

Abstract
Homoisocitrate dehydrogenase (HIcDH) catalyzes the NAD+-dependent oxidative decarboxylation of HIc to α-ketoadipate, the fourth step in the α-aminoadipate pathway responsible for the de novo synthesis of L-lysine in fungi. A mechanism has been proposed for the enzyme that makes use of a Lys-Tyr pair as acid-base catalysts, with Lys acting as a base to accept a proton from the α-hydroxyl of homoisocitrate, and Tyr acting as an acid to protonate the C3 of the enol of α-ketoadipate in the enolization reaction. Three conserved aspartate residues, D243, D267 and D271, coordinate Mg2+, which is also coordinated to the α-carboxylate and α-hydroxyl of homoisocitrate. On the basis of kinetic isotope effects, it was proposed that a conformational change to close the active site and organize the active site for catalysis contributed to rate limitation of the overall reaction of the Saccharomyces cerevisiae HIcDH (Lin, Y., Volkman, J., Nicholas, K. M., Yamamoto, T., Eguchi, T., Nimmo, S. L., West, A. H., and Cook, P. F. (2008) Biochemistry 47, 4169–4180.). In order to test this hypothesis, site-directed mutagenesis was used to change D271, a metal ion ligand and binding determinant for MgHIc, to N. The mutant enzyme was characterized using initial rate studies. A decrease of 520-fold was observed in V and V/KMgHIc, suggesting the same step(s) limit the reaction at limiting and saturating MgHIc concentrations. Solvent kinetic deuterium isotope effects (SKIE) and viscosity effects are consistent with a rate-limiting pre-catalytic conformational change at saturating reactant concentrations. In addition, at limiting MgHIc, an inverse (SKIE) of 0.7 coupled to a significant normal effect of viscosogen (2.1) indicates equilibrium binding of MgHIc prior to the rate-limiting conformational change. The maximum rate exhibits a small partial change at high pH suggesting a pH-dependent conformational change, while V/KMgHIc exhibits the same partial change observed in V, and a decrease at low pH with a pKa of 6 reflecting the requirement for the unprotonated form of MgHIc to bind to enzyme. However, neither parameter reflects the pH dependence of the chemical reaction. This pH independence of the chemical reaction over the range 5.5–9.5 is consistent with the much slower conformational change that would effectively perturb the observed pK values for catalytic groups to lower and higher pH. In other words, the pH dependence of the chemical reaction will only be observed when chemistry becomes slower than the rate of the conformational change. Data support the hypothesis of the existence of a pre-catalytic conformational change coupled to the binding of MgHIc.
Homoisocitrate dehydrogenase (3-carboxy-2-hydroxyadipate dehydrogenase, EC 1.1.1.87) (HIcDH1) catalyzes the fourth step of the AAA pathway, the NAD+-dependent conversion of homoisocitrate to α-ketoadipate (1). The dehydrogenase is a member of the family of pyridine dinucleotide-dependent β-hydroxyacid oxidative decarboxylating dehydrogenases, specifically the family that has (R)-β-hydroxyacid substrates, including isocitrate dehydrogenase (ICDH) among others (2). The second subfamily has (S)-β-hydroxyacid substrates, and includes only the malic enzymes. All of the enzymes except 6-phosphogluconate dehydrogenase (6-PGDH), require a divalent metal ion to catalyze the reactions (3). Tartrate dehydrogenase (4) and HIcDH from Saccharomyces cerevisiae (ScHIcDH) also requires a potassium ion as an activator, for optimal binding of NAD+ (5). Superposition of available structures of the malic enzyme, isopropylmalate dehydrogenase (IPMDH), IcDH, and HIcDH show a similar overall geometry of residues in the substrate and metal ion binding sites. A Lys (general base)-Tyr (general acid) pair is conserved among these enzymes (6). The similar structural geometry in the active site suggests a similar general chemical mechanism (7–11).
On the basis of a multiple sequence alignment and structures of the active site of the enzymes in the pyridine dinucleotide-dependent β-hydroxyacid oxidative decarboxylating dehydrogenase family, there are three aspartate residues conserved in the active sites of these enzymes (6). The aspartate residues are completely conserved among all HIcDHs sequenced to data, and are also conserved across the family of pyridine nucleotide-dependent oxidative decarboxylases including malic enzyme (6). Two to three Asp residues coordinate the metal ion in the active site of the R-hydroxyacid oxidative dehydrogenases. The three Asp residues are also conserved in the active site of ScHIcDH. Recent binary complex structures of HIcDH from Schizosaccharomyces pombe (12) and Thermus thermophilus (13) show the location of acidic residues in the active site in relationship to a bound substrate analog. Sequence alignment of the HIcDH from S. pombe, S. cerevisiae, and T. thermophilus indicates the two fungal enzymes, as expected, are most closely related, Fig. 1. The S. pombe enzyme exhibits 61% sequence identity to the enzyme from S. cerevisiae, while the enzyme from T. thermophilus exhibits 49% sequence identity. The active site is composed of residues from two subunits with one of three aspartate residues (D232′) and the catalytic lysine (K196′) donated by the one subunit, and these residues are identical to D243 and K206 in the ScHIcDH. Figure 2 shows a view of the active site of the S. pombe enzyme with the tripeptide glyglygly bound (12). The tripeptide is bound close to the NADP(H)-binding site, and the substrate-binding site is above in the figure. The active site is composed of residues from two subunits as shown by prime and non-prime residues in Fig. 2. Bulfer and colleagues (12) modeled MgIc into the S. pombe apo-enzyme structure, which suggested that Mg2+ is coordinated to D260 (D271 in ScHIcDH), D232′ (D243 in ScHIcDH) and D256 (D267 in ScHIcDH), as well as the α-carboxylate and α-hydroxyl of Ic, while R97 (R129 in ScHIcDH) and R126 (R148 in ScHIcDH) interact with the C1 and C6 carboxylates of the substrate. In other enzymes of the class of R-hydroxyacid oxidative decarboxylases, including IPMDH (14), IcDH (8), and TDH (4), metal ion is coordinated to two of three Asp residues. The catalytic lysine, K196′ (K206 in ScHIcDH) and tyrosine are in the vicinity of C2 and C3 of Ic (12). With glyglygly bound in the site, interaction is observed with R97 and R126, but none of the other residues discussed above.
Figure 1.

Alignment of the protein sequences of ScHIcSH, SpHIcDH, and TtHIcDH. The multiple sequence alignment was carried out using ClustalW2 (version 2.1) (23) and the figure was prepared using Boxshade. The black-shadowed residues are identical, while those that are gray-shadowed are conservative substitutions. The (*) shown below the alignment show likely metal ion ligands, D243, D267, D271, and the general base, K206, and general acid, Y150, residues in the ScHICDH sequence.
Figure 2.
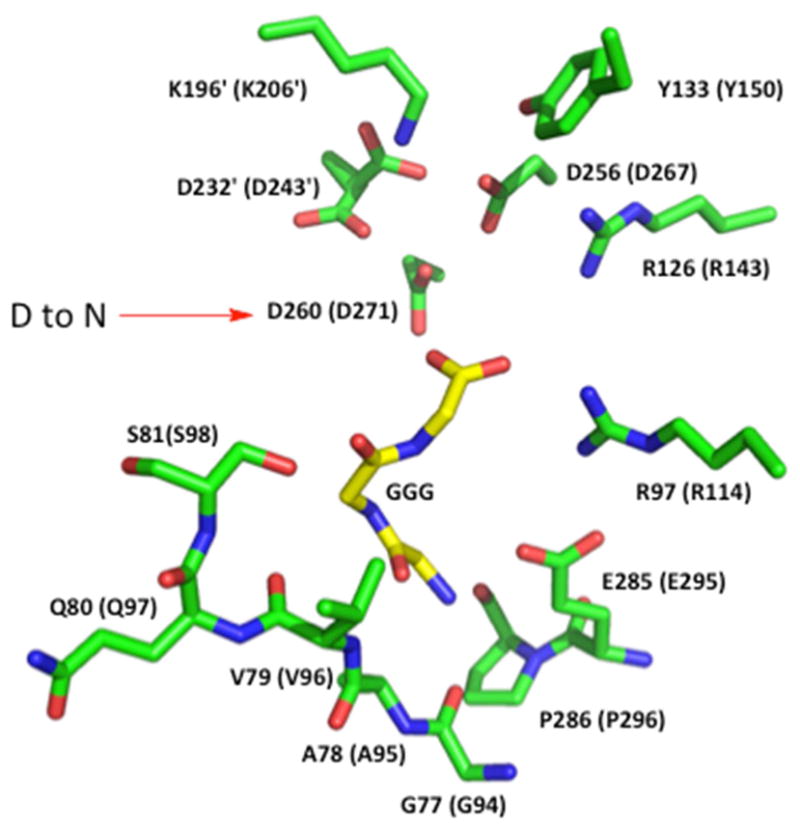
Active site of the HIcDH enzyme from S. pombe. A view of the active site with the peptide glyglygly bound in the NADP(H) site. Residues are numbered according to the SpHIcDH sequence and the ScHIcDH sequence in (). The residues with a prime are contributed by another subunit. Carbon atoms are in green for protein and yellow for the GGG ligand, N atoms are blue and O atoms are red. The general base, K196, and general acid, Y133 are shown at the top of the figure with the putative aspartate metal ion ligands below. Two arginine residues donate hydrogen bonds to two of the carboxylates of Hic are shown to the right. The figure was prepared using the PyMOL Molecular Graphics System, Version 1.7.4 Schrödinger, LLC.
A chemical mechanism has been proposed for the ScHIcDH on the basis of the pH dependence of kinetic parameters, dissociation constants for competitive inhibitors, and isotope effects (10). Data suggest there are 2 groups acting as acid-base catalysts in the reaction. One residue with a pKa of 6.5–7.0 serves as the general base to accept a proton as the β-hydroxy acid is oxidized to the β-keto acid, and this residue participates in all three of the chemical steps, acting to shuttle a proton between the C2 hydroxyl and itself. The metal ion then acts as a Lewis acid to catalyze the decarboxylation of the β-ketoacid, with the general base donating a proton to the keto oxygen as the enol of α-ketoadipate is formed. A second residue with a pKa of 9.5 likely catalyzes the tautomerization step by donating a proton to the enol to give the final product (Scheme 1). On the basis of multiple-sequence alignment of enzymes in the same family, and site-directed mutagenesis, K206 and Y150 of ScHIcDH were proposed as a Lys-Tyr pair in the active site acting as the general base and general acid in the reaction, and this was confirmed by a complete analysis of the K206M and Y150F mutant enzymes (10,11). Data from primary deuterium isotope effect, solvent deuterium isotope effect, viscosity effects, and multiple-substrate/13C isotope effects with MgHIc as the substrate suggested that the decarboxylation step contributes only slightly to rate limitation, and hydride transfer is not rate limiting. Furthermore, a slow conformational change required to close the site upon the binding of MgHIc prior to catalysis was proposed (10). With the slow substrate isocitrate, however, hydride transfer and decarboxylation steps contribute to rate limitation, and the decarboxylation step is the slower of the two (10).
Scheme 1.

Proposed Chemical Mechanism of ScHIcDH. I: Enzyme is in an open or partial open form upon binding of NAD+, and a conformational change closes the active site and positions reactants for catalysis, upon binding of MgHIc. The [ ]* reflects the closed form of the enzyme with all of the catalytic steps taking place in the closed form. II: Hydride transfer is facilitated by K206 acting as a general base. The conserved aspartate metal ion ligands are shown in this first figure only and are assumed in the remaining figues (II–V). III: Decarboxylation occurs with the metal ion serving as a Lewis acid and K206 acting as a general acid to donate a proton. IV: The general base (K206) and general acid (Y150) catalyze the enol-keto tautomerization to give α-ketoadipate (V). VI: A conformational change occurs to release α-ketoadipate.
As suggested above, a conformational change that closes the site, organizes it for catalysis, and is likely coupled to the binding of MgHIc (induced conformational change) has been proposed (Scheme 1). The metal ion in the active site is coordinated to several conserved aspartate residues, and the aspartates are thus ligands for substrate binding. An effect on substrate binding might be expected to alter the rate of the conformational change to close the site. In this study, we used site-directed mutagenesis to change D271, a MgHIc ligand, to N, and the mutant enzyme was then fully characterized by initial velocity, pH-rate profiles, and solvent deuterium kinetic isotope effect experiments. Data are consistent with the proposed induced conformational change upon binding of MgHIc.
MATERIALS AND METHODS
Chemicals
3-Acetylpyridine adenine dinucleotide 3′-phosphate (3-AcPyrADP), KAc and Mg(Ac)2 were from Sigma. The buffers Mes, Bis-Tris, Hepes, and Ches were purchased from Research Organics, Inc. β-NAD+, LB broth medium, and LB agar were from U.S.B. The Ni-NTA agarose resin was purchased from Qiagen. The IPTG, dNTPs, NdeI, XhoI, DpnI and Turbo Pfu polymerase were purchased from Invitrogen. Ampicillin was purchased from Fisher Biotech. For plasmid purification, the Nucleobond AX kit (The Next Group, Inc.) was used. D2O (99 atom % D) and DCl (99.5 atom % D) were from Cambridge Isotopes Laboratories, Inc. 2(R),3(S)-Homoisocitric acid was synthesized as previously reported (15). Thiahomoisocitrate ((2S,3S)-(-)-thiahomisocitrate (16); C4 of Hic is replaced by S) was the generous gift of Tadashi Eguchi, Department of Chemistry and Materials Science, Tokyo Institute of Technology, Tokyo, Japan. All other chemicals and reagents were obtained from commercial sources, were the highest purity available, and were used as purchased.
Preparation of the D271N mutant enzyme
A plasmid harboring the ScHIcDH gene (17) was purified as a template for mutagenesis, and the D271N mutation was created using the QuickChange site-directed mutagenesis kit from Stratagene. The nucleotide primers used to generate the D271N mutant enzyme were from Invitogen. Forward and reverse primers are as follows: D271Nf, 5′-GATATATTATCTAACGGTGCTGCTGC; D271Nr, 3′-GCAGCAGCACCGTTAGATAATATATC. The mutated codon is shown in bold.
The PCR amplification protocol was as follows: one cycle of initial denaturation at 95° C for 4 minutes, followed by 20 cycles of denaturation at 95° C for 1 minute, annealing at 57° C for 1 minute, and extension at 68° C for 7 minutes. Sequencing of the D271N mutant gene was carried out by the Laboratory for Genomics and Bioinformatics at the University of Oklahoma Health Science Center in Oklahoma City, and the BLAST program was used to compare the sequence of the mutant gene to that of WT ScHIcDH. The successfully mutated plasmid was transformed into BL-21 (DE3) competent cells for expression.
Expression and purification of D271N ScHIcDH
D271N BL-21 (DE3) cells were pre-incubated in 50 mL of LB medium, which contained 25 μL of ampicillin (1g/mL) at 37° C overnight. Cells were then transferred to 1 L of LB medium and growth was continued until an O.D. of 0.6 was reached. IPTG was then added to a final concentration of 1 mM, and after a 12 h induction period at 25° C, cells were harvested by centrifugation at 8,000g. Cells were then lysed in 20 mM Tris-HCl (pH 7.5), 500 mM NaCl, 10 mM MgCl2, and 2 mM β-ME by sonication on ice using a MISONIX sonicator XL device for 5 minutes (15 s pulse followed by 15 s rest). Centrifugation at 10,000g was then performed and the supernatant was collected. The supernatant was loaded onto a 5.5 × 2 cm Ni-NTA column equilibrated with 20 mM Tris-HCl, 500 mM NaCl, and 10 mM MgCl2. The same buffer containing imidazole at concentrations of 20, 30, 90, 120, 150, 200, 300, and 500 mM was used to elute the His-tagged enzyme; the D271N mutant HIcDH eluted at 150–200 mM imidazole. The enzyme solution was stored at −80° C in the elution buffer with 10 % of glycerol. The protein concentration was determined according to Bradford using the Bio-Rad protein assay kit with bovine serum albumin as a standard (18).
Enzyme Assay
The activity of HIcDH was measured by monitoring the production of NADH at 340 nm using a Beckman DU 640 spectrophotometer. All assays were carried out at 25° C. A unit of enzyme activity is defined as the amount of enzyme catalyzing the production of 1 μmol of NADH/min at 25° C. Rate measurements were carried out in a total volume of 500 μL in 1 mL quartz cuvettes with a 1 cm light path. The assay solution contained the following: 100 mM Hepes, pH 7.5, 100 mM KAc, 10 mM Mg(Ac)2, 5–103 μM HIc, and 1 mM NAD+. The concentration of MgHIc was calculated using the estimated Kd of 11 ± 2 μM for the Mg-HIc complex (17). Reactions were initiated by adding an appropriate amount of enzyme, and the initial linear portion of the time course was used to calculate the initial velocity.
Initial Velocity Studies
In the direction of NADH formation, the initial rate was measured as a function of MgHIc concentration (2.5 μM, 5 μM, 8 μM, and 50 μM) at the different fixed concentrations of NAD+ (25 μM, 35 μM, 60 μM, and 200 μM). The concentration of KAc and Mg(Ac)2 were maintained constant at 100 mM and 10 mM, respectively.
For dead-end inhibition studies initial rates were measured at different concentrations of one reactant, with the concentration of the other reactant fixed at its Km, and at different fixed concentrations of the inhibitor (Ki, 2Ki and 4Ki), including zero. For both inhibitors, 3-AcPyrADP and thiahomoisocitrate, an initial estimate of Ki for the inhibitor was obtained by fixing substrates at their respective Km, and measuring the rate as a function of inhibitor concentration. An appKi was then estimated by plotting 1/v versus I, obtaining the value of I at 1/v equal zero, and dividing the value by 2.
pH-rate Profiles
The initial rate was measured as a function of pH with the concentration of MgHIc varied and NAD+ maintained at saturation (~10 Km). The pH was maintained using the following buffers at 100 mM: Mes for pH 5.0–6.0, Bis-Tris for pH 6.0–7.0, Hepes for pH 7.0–8.0, Ches for pH 8.0–9.5. Sufficient overlap was allowed as buffers were changed to check for buffer effects; none were detected. The pH was recorded before and after initial velocity data were measured with observed changes limited to ±0.2 pH unit.
Effect of Solvent Deuterium and Viscosity
Initial velocities were measured in H2O and D2O. For rates measured in D2O, all the chemicals (Mes, KAc, and Mg(Ac)2) were dissolved in a small amount of D2O first, and lyophilized overnight to remove H2O and exchangeable protons. The lyophilized powders were then dissolved in D2O to get the desired concentration. NAD+ was prepared fresh by dissolving it in D2O to a stock concentration of 25 mM. The pD of 1 M Mes buffer was adjusted using DCl. A value of 0.4 was added to the pH meter reading to correct for the isotope effect on the electrode (19). Data were obtained by varying MgHIc at a fixed concentration (10 Km) of NAD+. The isotope effects were obtained by direct comparison of initial rates in H2O and D2O at pH(D) 6.5 which is in the pH-independent range of the V pH-rate profile. Reactions were initiated by adding 25 μL of the D271N mutant enzyme in H2O. The final percentage of D2O was ~95%.
To determine whether rates measured in D2O reflected the increased viscosity of the deuterated solvent, initial rates were measured in H2O at a relative viscosity of 1.24 at pH 6.5 and 25 °C. Glycerol at 9% (w/v) was included in the assays as the viscosogen, approximately equal to the viscosity of 100 % D2O (20). The amount of glycerol added was estimated from a standard curve of viscosity vs. % glycerol (21).
Data Processing
Initial velocity data were first analyzed graphically as double reciprocal plots of initial rates vs. substrate concentration to determine data quality and the proper rate equation for data fitting. Data were then analyzed using the appropriate rate equations, and the Marquardt-Levenberg algorithm supplied with the EnzFitter program from BIOSOFT (Cambridge, U.K.). Kinetic parameters and their corresponding standard errors were estimated using a simple weighting method.
Data obtained as a function of MgHIc concentration at different fixed levels of NAD+ were fitted using equation 1 for a sequential mechanism. Competitive and noncompetitive inhibition patterns were fitted to equation 2 and 3, respectively.
| (1) |
| (2) |
| (3) |
In equation 1, v and V are initial and maximum velocities, respectively, A and B are substrate concentrations, I is the inhibitor concentration, Ka and Kb are Michaelis constants for substrates A and B, and Kia is the dissociation constant for A from the EA complex. In equations 2–4, Kis and Kii represent the slope and intercept inhibition constants, respectively.
Data for the V pH-rate profile with a partial change at high pH were fitted using eq. 4, while data for the V/K pH-rate profile with a limiting slope of 1 at low pH and a partial change at high pH were fitted to equation 5.
| (4) |
| (5) |
In equation 5 and 6, y is the value of V and V/K at any pH, H is the hydrogen ion concentration, YL and YH are the constant values of V or V/K at low and high pH, respectively, while K1 and K2 are acid dissociation constants for the enzyme or substrate functional groups that must be unprotonated and protonated, respectively, for optimal binding and/or catalysis.
Data for solvent deuterium isotope effects on V and V/K were fitted using equation 6.
| (6) |
In equation 6, Fi is the fraction of D2O in the solvent and EV/K and EV are the solvent deuterium isotope effects minus 1 on V/K and V, respectively. Equations, with the exception of 5, are referenced in (22). Equation 5 was derived combining equation 4, which describes a partial change at high pH with an equation that describes an all-or-none change at low pH (YL/[1 + H/K1]).
Sequence Alignment
The multiple sequence alignment, Figure 1, was carried out using ClustalW2 (version 2.1) and the figure was prepared using Boxshade (23). The black-shadowed residues are identical, while those that are gray-shadowed are conservative substitutions. The (*) below the alignment identifies important active site residues, as discussed in the legend to Figure 1.
Preparation of the Structure Figure
The figure depicting the active site of the SpHIcDH, Fig. 2, was prepared using PyMOL, Molecular Graphics System, Version 1.7.4 Schrödinger, LLC. The labeling and color scheme are discussed in the legend to Fig. 2.
RESULTS
Expression and Purification of D271N Mutant ScHIcDH
The D271N mutant ScHIcDH was eluted from the Ni-NTA column using an imidazole gradient from 20 to 500 mM, and eluted most efficiently with 150–200 mM imidazole. The purity of the D271N mutant enzyme was ~98% on the basis of SDS-PAGE. The concentration of the D271N mutant enzyme was 2.5 mg/mL. The purified D271N mutant protein was separated into several aliquots with 10% glycerol in elution buffer and stored at −80° C. The mutant protein was stable under these conditions for more than 1 month.
Initial Velocity Studies
Initial rates of the D271N mutant enzyme were measured in the direction of oxidative decarboxylation. An initial velocity pattern was obtained by measuring the rate at varying concentrations of MgHIc and at different fixed concentrations of NAD+. Double-reciprocal plots intersected to the left of the ordinate, consistent with the sequential kinetic mechanism reported previously (17) (data not shown).
Replacing D271 with N results in a decrease of 520-fold in V/Et. The Km for MgHIc is essentially unchanged from WT, giving a 520-fold decrease in V/KMgHIcEt that reflects the change in V/Et. The Km for NAD+ decreases by 18-fold, resulting in an overall decrease of 26-fold in the value of V/KNADEt.
For the HIc reaction, 3-AcPyrADP and thiahomoisocitrate were chosen as the dead-end analogs of NAD+ and Hic, respectively. For the D271N mutant enzyme, 3-AcPyrADP is competitive versus NAD+ as predicted, but it is noncompetitive versus MgHIc. Thiahomoisocitrate shows competitive inhibition versus MgHIc, and is a noncompetitive inhibitor vs. NAD+. Initial rate kinetic parameters and inhibition constants for inhibitory analogs are compared to those of WT ScHIcDH in Tables 1 and 2.
Table 1.
Kinetic Parameters for WT ScHiCDH and the D271N Mutant Enzyme
| WTa | D271N | |
|---|---|---|
| V/Et (s−1) | 13 ± 1 | 0.026 ± 0.002 (−520) |
| V/KNADEt (M−1s−1) | (2.8 ± 0.2) × 104 | (1.08 ± 0.03) × 103 (−26) |
| V/KMgHIcEt (M−1s−1) | (3.1 ± 0.2) × 106 | (7.2 ± 0.2) × 103 (−520) |
| KNAD (mM) | 0.45 ± 0.08 | 0.025 ± 0.005 (−18) |
| KMgHIc (μM) | 4.2 ± 0.9 | 3.7 ± 0.7 (−1.1) |
| Ki MgHIc (μM) | 2 ± 1 | 5 ± 2 (+2.5) |
From reference 17. Values in () are fold change in the parameter.
Table 2.
Dead-end Inhibition Constants for WT ScHiCDH and the D271N Mutant Enzyme.
| Variable Reactant | Fixed Reactant | Inhibitor | Kis (mM) | Kii (mM) | Inhibition Pattern | Predicted for RER | |
|---|---|---|---|---|---|---|---|
| D271N | MgHIc | NAD+ | 3-AcPyrADP | 0.27 ± 0.08 | 0.40 ± 0.07 | NC | NC |
| NAD+ | MgHIc | 3-AcPyrADP | 0.34 ± 0.05 | -- | C | C | |
| MgHIc | NAD+ | ThiaHIc | 20 ± 3 | -- | C | C | |
| NAD+ | MgHIc | ThiaHIc | 32 ± 13 | 54 ± 17 | NC | NC | |
| Predicted for ssR | |||||||
| WTa | MgHIc | NAD+ | 3-AcPyrADP | 5.0 ± 0.5 | 9.0 ± 0.5 | NCb | NC |
| NAD+ | MgHIc | 3-AcPyrADP | 2.5 ± 0.4 | -- | C | C | |
| MgHIc | NAD+ | ThiaHIc | 0.14 ± 0.02 | -- | C | C | |
| NAD+ | MgHIc | ThiaHIc | _ | 0.07 ± 0.01 | UC | UC |
From reference 17 (reference 10 for ThiaHIc).
3-AcPyrADP is C vs. MgHIc at low concentrations and UC at high concentrations, a result of the proposed steady state random mechanism (17); the pathway with NAD+ adding first is slower than that with MgHIc adding first. The net result is a NC pattern if the mechanism were rapid equilibrium as in the case of the D271N mutant enzyme.
pH-rate Profiles
The pH dependence of kinetic parameters reflects the optimum protonation state of enzyme and/or reactant functional groups required for proper conformation, optimum binding, and/or catalysis. Initial velocity patterns were obtained at pH 5.4, 7.5 and 9.5 by measuring the initial rates at varied NAD+ concentration and at different fixed levels of MgHIc (at a saturating level of Mg2+). These patterns provide information about the kinetic mechanism as a function of pH and provide estimates of Km values at three pH’s, making it easier to determine substrate concentration ranges to be used as a function of pH. No change in kinetic mechanism was observed over the pH range used.
The pH dependence of kinetic parameters obtained for the D271N mutant enzyme with HIc as the substrate is shown in Figures 3 and 4. The V/Et pH-rate profile is pH-independent from pH 5.4 to 7.0 and decreases to a lower constant with a partial change above a pKa of 7.75 (Figure 3). The pH-independent values of V/Et at high and low pH are 0.040 ± 0.004 s−1 and 0.013 ± 0.002 s−1, respectively (0.31% and 0.10% of the V/Et of wild-type). The V/KMgHIcEt pH-rate profile, Fig. 4 is pH-dependent from pH 5.4 to 7.0 with a slope of 1 and exhibits the requirement for a group that must be unprotonated for optimal activity and/or binding; the observed pKa is 5.9. In addition, V/KMgHIcEt is also pH-dependent from pH 7.5 to 9.5, exhibits a partial change, giving a pKa of about 7.3. The pH-independent value of V/KMgHIcEt is about (1.4 ± 0.2) × 104 M−1 s−1, (0.45% of the V/KMgHIcEt of wild-type). The high pH-independent value of V/KMgHIcEt is 86 ± 6 M−1 s−1.
Figure 3.

pH dependence of V/Et for the oxidative decarboxylation reaction of the D271N mutant of ScHIcDH with Hic as the substrate. Initial velocity data were measured at 25 °C. The points are experimentally determined values, while the curve is theoretical based on a fit of the data using eq. 4.
Figure 4.

pH dependence of V/KMgHIcEt for the oxidative decarboxylation reaction of the D271N mutant of ScHIcDH with Hic as the substrate. Initial velocity data were measured at 25 °C. The points are experimentally determined values, while the curve is theoretical based on a fit of the data using eq. 5. The two points above the curve below pH 6 had lower precision than the rest of the data and were weighted lower for the final fit.
Solvent Deuterium Kinetic Isotope Effects and Effect of Viscosity
The D271N mutant enzyme exhibits a normal solvent deuterium isotope effect on V of 2.4. However, D2O(V/KMgHIc) is inverse with a value of 0.7 ± 0.2.
The ratio of V and V/K in water in the absence and presence of 9% glycerol, η(V) and η(V/K), are 1.2 ± 0.3 and 2.1 ± 0.7. Thus, it appears that there is no effect of viscosity at saturating reactant concentration and the SKIE of 2.4 reflects proton(s) in flight in the rate-determining transition state(s). On the other, there is a significant effect of viscosity at limiting Hic, and the reaction under these conditions must include a significant diffusional component.
DISCUSSION
Kinetic Mechanism
The double reciprocal plot obtained by measuring the initial rate as a function of MgHIc concentrations and different fixed concentrations of NAD+ intersects to the left of the ordinate. Data are consistent with a sequential kinetic mechanism for the D271N mutant enzyme. Additional information is provided by the dead-end inhibition studies; a dead-end inhibitor is a substrate analogue that normally resembles a reactant in structure and competes with a reactant for its binding site(s) on the enzyme, but cannot undergo the chemical transformation (22). In this study, 3-AcPyrADP was used as an analogue of NAD+ and thiahomoisocitrate was used as an analogue of Hic.. The inhibitors were competitive with the substrate it mimics, i.e., 3-AcPyrADP is competitive vs. NAD+ and thiahomoisocitrate is competitive vs. HIc. 3-AcPyrADP is a competitive inhibitor of the WT enzyme (17), and thiahomoisocitrate is a strong competitive inhibitor against HIc for the wild-type enzyme (10). However, they show noncompetitive inhibition versus the other reactants in both of the cases. Results are consistent with a rapid equilibrium random kinetic mechanism with 3-AcPyrADP binding to E and E-MgHIc, and thiahomoisocitrate binding to E and E-NAD+. A rapid equilibrium random kinetic mechanism has also been observed for the wild-type enzyme with isocitrate as the substrate; the enzyme with isocitrate is ~200-time slower than with HIc (17).
Previous studies suggested the rate of the WT enzyme reaction at limiting concentrations of MgHIc is limited by a conformational change triggered by the binding of MgHIc. This conformational change is likely required for the proper orientation of substrate and active site residues, i.e., generation of the catalytically competent Michaelis complex. At saturating reactant concentrations, tautomerization of the enol of α-ketoadipate coupled to a conformational to open the site and release products, was proposed as the limiting step (10). The net rate constant for the chemical steps is ~10-fold faster than that of the conformational change required to close the site upon binding of MgHIc (10). The 520-fold decrease in V/KMgHIcEt and V/Et observed for D271N mutant enzyme compared to WT indicates the importance of D271 in the ScHIcDH mechanism. The 520-fold decrease in the two limiting rate constants suggests: 1) if rate-limiting steps are different at limiting and saturating reactant concentration, the rates of the steps must fortuitously be equal; 2) the same step(s) limit the overall reaction at limiting and saturating reactant concentrations. The latter explanation is the more likely of the two. This aspect will be considered further below.
Interpretation of Isotope Effects
With HIc as the substrate for the oxidative decarboxylation reaction catalyzed by wild type ScHIcDH, the observed values of the solvent deuterium kinetic isotope effects, D2O(V) and D2O(V/KHIc), were 2.6 and 1.3, respectively (10). The V/K for homoisocitrate includes all steps from binding of reactant to the release of CO2 in the decarboxylation step (I to III in Scheme 1), while V includes all steps from the E-NAD-homoisocitrate complex to release of products (II to V in Scheme I). Given an inverse effect on V/K and a normal effect on V, these effects must be derived from steps that are not common to the limiting rate constants. A significant effect of viscosity is observed on V, but lower than the effect of solvent deuterium. Thus, a diffusional component is observed as part of the effect on V and a significant SKIE is also observed; the measured SKIE is 2.5, while the viscosity effect is only 1.5 (the SKIE corrected for viscosity is about 2). The normal effect on V likely reflects the ketonization of the enol of α-ketoadipate, and this is coupled to the conformational change to open the site and release products or close the site prior to catalysis, consistent with the slower rate in the presence of viscosogen indicative of a contribution from a diffusion-controlled process. The very small SKIE on V/K (1.3) coupled to the large inverse effect of viscosogen (0.6) is consistent with a slow conformational change to give a more active enzyme in the presence of glycerol (10).
Interpretation of the SKIE data for D271N must take into account the identical decrease in the limiting rate constants, V and V/KMgHIc. The D271N mutant enzyme exhibits a normal solvent deuterium kinetic isotope effect of 2.4 on V, identical to that observed for the wild type enzyme, but with a lower effect of viscosogen. An inverse isotope effect of 0.73 is observed on V/KMgHIc, together with a large effect of viscosity indicative of a significant diffusional component associated with the slow step. Data suggest the limiting step is likely binding of substrate coupled to a slow conformational change to close the site and generate the active Michaelis complex. The solvent deuterium kinetic isotope effects reflect a slow closing of the active site prior to catalysis at limiting and saturating concentrations of reactant. The lack of an inverse SKIE on V is expected since MgHIc is enzyme-bound under V conditions.
Interpretation of pH-rate Profiles
Wild-type ScHIcDH exhibits a steady-state random kinetic mechanism with a preferred order of addition of MgHIc prior to NAD+ (17). On the basis of the proposed chemical mechanism of WT, a conformational change is required to close the active site upon the binding of MgHIc prior to catalysis (9). The conformational change is rate-limiting at limiting MgHIc, and contributes to rate-limitation at saturating reactant concentrations. The dominant species present when V/KMgHIcEt is measured is E-NAD and MgHIc. The pH dependence under these conditions will reflect groups on reactant and/or enzyme important for binding and/or catalysis. The pH dependence of V will reflect groups on the enzyme important for catalysis (22).
For wild-type ScHIcDH, a chemical mechanism has been proposed in which a general base with a pKa of 7.1 acts to accept the proton from 2-hydroxyl of HIc and a general acid with a pKa of 9.5 acting to donate a proton to C3 once the decarboxylation has occurred. With MgHIc as the substrate, the pKa of 9.5 is only observed in the V pH-rate profile, suggesting that tautomerization of the enol of α-ketoadipic acid contributes to rate limitation at high pH and saturating reactant concentrations (10). A hollow is observed in the V pH-rate profile indicating substrate and proton are sticky in the HE-NAD-MgHIc ternary complex (10).
The pH-rate profile for V/KMgHIcEt of wild-type enzyme exhibits a decrease at low pH with a slope of 2 reflecting two groups, giving an average pKa value of 6.0. One of the groups likely reflects the protonation of Hic, which has a pKa of 5.8 (10); when MgHIc is protonated it does not bind to enzyme. The second of the pKa values likely reflects the catalytic general base. K206 and Y150 were proposed as general base and general acid in the mechanism. The V/Et pH-rate profile for the Y150F and K206M mutant enzymes exhibit pKa values of 6 and 9.5, respectively, reflecting K206 and Y150, in the mutant enzymes (11).
Elimination of the ionizable side chain of D271 results in pH independence of V/Et at low pH, with a slight decrease to a lower constant value at high pH giving a pKa of about 7.8. Neither of the pKas observed for the WT enzyme are observed. The V/KMgHIc pH-rate profile for D271N exhibits a single pKa of 6 at low pH, and also exhibits the partial change observed in the V profile at high pH, giving a pKa of about 7.8. The dominant enzyme species under V and V/K conditions for D271N is the E-NAD-MgHIc which slowly isomerizes to the active E*-NAD-MgHIc. The rate of the isomerization is pH-independent and much slower than the catalytic steps. As a result the rate will not decrease at low and high pH until the rate of the catalytic steps becomes equal to or less than the rate of the isomerization. The 520-fold decrease translates to about 2.7 log units, suggesting that the effective pKas of 6 and 9.5 will be perturbed to lower and higher pH by 2.7 pH units. The pKa of 6 observed in the V/K pH-rate profile reflects protonation of Hic, which must be unprotonated for optimum binding and catalysis. The partial change observed in the V and V/K pH-rate profiles likely reflects a pH-dependent conformational change. The group responsible and reason for this conformational change will have to await further study.
Conclusions
Data obtained from initial rate studies and isotope effects suggest a rate-limiting pre-catalytic conformational change. At limiting MgHIcDH, the conformational change includes the binding of MgHIc. The 520-fold decrease in the rate likely derives from a decrease in the rate of the conformational change, which becomes much slower than the rate of the chemical steps. As a result, the pH dependence of kinetic parameters does not reflect chemistry, rather the pH-independence of the conformational change. Data support the hypothesis that the pre-catalytic conformational change is coupled to the binding of MgHIc.
Replacing D271 with N in homoisocitrate dehydrogenase gave 500-fold lower activity
The same slow step limits at high and low homoisocitrate concentration
The pH independence of V/Et indicates chemistry is not slow
The effect of solvent deuterium suggests a slow pre-catalytic conformational change
Acknowledgments
This work was supported by a grant (GM 071417) from the National Institutes of Health (to P.F.C. and A.H.W), and the Grayce B. Kerr Endowment to the University of Oklahoma to support the research of P.F.C.
Footnotes
Abbreviations: AAA, α-aminoadipate; 3-AcPyrADP, 3-acetylpyridine adenine dinucleotide 3′-phosphate; NAD, nicotinamide adenine dinucleotide (the + charge is omitted for convenience); HIcDH, homoisocitrate dehydrogenase (prefix identifies the species of origin of the enzyme, e.g., Sc represents Saccharomyces cereviae); 6PGDH, 6 phosphogluconate dehydrogenase; IcDH, isocitrate dehydrogenase; IPMDH, isopropylmate dehydrogenase; Hic, homoisocitrate; Mes, 2-(N-morpholino)-ethanesulfonic acid; Bis-Tris, bis(2-hydroxyethyl)aminotris(hydroxymethyl)methane; Tris, tris(hydroxymethyl)aminomethane; Ches, 2-(N-cyclohexylamino)-ethanesulfonic acid; Hepes, 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid; IPTG, isopropyl β-D-thiogalactopyranoside; Ac, acetate; βME, β-mercaptoethanol; WT, wild type; DCl, deuterium chloride; NaOD, sodium deuterioxide; D2O, deuterium oxide; Ni-NTA, nickel-nitrilotriacetic acid; PAGE, polyacrylamide gel electrophoresis
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xu H, Andi B, Qian J, West AH, Cook PF. The α-aminoadipate pathway for lysine biosynthesis in fungi. Cell Biochemistry and Biophysics. 2006;45:43–64. doi: 10.1385/CBB:46:1:43. [DOI] [PubMed] [Google Scholar]
- 2.Karsten WE, Cook PF. Pyridine nucleotide dependent α-hydroxyacid oxidative decarboxylases: An overview. Protein Pept Lett. 2000;7:281–286. [Google Scholar]
- 3.Dyson JED, Dorazio RE, Hanson WH. Sheep liver 6-phosphogluconate dehydrogenase: Isolation procedure and effect of pH, ionic-strength, and metal-ions on kinetic parameters. Arch Biochem Biophys. 1973;154:623–635. doi: 10.1016/0003-9861(73)90017-9. [DOI] [PubMed] [Google Scholar]
- 4.Malik R, Viola RE. Structural characterization of tartrate dehydrogenase: a versatile enzyme catalyzing multiple reactions. Acta Crystallogr D Biol Crystallogr. 2010;66:673–684. doi: 10.1107/S0907444910008851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin Y, West AH, Cook PF. Potassium activates homoisocitrate dehydrogenase by increasing the affinity for NAD. Biochemistry. 2008;47:10809–10815. doi: 10.1021/bi801370h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aktas DF, Cook PF. A Lysine-Tyrosine pair carries out acid-base chemistry in the metal ion-dependent pyridine dinucleotide-Linked β-hydroxyacid oxidative decarboxylases. Biochemistry. 2009;48:3565–3577. doi: 10.1021/bi8022976. [DOI] [PubMed] [Google Scholar]
- 7.Grodsky NB, Soundar S, Colman RF. Evaluation by site-directed mutagenesis of aspartic acid residues in the metal site of pig heart NADP-dependent isocitrate dehydrogenase. Biochemistry. 2000;39:2193–2200. doi: 10.1021/bi9919753. [DOI] [PubMed] [Google Scholar]
- 8.Hurley JH, Dean AM, Koshland DE, Stroud RM. Catalytic mechanism of NADP+-dependent isocitrate dehydrogenase: Implications from the structures of magnesium isocitrate and NADP+ complexes. Biochemistry. 1991;30:8671–8678. doi: 10.1021/bi00099a026. [DOI] [PubMed] [Google Scholar]
- 9.Karsten WE, Liu DL, Rao GSJ, Harris BG, Cook PF. A catalytic triad is responsible for acid-base chemistry in the Ascaris suum NAD-malic enzyme. Biochemistry. 2005;44:3626–3635. doi: 10.1021/bi047826o. [DOI] [PubMed] [Google Scholar]
- 10.Lin Y, Volkman J, Nicholas KM, Yamamoto T, Eguchi T, Nimmo SL, West AH, Cook PF. Chemical mechanism of homoisocitrate dehydrogenase from Saccharomyces cerevisiae. Biochemistry. 2008;47:4169–4180. doi: 10.1021/bi702361j. [DOI] [PubMed] [Google Scholar]
- 11.Lin Y, West AH, Cook PF. Site-directed mutagenesis as a probe of the acid-base catalytic mechanism of homoisocitrate dehydrogenase from Saccharomyces cerevisiae. Biochemistry. 2009;48:7305–7312. doi: 10.1021/bi900175z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulfer SL, Hendershot JM, Trievel RC. Crystal structure of homoisocitrate dehydrogenase from Schizosaccharomyces pombe. Wiley Online Library, Proteins, Structure Note. 2011:661–666. doi: 10.1002/prot.23231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nango E, Yamamoto T, Kumasaka T, Eguchi T. Structure of Thermus thermophilus homoisocitrate dehydrogenase in complex with a designed inhibitor. J Biochem. 2011;150:607–614. doi: 10.1093/jb/mvr097. [DOI] [PubMed] [Google Scholar]
- 14.Imada K, Inagaka K, Matsunami H, Tanaka H, Tanaka N, Namba K. Structure of 3-isoprpylmalate dehydrogenase in complex with 3-isopropylmalate at 2.0 angstrom resolution: The role of Glu88 in the unique substrate recognition mechanism. Struct Folding Design. 1998;6:971–982. doi: 10.1016/s0969-2126(98)00099-9. [DOI] [PubMed] [Google Scholar]
- 15.Ma G, Palmer DRJ. Improved asymmetric syntheses of (2R)-(3S)-homocitrate and (2R, 3S)-homoisocitrate, intermediates in the α-aminoadipate pathway of fungi. Tetrahedron Lett. 2000;41:9209–9212. [Google Scholar]
- 16.Yamamoto T, Eguchi T. Thiahomoisocitrate: A highly potent inhibitor of homoisocitrate dehydrogenase involved in the α-aminoadipate pathway. Bioorganic & Medicinal Chemistry. 2008;16:3372–3376. doi: 10.1016/j.bmc.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 17.Lin Y, Alguindigue SS, Volkman J, Nicholas KM, West AH, Cook PF. The complete kinetic mechanism of homoisocitrate dehydrogenase from Saccharomyces cerevisiae. Biochemistry. 2007;46:890–898. doi: 10.1021/bi062067q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 19.Schowen KB, Schowen RL. Solvent isotope effects on enzyme systems. Methods Enzymol. 1982;87:551–606. [PubMed] [Google Scholar]
- 20.Quinn DM, Sutton LD. In: Enzyme Mechanism from Isotope Effects. Cook PF, editor. CRC Press; Boca Raton, FL: 1991. pp. 73–126. [Google Scholar]
- 21.Bazelyansky M, Robey E, Kirsch JF. Fractional diffusion-limited component of reactions catalyzed by acetylcholinesterase. Biochemistry. 1986;25:125–130. doi: 10.1021/bi00349a019. [DOI] [PubMed] [Google Scholar]
- 22.Cook PF, Cleland WW. Enzyme kinetics and mechanism. Garland Press; London: 2007. [Google Scholar]
- 23.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]