

Abstract
While Kisspeptin was initially found to function as a metastasis suppressor, after identification of its receptor KissR1 and their expression profile in tissues like the hypothalamus and adrenals, kisspeptin and Kiss1R were predominantly assigned endocrine functions, including regulating puberty and fertility through their actions on hypothalamic gonadotropin releasing hormone production. More recently, an alter ego for kisspeptin has emerged, with a significant role in regulating glucose homeostasis, insulin secretion, as well as food intake and body composition, with lack of kisspeptin signaling resulting in reduced locomotor activity and increased adiposity. This review will highlight these recent observations of kisspeptin's role in metabolism and the several key questions that have emerged and that need to be addressed in the future.
Keywords: Kisspeptin, glucagon, insulin, satiety, obesity, sexual dimorphism
Introduction
Screening to identify molecules responsible for suppressing metastasis, over 18 years ago, led to the initial isolation of a novel cDNA enriched in non-metastatic melanoma cells, named KISS1 (1). Its product, a 54 amino acid protein, was found to suppress metastatic potential and was named metastin (2). Subsequent studies described its metastasis-suppressive activity in a variety of cancers, actions that it exerts through binding and activating a specific Gq/G11-protein-coupled receptor (GPCR) (3, 4). The GPCR GPR54 (later renamed to kisspeptin1 receptor = KISS1R) was identified to bind and transmit the cellular action of secreted kisspeptins (5) that were then considered to be a promising new target in cancer therapy (3).
The Kiss1 cDNA encodes a 154 amino acid pre-propeptide that is C-terminally amidated and proteolytically processed, yielding not only kisspeptin-54 (KP54, = metastin) but also three C-terminal fragments, namely kisspeptin-14 (KP14), kisspeptin-13 (KP13) and kisspeptin-10 (KP10). All of these products share the same C-terminal 10 amino acid amidated sequence and belong to the larger family of RF-amide peptides. The KP10 sequence is sufficient to bind and activate KISS1R. Kisspeptin1 is the mammalian form of the peptide (KP54), which also occurs in non-mammalian organisms, where it is called Kisspeptin2 (6).
Kisspeptin and Kiss1r expression have been reported in a variety of tissues. The tissue distribution of Kiss1 and Kiss1r often localize to the same cells. In rodents Kiss1 and Kiss1r expression are highest in placenta and the central nervous system, where the highest levels are detected in the hypothalamus and pituitary but also in brainstem, cortex, and cerebellum (5, 7, 8). Expression of both Kiss1 and Kiss1r have been reported in rodent adipose tissue, pancreas, liver, small intestine, peripheral blood lymphocytes, testes, lymph nodes, as well as in in human aorta, coronary artery and umbilical vein (2, 8-10). These reports need to be taken with caution since the specificity of reagents used in some of these studies may be cross reacting with the large number of peptides with a C-terminally amidated motif (RF-amide peptides).
Soon after the initial observations as a metastasis suppressor, kisspeptin's fate turned and its biological journey switched from cancer biology to an entirely different path. Kiss1R signals through stimulation of phospholipase C and plays a key role in the neuroendocrine control of the gonadotrophin axis. Kisspeptin stimulates the secretion of gonadotropins from the pituitary by stimulating the release of GnRH from the forebrain after the activation of Kiss1r, which is expressed by GnRH neurons. Kisspeptin is expressed abundantly in the arcuate nucleus (Arc) and the anteroventral periventricular nucleus (AVPV) of the forebrain. Both estradiol and testosterone regulate Kiss1 gene expression in Arc and AVPV; however, while estradiol and testosterone down-regulate Kiss1 mRNA in the Arc, they up-regulate Kiss1 expression in the AVPV. Thus, kisspeptin neurons in the Arc may participate in the negative feedback regulation of gonadotropin secretion, whereas kisspeptin neurons in the AVPV may contribute to generating the preovulatory gonadotropin surge in the female. Hypothalamic levels of Kiss1 and GPR54 mRNA increase dramatically at puberty, suggesting that kisspeptin signaling mediate the neuroendocrine events that trigger the onset of puberty (11). KISS1R was found to regulate puberty and fertility, and inactivating mutations in the KISS1R were found to be associated with reduced or absent fertility (12, 13). Subsequently, inactivating mutations in Kiss1 were also described to be associated with a subfertile phenotype (14). Moreover, functionally activating mutations of KISS1R are associated with precocious puberty (15, 16). A number of recent reviews highlight exciting new findings on kisspeptin regulation of reproduction (3, 11, 17).
More recent work assigns kisspeptin roles in regulating glucose homeostasis, locomotor activity and body weight control (18, 19). The present article will discuss the recent surprising and exciting findings of kisspeptin in metabolism regulation, their potential implications in understanding disease and outline new questions that will direct the path of kisspeptin's ever continuing journey.
Liver derived kisspeptin participates in islet hormone crosstalk
A long-standing question in pancreatic islet biology and regulation of insulin secretion has been whether and how the two principal islet hormones, insulin from ß-cells and glucagon from α-cells, may regulate each-others' secretion. Teleologic considerations of the opposing effects of each hormone on blood glucose levels would posit that 1) insulin might suppress glucagon secretion from α-cells and 2) that glucagon may suppress insulin secretion from ß-cells.
In the first case, several convincing observations indicate that insulin exerts direct effects on glucagon secretion. The absence of insulin, as is seen in rodent models of type 1 diabetes (T1DM) and humans with T1DM is accompanied by very high levels of circulating glucagon (20). Further, treatment of insulin-deficient hyperglycemic mice with insulin, result in reversal of hyperglucagonemia, while simply reversing the hyperglycemic state in the absence of insulin replacement does not suppress glucagon secretion (21). Insulin receptors are present on glucagon-producing α-cells, and when these insulin receptors are selectively and conditionally ablated in mice using the CRE-loxP technology, hyperglucagonemia is observed (22). Collectively, these observations indicate that insulin may be acting directly via its receptor on α-cells to suppress glucagon secretion.
Conversely, whether glucagon directly influences insulin secretion from ß-cells has remained somewhat unclear. Subsets of humans with type 2 diabetes mellitus (T2DM) exhibit elevated glucagon levels and insufficient insulin secretion to control glucose levels. Moreover, individuals at high risk for developing T2DM exhibit elevated glucagon levels prior to being diagnosed with T2DM (23).
In an experimental system of chronic intravenous glucose infusions in rats, a compensatory increase in insulin secretion occurs to control glycemia. Remarkably however, after prolonged glucose infusion, insulin secretion wanes, coincident with increasing glucagon levels. Importantly, subsequent treatment of these mice with glucagon neutralizing antibodies is accompanied by a recovery of insulin secretion and normalization of glucose homeostasis, despite continued exogenous glucose infusions (24). At first sight these observations would indicate that glucagon might directly act on ß-cells to suppress insulin secretion. However, direct in vitro testing of glucagon action on isolated ß-cells has not provided conclusive evidence for a direct effect of glucagon on ß-cells. In vitro glucagon treatment of rat islets stimulates the inducible cyclic AMP element repressor (ICER) in ß-cells, which theoretically would suppress insulin biosynthesis (25). However, conditional ablation of the glucagon receptor in ß-cells in mice would be the most direct approach to examine whether glucagon exerts any effects directly on ß-cells to influence insulin secretion in vivo. Such studies have not been reported thus far.
The G protein Gas-coupled glucagon receptor is expressed on hepatocytes, where its activation rapidly stimulates cyclic AMP (cAMP) production (26). Cyclic AMP signaling in turn binds to the regulatory subunit of the protein kinase A (PKA) holoenzyme, thereby releasing the PKA catalytic subunit (27), and activating the PKA signaling cascade and transcription of cyclic AMP response element binding protein (CREB) response genes. Among these genes are the well-characterized genes of the gluconeogenesis program (28-30).
Thus, selective activation of liver PKA-dependent signaling stimulates gluconeogenesis, leading to hyperglycemia, which would be expected to stimulate insulin secretion from ß-cells. Paradoxically however, when this experiment was initially conducted in transgenic mice over-expressing PKA catalytic subunit in hepatocytes, insulin secretion was suppressed (31).
In combination, the above-summarized observations suggest the possibility that glucagon may not be acting directly on ß-cells to regulate insulin secretion, but rather that glucagon may be acting indirectly via action on the liver, where activation of PKA-dependent signaling is relayed by a separate signal that reaches ß-cells and suppresses insulin secretion. The functional role for glucagon receptors on ß-cells thus remains unclear and will need further studies.
This possibility was tested by selectively ablating in vivo the PKA regulatory subunit 1a (Prkar1a) in the liver of adult mice- using the CRE-LoxP technology - with the aim of constitutively activating PKA catalytic activity. Mice harboring floxed prkar1a alleles were treated intravenously with adenovirus expressing CRE recombinase to ablate prkar1a and activate liver PKA signaling and the gluconeogenic program (= ΔL-Prkar1a mice) (18). This mouse model was compared to mice receiving exogenous intravenous glucose infusion to achieve similar levels of hyperglycemia (32). It is important to note that these studies were all performed after 4 days of hyperglycemia, thereby minimizing the possibility of so-called glucotoxic damage to beta cell function following prolonged exposure to high glucose levels.
Mice receiving intravenous glucose exhibited mildly impaired glucose tolerance and robust and significantly elevated insulin secretion in response to an intra-peritoneally administered glucose load (intraperitoneal tolerance test - ipGTT). In contrast, ΔL-Prkar1a mice exhibited, during an ipGTT, significantly impaired glucose tolerance, owing to significantly impaired insulin secretion. Most importantly, plasma from ΔL-Prkar1a suppressed insulin secretion from in vitro incubated wild-type mouse islets. Dilution of ΔL-Prkar1a plasma also reduced the suppression of insulin secretion (18). These observations clearly suggested that ΔL-Prkar1a mice harbor a circulating factor, which directly suppresses ß-cell response to glucose.
A gene expression array combined with bioinformatic analysis to identify secreted proteins in the liver of ΔL-Prkar1a mice and Prkar1a floxed controls was used in an attempt to identify this potential liver-secreted factor. Surprisingly, this approach yielded a single candidate gene, Kiss1, which was further confirmed by direct assessment of liver kisspeptin mRNA expression as well as protein levels by immunodetection to be stimulated in livers of ΔL-Prkar1a mice but not in Prkar1a floxed controls (18).
Consistent with the hypothesis that glucagon stimulates kisspeptin expression in the liver, in vitro treatment of primary hepatocytes with glucagon or by forskolin-induced increase in intracellular cAMP concentrations stimulated kisspeptin production. Further, in vivo glucagon treatment stimulated liver kisspeptin production in control mice but not in mice lacking the glucagon receptor on the liver (ΔL-Gcgr mice). Endogenous stimulation of glucagon secretion by fasting also increased liver kisspeptin synthesis in control (Gcgr floxed) but not ΔL-Gcgr mice. Analysis of the immediate upstream elements of the Kiss1 promoter reveals in mice two and in humans one cAMP response element binding protein (CREB) recognition site, and luciferase reporter constructs of the Kiss1 immediate upstream element respond to cAMP induction and CREB activation. Conversely, mutation and inactivation of the promoter CRE binding sites abolishes Kiss1 promoter activation by cAMP or during co-transfection of a dominant negative CREB expression vector in transient co-transfection studies (18).
Similar to observations made in genes of the gluconeogenic program, treatment of isolated mouse hepatocytes with glucagon stimulated kisspeptin production, while additional treatment with insulin dampened these effects of glucagon. Importantly however in vivo ablation of the liver insulin receptor (ΔL-Insr mice) did not result in an increase in Kiss1 expression, suggesting that insulin resistance is not an important mechanism of increased in vivo liver kisspeptin production. Furthermore, treatment of mice with synthetic KP10 or KP54 intraperitoneally simultaneously with glucose administration during an intraperitoneal glucose tolerance test (ipGTT) resulted in impaired glucose tolerance owing to markedly reduced glucose stimulated insulin secretion (18).
Thus, the observations above suggest that liver glucagon receptor activation on one hand stimulates insulin secretion by increased hepatic glucose production (HGP) and hyperglycemia, while on the other hand, liver glucagon action may inhibit insulin secretion by stimulating kisspeptin production (Figure 1).
Figure 1. The tri-hormonal endocrine circuit between the islet and liver and its interplay in glucose regulation.
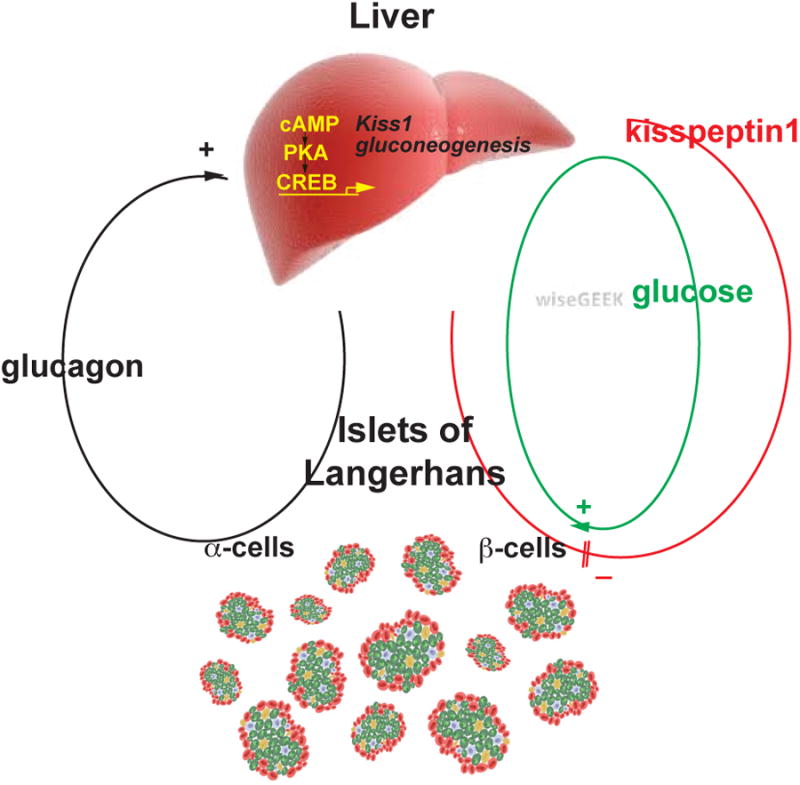
Glucagon - produced from alpha-cells in the Islets of Langerhans - stimulates in the liver cAMP - PKA signaling, which up-regulates transcription of the gluconeogenic gene program and of Kiss1. This leads to increased hepatic glucose production and hepatic glucose output and also to kisspeptin1 production. Glucose stimulates insulin secretion, whereas kisspeptin1 inhibits insulin secretion from pancreatic beta-cells. (Reprinted with permission from Song et al. 2014).
This idea was further tested by dissociating gluconeogenesis from kisspeptin production, in ΔL-Prkar1a mice. Adenovirus-mediated delivery of Kiss1-specific shRNA selectively inhibited of liver kisspeptin production while PKA activation of the hepatic glucose production remained elevated (18). However, ΔL-Prkar1a mice with kisspeptin knockdown showed - as compared to ΔL-Prkar1a mice treated with control adenovirus -improved glucose tolerance, owing to increased insulin secretion, indicating that in vivo subtraction of kisspeptin in mice with constitutively active HGP allows ß-cells to secrete more insulin in response to a glucose stimulus (18).
Functional kisspeptin1 receptor on pancreatic ß-cells
Kisspeptin and Kiss1R have both been described on pancreatic islet cells (33, 34). However, in the absence of appropriate cell specific Kiss1r ablated models, it has until recently been difficult to assess how kisspeptin may affect GSIS (see also below). Mouse islets express Kiss1r protein at high levels in ß-cells, whereas little Kiss1r immunoreactivity is found in α-cells.
Kiss1r effects on islet cell function was interrogated using the CRE-loxP system to generate mice with selective ablation of pancreatic Kiss1r using the pancreas-specific PDX-1 CRE driver mouse and the Kiss1R floxed mouse (ΔPanc-Kiss1r mouse) (18). At baseline fasting conditions, ΔPanc-Kiss1r exhibit no obvious phenotype and defect in GSIS, except mildly elevated fasting insulin levels. Conversely, intraperitoneal treatment with synthetic 10 nmol KP54 or KP10 suppressed GSIS in control mice but not from mice that lack pancreas Kiss1r. Consistent with these in vivo observations, kisspeptin (KP54) inhibits in a dose-dependent manner GSIS in vitro from islets isolated from control mice, but not from islets isolated from ΔPanc-Kiss1r mice. Furthermore, kisspeptin (KP54) also inhibits in a dose-dependent manner GSIS potentiation and cAMP production in islets that results from treatment with the potent incretin glucagon-like-peptide-1 receptor activator exendin-4 (18). These findings are consistent with the idea that Kiss1-Kiss1r action on ß-cells induces resistance to incretin action by suppressing cAMP synthesis in ß-cells - the latter effect is also seen when the galanin receptor, structurally closely related to Kiss1r - is activated on ß-cells (35).
Liver kisspeptin and diabetes mellitus
Are the findings made in the above-described genetically defined animal models with cell-specific gene manipulations, reflected in disease models of T2DM?
Liver from high fat diet (HFD) fed obese and glucose intolerant mice as well as a mouse model of T2DM (db/db mice) - when compared to controls exhibit increased liver kisspeptin expression as well as increased circulating plasma kisspeptin concentrations (18) In addition, liver biopsies taken from humans diagnosed with T2DM exhibit higher kisspeptin production, when compared to liver tissue from humans not diagnosed with T2DM. Accordingly, circulating kisspeptin levels in humans with T2DM are higher than in those free of T2DM. It is important to note, however, that liver kisspeptin expression in humans diagnosed with T2DM can be variable, even when samples are examined from donors not being treated with insulin or metformin. At this point, it remains unclear whether there exists a subgroup among humans with T2DM that exhibits elevated kisspeptin expression, and how this subgroup may be further characterized.
Both HFD-fed and db/db mice exhibit increased circulating glucagon levels, and treatment with a glucagon receptor antagonist in these mice reduces liver kisspeptin production and improved glucose homeostasis (18) Moreover, shRNA-mediated kisspeptin knockdown in the liver in HFD-fed and in db/db mice results in improved glucose tolerance owing to increased insulin secretion.
These findings collectively suggest that in T2DM, kisspeptin production is elevated in the liver, and that this increased kisspeptin production is likely secondary to increased glucagon levels. Furthermore, reducing liver kisspeptin production by shRNA mediated knockdown in rodent models mimicking T2DM de-represses insulin secretion.
The enigma of circulating kisspeptin concentrations
Previous attempts to assess a role for kisspeptin on insulin secretion have yielded conflicting results. While some investigators have reported that kisspeptin stimulates GSIS (9), others have reported the opposite (33, 34). A careful assessment of these diverging reports reveals that the concentrations for kisspeptin used in the various studies have been vastly different. In general, kisspeptin concentrations in the nanomolar range have been reported to suppress GSIS, while micromolar kisspeptin concentrations stimulate GSIS.
To directly address this controversy, islets from control (Kiss1r fl/fl) and islets from ΔPanc-Kiss1R mice were treated with different concentrations of KP10 or KP54. Consistent with previous observations, kisspeptin at nanomolar concentrations suppressed GSIS from control islets but not from islets lacking Kiss1R. In contrast, kisspeptin at micromolar concentrations stimulated GSIS even in the absence of Kiss1r (ΔPanc-Kiss1r islets) (18).
Based on these studies, it appears that at nanomolar concentrations kisspeptin -induced GSIS suppression is mediated by the known KISS1R. However at concentrations in the micromolar range, which appear supraphysiologic, kisspeptin stimulates GSIS independently of KISS1R. These studies suggest that at micromolar concentrations, kisspeptin action may occur through alternate receptors but not through the known bona fide Kiss1R. This possibility is further supported by a recent finding that kisspeptin action in the arcuate nucleus can be mediated by Kiss1r and, in the absence of Kiss1r, by the neuropeptide FF receptor (36).
The measurement of circulating kisspeptin concentrations in rodents and humans using commercially available methods has not been very reliable, due to large variations in the assay methods, their ranges of detection, and uncertainty about which forms of kisspeptin (i.e. KP10, KP15, KP54) are detected (4, 37, 38). Improved assays for measuring the different fragments of kisspeptin in biological fluids will be required to advance the field of kisspeptin-related studies in humans.
Because of these limitations, a bioassay of insulin secretion from isolated islets that are exposed to plasma samples from rodents and from humans was developed to assess GSIS suppression by circulating factors (18). This bioassay assesses functional kisspeptin activity on insulin secretion rather than kisspeptin concentrations. Using synthetic KP54 or KP10 as standards in this bioassay, reveals that high kisspeptin levels in biological samples from humans and experimental mice lie in the nanomolar range and not in the micromolar range (18).
A recent report of in vivo treatment of cynomongolous monkeys showed that a bolus dose of kisspeptin potentiates GSIS, rather than suppression of insulin secretion (39). No circulating kisspeptin concentrations are available from these studies and it remains unclear what the mechanism of increased kisspeptin-induced GSIS may be in this animal model. Further, intravenous glucagon administration followed by measurement of extent of C-peptide elevation is an established clinical test to assess insulin secretion reserve from pancreatic beta-cells (40). The underlying mechanism of glucagon on stimulating insulin secretion during this clinical test is unclear, and has been presumed to be secondary to increased circulating glucose levels caused by glucagon action on hepatic glucose output (i.e. glycogenolysis and gluconeogenesis). Whether kisspeptin - also stimulated by glucagon – may in fact dampen overall insulin secretion during such tests remains at present unclear.
Kisspeptin receptor ablation results in obesity and glucose intolerance in female mice
The studies described above have clearly delineated an important role for KISS1 signaling in the regulation of insulin secretion from the pancreatic islets. Evidence for a broader role for Kiss1r signaling in the regulation of metabolism comes from detailed analysis of the Kiss1r knock-out (KO) mouse (19). These studies confirmed Kiss1R mediated signaling is essential for regulating reproductive function, but reported dramatic differences in weight, body composition and glucose metabolism. In addition, there were striking differences in the phenotype observed between males and females that raise a host of new questions (Figure 2).
Figure 2. Kisspeptin receptor knockout mice reveal a regulatory role for Kiss1R signaling in activity and body composition.

Top panel: Kisspeptin receptor-defective mice exhibit infertility and increased fat mass but show sexual dimorphism with respect to total body weight, lean body mass and glucose tolerance. Female Kiss1R KO mice show more pronounced metabolic dysfunction with obesity and glucose intolerance.
Bottom panel: Increased weight gain in female Kiss1R KO mice results from profoundly reduced locomotor activity in the face of slightly reduced food intake and reduced energy expenditure while thyroid hormone production is not disturbed. (Summarized from Tolson et al. 2014)
Previous studies had not noted a difference in body weight between Kiss1 and Kiss1r KO mice and WT controls (19). However, these studies had only monitored body weight until 7 weeks of life. Measuring the weights beyond 7 weeks revealed a significant gain in weight by female mice beginning at 8 weeks that grew more pronounced as the mice aged (Kiss1r KO mice were 30% heavier than controls;). Tolson et al. reported that while both males and females exhibited an increase in body fat percentage, only females were found to have significant weight gain and impaired glucose tolerance (19). Additional metabolic testing needs to be performed to ascertain why increased adiposity in male Kiss1r KO males, relative to controls, was not associated with impaired glucose tolerance as it was for females
To further understand the underlying mechanism contributing to this metabolic profile in female KO mice, comprehensive lab animal phenotyping (CLAMS) was performed and demonstrated that the female mice were not hyperphagic, and in fact they ate less than control mice. However, energy expenditure was greatly reduced. While the reduced energy expenditure was attributed in large part to dramatically reduced locomotor activity (19), thyroid hormone activation of basal metabolic rate could also contribute to increased energy expenditure. However, circulating thryoxine (T4) levels did not differ between Kiss1r KO and control mice. Other potential sources of decreased energy expenditure, including impaired thyoroxine deiodination to active T3, autonomic regulation of brown adipose tissue have not yet been fully explored.
Because of the well-established obesity observed in estrogen-deficient females (41-44) the authors sought to determine whether weight gain in Kiss1r KO mice was related to low estradiol (E2) levels resulting from impaired Kiss1 signaling in GnRH neurons, or due to impaired Kiss1 signaling independently of E2 production. Ovariectomized - and thus E2 deficient - Kiss1r KO female mice continued to exhibit reduced energy expenditure, increased weight gain, fat mass accumulation and impaired glucose homeostasis relative to ovariectomized controls (19). Thus, lack of Kiss1R signaling, independently of changes in E2 levels, contributes to satiety regulation and energy homeostasis. Interestingly, gonadectomized (GNX) male Kiss1r KO mice did not exhibit increased fat mass relative to controls, as was seen in gonad intact males, suggesting that the effects on fat accumulation in males may be secondary to reduced testosterone levels (19).
While the sources and targets of Kiss1 contributing to locomotion and satiety regulation were not revealed by these studies, they do suggest that new avenues of exploration are required to understand how kisspeptin signaling contributes to regulation of body composition and to the sex-specific differences that are observed in Kiss1r KO mice. Further studies also need to be performed to determine whether there are sex differences in kisspeptin action at the level of the ß-cell since the data reported in Song et al. (18) were exclusively reported for male mice.
Concluding remarks
A variety of roles for kisspeptin have been assigned in cancer metastasis, fertility and puberty regulation and most recently in regulating insulin secretion and glycemia as well as control of feeding behavior, locomotor activity and energy expenditure. In addition functions for kisspeptin are described in ovarian follicular maturation, embryo implantation, sperm capacitation, and placentation (45-48). These different functions of kisspeptin, at present appear at best only loosely connected. How the various functions of kisspeptin are functionally interconnected and how in aggregate they serve an individual organism to maintain homeostasis still remains to be fully understood, require several key questions to be addressed (see Outstanding Questions Box) and will require extensive additional studies.
Trends Box.
Kisspeptin and its Class I G-protein coupled receptor Kiss1R regulate a) puberty and fertility but also b) glucose homeostasis, locomotor activity and body weight control
Glucagon stimulates liver kisspeptin production. Kisspeptin inhibits insulin secretion via activating Kiss1R located on pancreatic beta-cells.
In the absence of Kiss1R signaling, locomotor activity is profoundly reduced accompanied by slight reduction in food intake and energy expenditure, leading to adiposity
Absence of Kiss1R signaling results in increased body weight and impaired glucose homeostasis in female but not male mice.
Outstanding questions.
Does peripherally produced kisspeptin regulate hypothalamic gonadotropin releasing hormone production, puberty onset or fertility?
Does centrally produced kisspeptin circulate and influence insulin secretion?
Does kisspeptin signaling in the central nervous system influence locomotor activity and satiety regulation and by which neuronal circuitry?
Does peripheral nutritional status via kisspeptin influence fertility regulation in the central nervous system. How does this mechanism interplay with leptin's regulation of fertility?
Which assay will most reliably allow measurement of circulating kisspeptin levels?
Are the observations made in experimental rodent models reflected in humans and is there a role for kisspeptin antagonist treatment for diabetes mellitus?
Which neuronal and peripheral Kiss1R-mediated actions modulate satiety and energy homeostasis. What is the basis for the sexual dimorphic effects of Kiss1R ablation on body weight control?
What is the metabolic role of kisspeptin that is produced in the placenta during gestation?
Acknowledgments
This work is supported by funding from the National Institutes of Health: DK101591, DK081472, DK079637
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee JH, Welch DR. Identification of highly expressed genes in metastasis-suppressed chromosome 6/human malignant melanoma hybrid cells using subtractive hybridization and differential display. Int J Cancer. 1997;71:1035–44. doi: 10.1002/(sici)1097-0215(19970611)71:6<1035::aid-ijc20>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 2.Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, Terao Y, Kumano S, Takatsu Y, Masuda Y, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001;411:613–7. doi: 10.1038/35079135. [DOI] [PubMed] [Google Scholar]
- 3.Bhattacharya M, Babwah AV. Kisspeptin: beyond the brain. Endocrinology. 2015;156:1218–27. doi: 10.1210/en.2014-1915. [DOI] [PubMed] [Google Scholar]
- 4.Cetkovic A, Miljic D, Ljubic A, Patterson M, Ghatei M, Stamenkovic J, Nikolic-Djurovic M, Pekic S, Doknic M, Glisic A, et al. Plasma kisspeptin levels in pregnancies with diabetes and hypertensive disease as a potential marker of placental dysfunction and adverse perinatal outcome. Endocr Res. 2012;37:78–88. doi: 10.3109/07435800.2011.639319. [DOI] [PubMed] [Google Scholar]
- 5.Kotani M, Detheux M, Vandenbogaerde A, Communi D, Vanderwinden JM, Le Poul E, Brezillon S, Tyldesley R, Suarez-Huerta N, Vandeput F, et al. The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J Biol Chem. 2001;276:34631–6. doi: 10.1074/jbc.M104847200. [DOI] [PubMed] [Google Scholar]
- 6.Kitahashi T, Ogawa S, Parhar IS. Cloning and expression of kiss2 in the zebrafish and medaka. Endocrinology. 2009;150:821–31. doi: 10.1210/en.2008-0940. [DOI] [PubMed] [Google Scholar]
- 7.Lee JH, Welch DR. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997;57:2384–7. [PubMed] [Google Scholar]
- 8.Muir AI, Chamberlain L, Elshourbagy NA, Michalovich D, Moore DJ, Calamari A, Szekeres PG, Sarau HM, Chambers JK, Murdock P, et al. AXOR12, a novel human G protein-coupled receptor, activated by the peptide KiSS-1. J Biol Chem. 2001;276:28969–75. doi: 10.1074/jbc.M102743200. [DOI] [PubMed] [Google Scholar]
- 9.Hauge-Evans AC, Richardson CC, Milne HM, Christie MR, Persaud SJ, Jones PM. A role for kisspeptin in islet function. Diabetologia. 2006;49:2131–5. doi: 10.1007/s00125-006-0343-z. [DOI] [PubMed] [Google Scholar]
- 10.Mead EJ, Maguire JJ, Kuc RE, Davenport AP. Kisspeptins are novel potent vasoconstrictors in humans, with a discrete localization of their receptor, G protein-coupled receptor 54, to atherosclerosis-prone vessels. Endocrinology. 2007;148:140–7. doi: 10.1210/en.2006-0818. [DOI] [PubMed] [Google Scholar]
- 11.Oakley AE, Clifton DK, Steiner RA. Kisspeptin signaling in the brain. Endocr Rev. 2009;30:713–43. doi: 10.1210/er.2009-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100:10972–6. doi: 10.1073/pnas.1834399100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seminara SB, Messager S, Chatzidaki EE, Thresher RR, Acierno JS, Jr, Shagoury JK, Bo-Abbas Y, Kuohung W, Schwinof KM, Hendrick AG, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–27. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 14.d'Anglemont de Tassigny X, Fagg LA, Dixon JP, Day K, Leitch HG, Hendrick AG, Zahn D, Franceschini I, Caraty A, Carlton MB, et al. Hypogonadotropic hypogonadism in mice lacking a functional Kiss1 gene. Proc Natl Acad Sci U S A. 2007;104:10714–9. doi: 10.1073/pnas.0704114104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silveira LG, Noel SD, Silveira-Neto AP, Abreu AP, Brito VN, Santos MG, Bianco SD, Kuohung W, Xu S, Gryngarten M, et al. Mutations of the KISS1 gene in disorders of puberty. J Clin Endocrinol Metab. 2010;95:2276–80. doi: 10.1210/jc.2009-2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teles MG, Bianco SD, Brito VN, Trarbach EB, Kuohung W, Xu S, Seminara SB, Mendonca BB, Kaiser UB, Latronico AC. A GPR54-activating mutation in a patient with central precocious puberty. N Engl J Med. 2008;358:709–15. doi: 10.1056/NEJMoa073443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanchez-Garrido MA, Tena-Sempere M. Metabolic control of puberty: roles of leptin and kisspeptins. Horm Behav. 2013;64:187–94. doi: 10.1016/j.yhbeh.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 18.Song WJ, Mondal P, Wolfe A, Alonso LC, Stamateris R, Ong BW, Lim OC, Yang KS, Radovick S, Novaira HJ, et al. Glucagon regulates hepatic kisspeptin to impair insulin secretion. Cell Metab. 2014;19:667–81. doi: 10.1016/j.cmet.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tolson KP, Garcia C, Yen S, Simonds S, Stefanidis A, Lawrence A, Smith JT, Kauffman AS. Impaired kisspeptin signaling decreases metabolism and promotes glucose intolerance and obesity. J Clin Invest. 2014;124:3075–9. doi: 10.1172/JCI71075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller WA, Faloona GR, Unger RH. Hyperglucagonemia in diabetic ketoacidosis. Its prevalence and significance. Am J Med. 1973;54:52–7. doi: 10.1016/0002-9343(73)90083-1. [DOI] [PubMed] [Google Scholar]
- 21.Dumonteil E, Magnan C, Ritz-Laser B, Meda P, Dussoix P, Gilbert M, Ktorza A, Philippe J. Insulin, but not glucose lowering corrects the hyperglucagonemia and increased proglucagon messenger ribonucleic acid levels observed in insulinopenic diabetes. Endocrinology. 1998;139:4540–6. doi: 10.1210/endo.139.11.6294. [DOI] [PubMed] [Google Scholar]
- 22.Kawamori D, Kurpad AJ, Hu J, Liew CW, Shih JL, Ford EL, Herrera PL, Polonsky KS, McGuinness OP, Kulkarni RN. Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab. 2009;9:350–61. doi: 10.1016/j.cmet.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.D'Alessio D. The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes Metab. 2011;13(Suppl 1):126–32. doi: 10.1111/j.1463-1326.2011.01449.x. [DOI] [PubMed] [Google Scholar]
- 24.Jamison RA, Stark R, Dong J, Yonemitsu S, Zhang D, Shulman GI, Kibbey RG. Hyperglucagonemia precedes a decline in insulin secretion and causes hyperglycemia in chronically glucose-infused rats. Am J Physiol Endocrinol Metab. 2011;301:E1174–83. doi: 10.1152/ajpendo.00175.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussain MA, Daniel PB, Habener JF. Glucagon stimulates expression of the inducible cAMP early repressor and suppresses insulin gene expression in pancreatic beta-cells. Diabetes. 2000;49:1681–90. doi: 10.2337/diabetes.49.10.1681. [DOI] [PubMed] [Google Scholar]
- 26.Dentin R, Liu Y, Koo SH, Hedrick S, Vargas T, Heredia J, Yates J, 3rd, Montminy M. Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature. 2007;449:366–9. doi: 10.1038/nature06128. [DOI] [PubMed] [Google Scholar]
- 27.Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–69. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 28.Brindle PK, Montminy MR. The CREB family of transcription activators. Curr Opin Genet De. 1992;2:199–204. doi: 10.1016/s0959-437x(05)80274-6. [DOI] [PubMed] [Google Scholar]
- 29.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–83. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 30.Montminy MR, Gonzalez GA, Yamamoto KK. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–8. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]
- 31.Niswender CM, Willis BS, Wallen A, Sweet IR, Jetton TL, Thompson BR, Wu C, Lange AJ, McKnight GS. Cre recombinase-dependent expression of a constitutively active mutant allele of the catalytic subunit of protein kinase. A Genesis. 2005;43:109–19. doi: 10.1002/gene.20159. [DOI] [PubMed] [Google Scholar]
- 32.Alonso LC, Yokoe T, Zhang P, Scott DK, Kim SK, O'Donnell CP, Garcia-Ocana A. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes. 2007;56:1792–801. doi: 10.2337/db06-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silvestre RA, Egido EM, Hernandez R, Marco J. Kisspeptin-13 inhibits insulin secretion without affecting glucagon or somatostatin release: study in the perfused rat pancreas. J Endocrinol. 2008;196:283–90. doi: 10.1677/JOE-07-0454. [DOI] [PubMed] [Google Scholar]
- 34.Vikman J, Ahren B. Inhibitory effect of kisspeptins on insulin secretion from isolated mouse islets. Diabetes Obes Metab. 2009;11(Suppl 4):197–201. doi: 10.1111/j.1463-1326.2009.01116.x. [DOI] [PubMed] [Google Scholar]
- 35.Tang G, Wang Y, Park S, Bajpayee NS, Vi D, Nagaoka Y, Birnbaumer L, Jiang M. Go2 G protein mediates galanin inhibitory effects on insulin release from pancreatic beta cells. Proc Natl Acad Sci U S A. 2012;109:2636–41. doi: 10.1073/pnas.1200100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu X, Herbison A. Kisspeptin regulation of arcuate neuron excitability in kisspeptin receptor knockout mice. Endocrinology. 2015;156:1815–27. doi: 10.1210/en.2014-1845. [DOI] [PubMed] [Google Scholar]
- 37.Akinci A, Cetin D, Ilhan N. Plasma kisspeptin levels in girls with premature thelarche. J Clin Res Pediatr Endocrinol. 2012;4:61–5. doi: 10.4274/jcrpe.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horikoshi Y, Matsumoto H, Takatsu Y, Ohtaki T, Kitada C, Usuki S, Fujino M. Dramatic elevation of plasma metastin concentrations in human pregnancy: metastin as a novel placenta-derived hormone in humans. J Clin Endocrinol Metab. 2003;88:914–9. doi: 10.1210/jc.2002-021235. [DOI] [PubMed] [Google Scholar]
- 39.Wahab F, Riaz T, Shahab M. Study on the effect of peripheral kisspeptin administration on basal and glucose-induced insulin secretion under fed and fasting conditions in the adult male rhesus monkey (Macaca mulatta) Horm Metab Re. 2011;43:37–42. doi: 10.1055/s-0030-1268458. [DOI] [PubMed] [Google Scholar]
- 40.Sarlund H, Siitonen O, Laakso M, Pyorala K. Repeatability of C-peptide response in glucagon stimulation test. Acta Endocrinol (Copenh) 1987:114, 515–8. doi: 10.1530/acta.0.1140515. [DOI] [PubMed] [Google Scholar]
- 41.Butera PC. Estradiol and the control of food intake. Physiol Behav. 2010;99:175–80. doi: 10.1016/j.physbeh.2009.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eckel LA. The ovarian hormone estradiol plays a crucial role in the control of food intake in females. Physiol Behav. 2011;104:517–24. doi: 10.1016/j.physbeh.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao Q, Horvath TL. Cross-talk between estrogen and leptin signaling in the hypothalamus. Am J Physiol Endocrinol Metab. 2008;294:E817–26. doi: 10.1152/ajpendo.00733.2007. [DOI] [PubMed] [Google Scholar]
- 44.Mauvais-Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev. 2013;34:309–38. doi: 10.1210/er.2012-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Calder M, Chan YM, Raj R, Pampillo M, Elbert A, Noonan M, Gillio-Meina C, Caligioni C, Berube NG, Bhattacharya M, et al. Implantation failure in female Kiss1-/- mice is independent of their hypogonadic state and can be partially rescued by leukemia inhibitory factor. Endocrinology. 2014;155:3065–78. doi: 10.1210/en.2013-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaytan F, Gaytan M, Castellano JM, Romero M, Roa J, Aparicio B, Garrido N, Sanchez-Criado JE, Millar RP, Pellicer A, et al. KiSS-1 in the mammalian ovary: distribution of kisspeptin in human and marmoset and alterations in KiSS-1 mRNA levels in a rat model of ovulatory dysfunction. Am J Physiol Endocrinol Metab. 2009;296:E520–31. doi: 10.1152/ajpendo.90895.2008. [DOI] [PubMed] [Google Scholar]
- 47.Hsu MC, Wang JY, Lee YJ, Jong DS, Tsui KH, Chiu CH. Kisspeptin modulates fertilization capacity of mouse spermatozoa. Reproduction. 2014;147:835–45. doi: 10.1530/REP-13-0368. [DOI] [PubMed] [Google Scholar]
- 48.Taylor J, Pampillo M, Bhattacharya M, Babwah AV. Kisspeptin/KISS1R signaling potentiates extravillous trophoblast adhesion to type-I collagen in a PKC- and ERK1/2-dependent manner. Mol Reprod Dev. 2014;81:42–54. doi: 10.1002/mrd.22279. [DOI] [PubMed] [Google Scholar]