier L., Davignon J., Soini J., Pandolfo M., Laaksonen R. (2007) Gene set enrichment analyses revealed several affected pathways in Niemann-pick disease type C fibroblasts. DNA Cell Biol. 26, 665–671 [DOI] [PubMed] [Google Scholar]
- 39. Vogel C., Marcotte E. M. (2012) Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cologna S. M., Jiang X. S., Backlund P. S., Cluzeau C. V., Dail M. K., Yanjanin N. M., Siebel S., Toth C. L., Jun H. S., Wassif C. A., Yergey A. L., Porter F. D. (2012) Quantitative proteomic analysis of Niemann-Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS One 7, e47845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Byun K., Kim J., Cho S. Y., Hutchinson B., Yang S. R., Kang K. S., Cho M., Hwang K., Michikawa M., Jeon Y. W., Paik Y. K., Lee B. (2006) Alteration of the glutamate and GABA transporters in the hippocampus of the Niemann-Pick disease, type C mouse using proteomic analysis. Proteomics 6, 1230–1236 [DOI] [PubMed] [Google Scholar]
- 42. Krumova P., Weishaupt J. H. (2013) Sumoylation in neurodegenerative diseases. Cell Mol. Life Sci. 70, 2123–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Setsuie R., Wada K. (2007) The functions of UCH-L1 and its relation to neurodegenerative diseases. Neurochem. Int. 51, 105–111 [DOI] [PubMed] [Google Scholar]
- 44. Kumar P., Pradhan K., Karunya R., Ambasta R. K., Querfurth H. W. (2012) Cross-functional E3 ligases Parkin and C-terminus Hsp70-interacting protein in neurodegenerative disorders. J. Neurochem. 120, 350–370 [DOI] [PubMed] [Google Scholar]
- 45. Goldberg A. L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899 [DOI] [PubMed] [Google Scholar]
- 46. Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. (2008) Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 47. Yu T., Chung C., Shen D., Xu H., Lieberman A. P. (2012) Ryanodine receptor antagonists adapt NPC1 proteostasis to ameliorate lipid storage in Niemann-Pick type C disease fibroblasts. Hum. Mol. Genet. 21, 3205–3214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McDonough H., Patterson C. (2003) CHIP: a link between the chaperone and proteasome systems. Cell Stress Chaperones 8, 303–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Klemm E. J., Spooner E., Ploegh H. L. (2011) Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem. 286, 37602–37614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarkar S., Carroll B., Buganim Y., Maetzel D., Ng A. H., Cassady J. P., Cohen M. A., Chakraborty S., Wang H., Spooner E., Ploegh H., Gsponer J., Korolchuk V. I., Jaenisch R. (2013) Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 5, 1302–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tretter L., Chinopoulos C., Adam-Vizi V. (1997) Enhanced depolarization-evoked calcium signal and reduced [ATP]/[ADP] ratio are unrelated events induced by oxidative stress in synaptosomes. J. Neurochem. 69, 2529–2537 [DOI] [PubMed] [Google Scholar]
- 52. Ikonen E. (2008) Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 125–138 [DOI] [PubMed] [Google Scholar]
- 53. Porter F. D., Scherrer D. E., Lanier M. H., Langmade S. J., Molugu V., Gale S. E., Olzeski D., Sidhu R., Dietzen D. J., Fu R., Wassif C. A., Yanjanin N. M., Marso S. P., House J., Vite C., Schaffer J. E., Ory D. S. (2010) Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med. 2, 56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Johansson M., Lehto M., Tanhuanpaa K., Cover T. L., Olkkonen V. M. (2005) The oxysterol-binding protein homologue ORP1L interacts with Rab7 and alters functional properties of late endocytic compartments. Mol. Biol. Cell 16, 5480–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wollmer M. A., Sleegers K., Ingelsson M., Zekanowski C., Brouwers N., Maruszak A., Brunner F., Huynh K. D., Kilander L., Brundin R. M., Hedlund M., Giedraitis V., Glaser A., Engelborghs S., De Deyn P. P., Kapaki E., Tsolaki M., Daniilidou M., Molyva D., Paraskevas G. P., Thal D. R., Barcikowska M., Kuznicki J., Lannfelt L., Van Broeckhoven C., Nitsch R. M., Hock C., Papassotiropoulos A. (2007) Association study of cholesterol-related genes in Alzheimer's disease. Neurogenetics 8, 179–188 [DOI] [PubMed] [Google Scholar]
- 56. Caramelli P., Nitrini R., Maranhao R., Lourenco A. C., Damasceno M. C., Vinagre C., Caramelli B. (1999) Increased apolipoprotein B serum concentration in Alzheimer's disease. Acta Neurol. Scand. 100, 61–63 [DOI] [PubMed] [Google Scholar]
- 57. Penning T. M. (1997) Molecular endocrinology of hydroxysteroid dehydrogenases. Endocr. Rev. 18, 281–305 [DOI] [PubMed] [Google Scholar]
- 58. Kishimoto Y., Hiraiwa M., O'Brien J. S. (1992) Saposins: structure, function, distribution, and molecular genetics. J. Lipid Res. 33, 1255–1267 [PubMed] [Google Scholar]
- 59. Tamargo R. J., Velayati A., Goldin E., Sidransky E. (2012) The role of saposin C in Gaucher disease. Mol. Genet. Metab. 106, 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zampieri S., Mellon S. H., Butters T. D., Nevyjel M., Covey D. F., Bembi B., Dardis A. (2009) Oxidative stress in NPC1 deficient cells: protective effect of allopregnanolone. J. Cell Mol. Med. 13, 3786–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tint G. S., Pentchev P., Xu G., Batta A. K., Shefer S., Salen G., Honda A. (1998) Cholesterol and oxygenated cholesterol concentrations are markedly elevated in peripheral tissue but not in brain from mice with the Niemann-Pick type C phenotype. J. Inherit. Metab. Dis. 21, 853–863 [DOI] [PubMed] [Google Scholar]
- 62. Zhang J. R., Coleman T., Langmade S. J., Scherrer D. E., Lane L., Lanier M. H., Feng C., Sands M. S., Schaffer J. E., Semenkovich C. F., Ory D. S. (2008) Niemann-Pick C1 protects against atherosclerosis in mice via regulation of macrophage intracellular cholesterol trafficking. J. Clin. Invest. 118, 2281–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chong P. K., Gan C. S., Pham T. K., Wright P. C. (2006) Isobaric tags for relative and absolute quantitation (iTRAQ) reproducibility: Implication of multiple injections. J. Proteome Res. 5, 1232–1240 [DOI] [PubMed] [Google Scholar]
- 64. Alvarez A. R., Klein A., Castro J., Cancino G. I., Amigo J., Mosqueira M., Vargas L. M., Yevenes L. F., Bronfman F. C., Zanlungo S. (2008) Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 22, 3617–3627 [DOI] [PubMed] [Google Scholar]
- 65. Klein A., Maldonado C., Vargas L. M., Gonzalez M., Robledo F., Perez de Arce K., Munoz F. J., Hetz C., Alvarez A. R., Zanlungo S. (2011) Oxidative stress activates the c-Abl/p73 proapoptotic pathway in Niemann-Pick type C neurons. Neurobiol Dis 41, 209–218 [DOI] [PubMed] [Google Scholar]
- 66. Flynn J. M., Melov S. (2013) SOD2 in mitochondrial dysfunction and neurodegeneration. Free Radic. Biol. Med. 62, 4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Conrad M., Jakupoglu C., Moreno S. G., Lippl S., Banjac A., Schneider M., Beck H., Hatzopoulos A. K., Just U., Sinowatz F., Schmahl W., Chien K. R., Wurst W., Bornkamm G. W., Brielmeier M. (2004) Essential role for mitochondrial thioredoxin reductase in hematopoiesis, heart development, and heart function. Mol. Cell. Biol. 24, 9414–9423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Manevich Y., Fisher A. B. (2005) Peroxiredoxin 6, a 1-Cys peroxiredoxin, functions in antioxidant defense and lung phospholipid metabolism. Free Radic. Biol. Med. 38, 1422–1432 [DOI] [PubMed] [Google Scholar]
- 69. Aqul A., Liu B., Ramirez C. M., Pieper A. A., Estill S. J., Burns D. K., Liu B., Repa J. J., Turley S. D., Dietschy J. M. (2011) Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 31, 9404–9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Pipalia N. H., Cosner C. C., Huang A., Chatterjee A., Bourbon P., Farley N., Helquist P., Wiest O., Maxfield F. R. (2011) Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 108, 5620–5625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fu R., Wassif C. A., Yanjanin N. M., Watkins-Chow D. E., Baxter L. L., Incao A., Liscum L., Sidhu R., Firnkes S., Graham M., Ory D. S., Porter F. D., Pavan W. J. (2013) Efficacy of N-acetylcysteine in phenotypic suppression of mouse models of Niemann-Pick disease, type C1. Hum. Mol. Genet. 22, 3508–3523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally co-ordinated proteomics data submission and dissemination. Nat. Biotechnol. 30, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.