

Abstract
In this article, we aimed to review the literature on the clinics and management of nonfunctional pancreatic neuroendocrine tumors (NPNET). Pancreatic neuroendocrine tumors (PNET) are rare tumors with a <1/100,000 incidence and constitute approximately 2 to 10% of all pancreatic tumors. Nonfunctional PNETs are difficult to detect at early stages since they have no symptoms. Except those detected accidentally during different diagnoses, the majority of PNETs are detected in the advanced stages, with symptoms related to tumor size or liver metastasis. We reviewed the studies published in the English medical literature through PubMed and summarized the clinical features and current approaches to the treatment and follow-up of the NPNET. The common imaging techniques used for the detection of tumor localization, size, locoregional, and metastatic involvement are contrasted computed tomography, magnetic resonance imaging, endoscopic ultrasonography, and somatostatin receptor scintigraphy. Surgical resection is the only curative treatment. However, in advanced locoregional disease and liver metastasis, interventive ablative therapies such as palliative reductive surgery, selective hepatic arterial embolization, radiofrequency ablation; and systemic therapies, such as peptide receptor radionuclide therapy, chemotherapy, somatostatin analogous therapy, interferon, VEGF inhibitor, and mTOR inhibitor may be used as symptom relieving or may improve progression-free survival and total survival. Current knowledge on NPNET shows that the treatment should be personalized considering the prognostic features and life expectancy of the patient.
Key words: Nonfunctional pancreatic neuroendocrine tumor, Surgical resection, Diagnosis criteria, Treatment options
Gastroenteropancreatic neuroendocrine tumors (GEP-NET) are produced as a result of the malignant transformation of the neuroendocrine cells that regulate the secretion and motility of the gastrointestinal tract. The incidence of GEP-NET was found to be 3.65/100.000, according to the 2000–2007 American SEER database, and approximately 7% of those were classified as (0.43/100,000) pancreatic neuroendocrine tumors (PNET).1 Approximately 2 to 10% of the pancreatic tumors consist of PNETs.2 Although it seems to be a rare tumor in clinics, a histologic PNET finding may be observed in 1 to 10% of the series in some autopsy studies.3 PNETs are commonly known as slow progressing tumors; however, their metastatic potential is high. In both population- and center-based studies, distant metastasis has been reported in 60 to 77% of the patients with PNET.4–6 The most frequent PNET-related metastasis is observed in the liver.
PNETs are classified as functional or nonfunctional according to their functions. Functional PNETs are labeled insulinoma, gastrinoma, or glucagonoma, according to the hormones they secrete, and may appear with symptoms related to excessive hormone secretion. Nonfunctional PNETs on the other hand, secrete some molecules such as chromogranin, neuron-specific enolase, pancreatic polypeptide, and ghrelin, but do not result in clinical manifestations related to hormone secretion. Since they show quiescent progression for a long time, they are detected in the advanced stages with symptoms related to the physical size of the tumor and metastasis. The main complaints observed in PNET are abdominal pain (35–78%), weight loss (20–35%), anorexia and nausea (45%), icterus (17–50%), intra-abdominal hemorrhage (4–20%), and mass observed in the examination (7–40%).7 Functional PNETS appear more frequently in previous studies, whereas recent studies include nonfunctional PNETS with rates approximating 90%.4,8 A retrospective trend study also shows a more than 2-fold increase in the incidence of nonfunctional PNETS compared to 16 years previous.9 Asymptomatic PNETS may have a role in this relative increase due to the advances in imaging techniques and related accidental detection of the tumors.
Hereditary Syndromes
Although PNETs are generally sporadic, they have been related to several hereditary syndromes, such as multiple endocrine neoplasia type I (MEN-1), von Hippel-Lindau (VHL), neurofibromatosis-1 (NF-1) and tuberous sclerosis. Concerning the MEN-1 patients, clinical findings are detected in 20 to 80% and microscopic PNET can be found in almost all patients.10 The majority of the MEN-1 related PNETs are of the nonfunctional type. PNETs are observed at a rate of 10 to 17% in von Hippel-Lindau (VHL) disease, and 98% of these are nonfunctional as well.10 Nonfunctional or functional PNETs may be observed although rare in NF-1 and tuberous sclerosis.
Tumor Classification and Grading Systems, and Prognosis
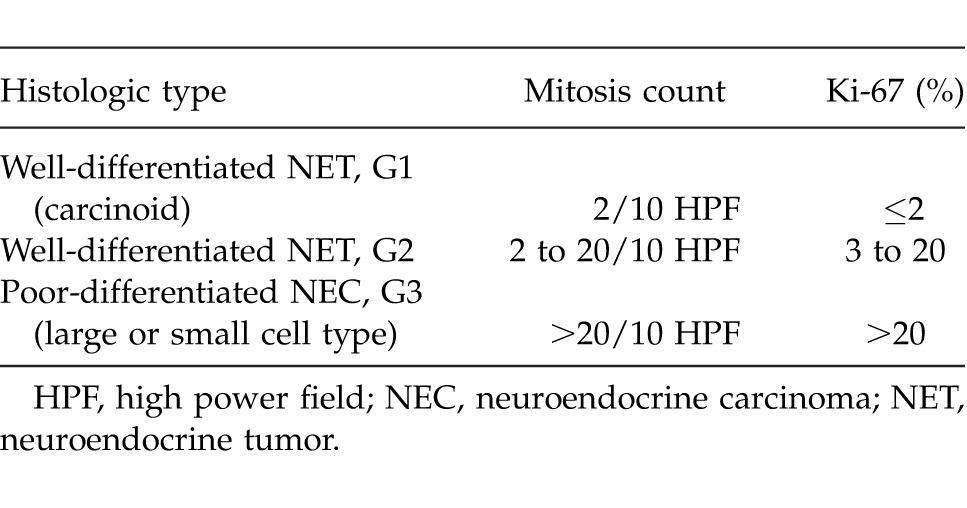
In terms of classification, it is irrelevant whether the PNETs are functional or nonfunctional. The well-differentiated neuroendocrine tumors and poorly-differentiated small or large cell carcinomas are separately evaluated in the 2010 gastrointestinal system tumor classification of WHO. Neuroendocrine tumors are graded as G1 (mitosis count, <2/10 HPF (high power field) and/or Ki-67, ≤2%) and G2 (mitosis count, 2–20/10 HPF and/or Ki-67, 3–20%) according to the mitosis count or Ki-67 proliferative index. All neuroendocrine carcinomas are graded as G3 (mitosis count, >20 and/or Ki-67 >20%; Table 1).11
Table 1.
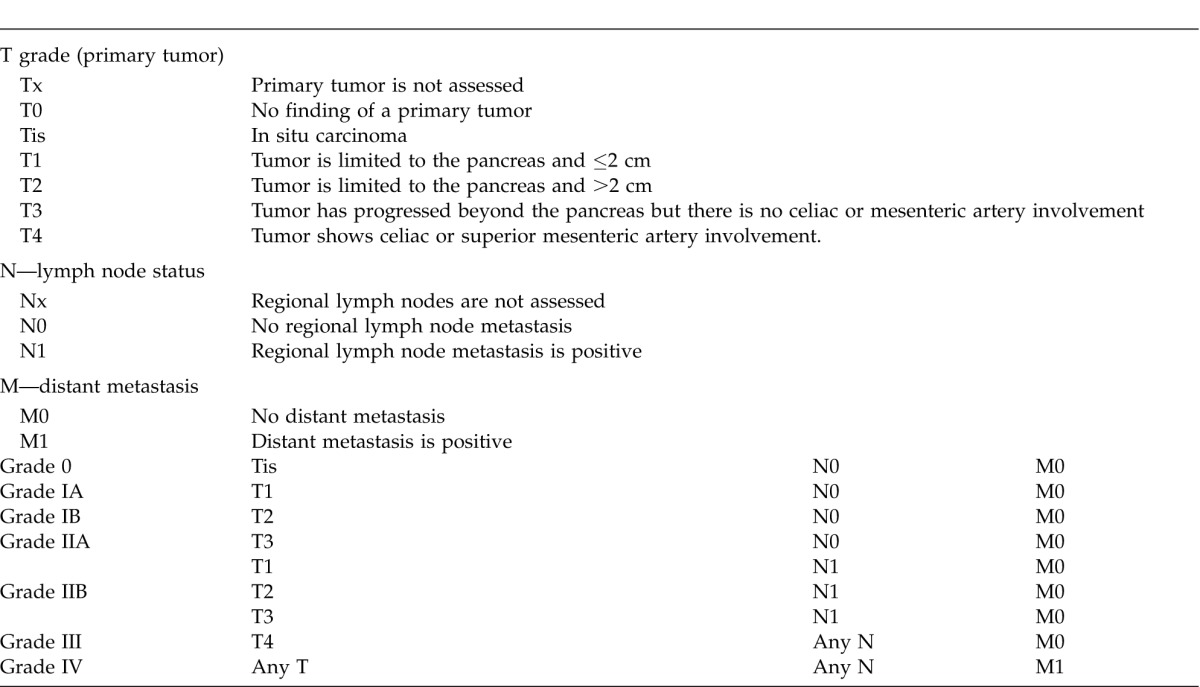
However, the European Neuroendocrine Tumor Society (ENETS)12 and American Joint Committee on Cancer (AJCC)13 have also suggested TNM-based PNET classification and grading systems in relation to prognosis (Table 2 and 3). Although ENETS and AJCC TNM classification systems are different in terms of some aspects, it has been proven in retrospective studies that both gave valid postulations for survival rates according to the grading systems.14,15 When the 2 TNM classification systems are compared regarding their prognoses, the 5-year total survival was found to be 100%, 88%, 85%, and 57% with ENETS and 92%, 84%, 81%, and 57% with AJCC for grades 1, 2, 3, and 4, respectively.15 Liver/bone metastasis is the most common factor affecting prognosis. Other factors may be counted as lymph node involvement, invasion depth, presence of certain histologic markers, necrosis, high serum alkaline phosphatase level, advanced age, and WHO and TNM scoring systems.16
Table 2.
ENETS TNM grading system for pancreatic tumors12

Table 3.
AJCC TNM grading system for pancreatic tumors13
Diagnostic Methods
Nonfunctional PNETs are generally large in size and have a heterogeneous structure. Irregular calcification, necrosis, and cystic changes may be observed. Local and vascular invasion and distant metastasis are commonly observed conditions. Confirmation of PNET necessitates Chromogranin A (CgA) and synaptophysin positivity in immunohistochemical staining in the histopathologic examination.7 Diagnosis is made upon the presence of the primary tumor and various imaging techniques for the evaluation of nodal or metastatic situations. Routine sectional imaging techniques such as computed tomography (CT) or magnetic resonance imaging (MRI), endoscopic ultrasonography (EUS), and somatostatin receptor scintigraphy (SRS) are the techniques that are routinely used in the diagnosis of the disease.
CT and MRI
Since PNETs have a hypervascular structure, the best imaging is obtained via contrast agent administration. The sensitivity of helical multiphasic contrasted CT is over 80%, and even 100% sensitivity has been reported for tumors larger than 3 cm.16,17 A typical pancreatic CT scan consists of the noncontrasted phase, contrasted arterial phase, and portal venous phase.18
The advantageous aspects of MRI may include nonexposure to ionized radiation and excellent soft tissue contrast resonance. Diagnostic PNET studies via MRI report 80% sensitivity and 100% specificity.19 The PNETs are visualized as hypervascular lesions of low T1 signal intensity or moderate T2 signal intensity in the MRI; larger necrotic tumors may give heterogeneous signal distribution.18 MRI has been reported to be more effective in detecting liver metastasis compared to CT or SRS.20
Endoscopic ultrasonography (EUS)
EUS is another method for the diagnosis of pancreatic lesions, especially for the lesions smaller than 1 cm. The sensitivity of EUS is reported to be 79 to 100%.21 Furthermore, EUS plays an important role in the early detection and follow-up of multifocal PNTs that are common among patients with MEN-1 and VHL syndromes.22
EUS-guided thin needle biopsy provides the preoperative histopathologic examination and plays an important role in the discrimination of guided nonfunctional PNETs from pancreatic adenocarcinomas and other pancreatic lesions.23
Somatostatin receptor scintigraphy (SRS)—Octreoscan
Most of the PNETs include somatostatin receptors on their cell membranes. Therefore, it is possible to visualize the PNET cells via radioactive-labeled somatostatin analogues. Indium 111 labeled DTPA-octreotide (a synthetic somatostatin analogue) is a routinely used agent in SRS; SRS is therefore known as octreoscan. SRS is highly effective in diagnosing nonfunctional PNETs, gastrinonomas and glucagonomas; however, it cannot effectively detect insulinoma or poorly-differentiated tumors since they have a weak somatostatin receptor expression.24 The advantage of SRS is that it can scan not only the abdominal region, but the whole body rapidly, with regard to the presence of a metastasis. In patients with suspicious extra hepatic metastasis, SRS may both confirm the diagnosis and affect the treatment plan.25 The accuracy rate of SRS has been increased by the usage of SPECT (single photon emission computed tomography).26
Biochemical marker scanning
Although no biomarker has been defined specific to PNETs, increase in the plasma levels of various nonspecific biomarkers may indicate the presence of a neuroendocrine tumor. The most facilitating marker among these is plasma CgA. The sensitivity and specificity of CgA in GEP-NETs are approximately 60% and 80%, respectively.27 Furthermore, increase in the plasma levels of CgA has been related to a progression in the disease and metastasis, and thus it may be used in the follow-up of the disease.27,28 In addition to CgA, markers such as pancreatic polypeptide (pp), plasma neuron-specific enolase, pancreastatin, and human chorionic gonadotropin α and β may be used in the diagnosis of GEP-NET. However, their sensitivities are less. In nonfunctional PNETs, concomitant usage of plasma CgA and pp markers may significantly increase the sensitivity of the test. In a study, the sensitivity of plasma CgA alone was shown to be 68% and the combination of CgA and pp has resulted in 93% sensitivity in the detection of nonfunctional PNETs.29
Surgical Treatment
The treatment of PNETs are planned according to several factors, such as the grade of the tumor, whether it is well or poorly differentiated, severity of the symptoms and the presence of a hereditary syndrome. The main curative treatment is surgical resection. In advanced locoregional disease and liver metastasis, palliative reductive surgery, other interventive therapies, and medical therapies may be used.
Primary tumor resection
Surgical resection is recommended for both the sporadic and MEN-1 related nonfunctional PNETs where the tumor size is greater than 2 cm, because the risk of locoregional and distant metastasis is increased.30,31 In a retrospective study based on the American SEER data base, the survival rates were demonstrated to increase significantly via aggressive treatment.32 In this study, tumor resection was recommended for 425 patients and performed on 310. Among those, the survival was 114 months, and among the remaining 115 with no resection, the survival was 35 months (P < 0.001). In the same study, the odds ratio of mortality was calculated to be 0.48 in the resection group compared to the nonresection group.32 However, surgical resection is controversial in patients with a tumor size smaller than 2 cm, having a benign appearance and showing slow progression. There are even studies suggesting aggressive treatment in nonfunctional and accidentally detected PNETs smaller than 2 cm due to the unknown metastatic potential.33 In some other studies, follow-up via EUS or MRI are recommended in such cases since resection-free survival is generally high.34,35 The risk/benefit ratio of surgical intervention, particularly in multifocal MEN-1 related PNETs with a known nature, should be carefully considered, because the risk of glucose intolerance and diabetes may be high in patients with MEN-1 and undertaken distal pancreatectomy.36
The surgical approach may be performed via organ protective enucleation or central pancreatectomy, or as open or laparoscopic techniques in patients with small and benign type tumors.31 The advantage of enucleation and central pancreatectomy is that they do not result in pancreatic failure in the long term. The disadvantages of the method are negative surgical border ambiguity and the absence of routine lymphadenectomy. Lymph node sampling may be necessary for patients with uncertain clinical or pathologic findings for potential malignancy.36
In cases where locoregional disease or with a tumor size larger than 2 cm are suspected, a distal pancreatectomy may be performed if the tumor is localized in the caudal region (with or without splenectomy); or pancreatic duodenectomy (Whipple's procedure) may be performed if the tumor is localized in the caput/corpus region, or lymph node dissection may be performed.31,36
Surgery in liver metastasis
The first treatment option in liver metastasis is surgical resection. However, the complete resection of the liver metastasis is usually not possible and the risk of recurrence is generally high. It may be performed when the hepatic involvement is less than 50% and at least 90% of the tumoral tissue can be resected, or in the palliative treatment of symptomatic diseases.31,36 It has been observed that cases with proper criteria curative or palliative surgical resection increased survival, despite high recurrence rates. In a retrospective series including 72 patients with liver invasive PNET, the morbidity and mortality rates following surgery were found to be 50% and 0% respectively, and 1- and 5-year survival rates were found to be 97.1% and 59.9%.37 Surgical resections of the primary pancreatic tumor and liver metastasis may be performed concomitantly or in consequent operations. However, in a double-center, retrospective study, the major complication (liver abscess) was more frequent in 2-step pancreatic duodenectomy and liver metastasis resections than concomitant resection (14.5% versus 7%, P < 0.05).38 In another recent study on patients with primary GEP-NET and bilobar liver metastasis, 2-step surgery was performed where the first step was the resection of the primary tumor and the involvement in 1 lobe. A period of 8 weeks was then allowed for the hypertrophy of the liver, after which the other lobe was resected.39 It was suggested that a radical surgical treatment may be performed in patients with high grade of liver involvement via this method, with acceptable morbidity rates.40
Liver transplants
Liver transplantations have been performed on some rare liver metastasis patients as a treatment option. However, this approach is not quite acceptable when the donor insufficiency and rapid recurrence rates are considered. It may be a last option in patients over 50 years of age, patients with a proven absence of extrahepatic metastasis via comprehensive whole body imaging techniques, and in patients with positive histologic properties (well-differentiated, Ki-67 < 5%) who are not candidates for resection and who have not responded to other therapies.40
Other Interventive Locoregional Treatments
Selective hepatic arterial embolization
Metastatic liver lesions are hypervascular and are fed by the hepatic artery. The hepatic tissue on the other hand is fed primarily by the portal vein. Hepatic arterial embolization aims to direct the tumoral lesions to ischemia without deteriorating the liver parenchyma. This method is used in liver metastases where surgical resection is not possible due to its diffuse and multifocal nature. The indicators of good prognosis are previous primary pancreatic tumor resection, less than 75% involvement of the liver, <5 cm tumor size and absence of extrahepatic metastasis.41 Embolization may be performed via gel foam powder infusion (single embolization), microspheres that secrete chemotherapeutic agents (chemoembolization), or microspheres that secrete radioactive isotope (radioembolization). There is no sufficient data demonstrating the superiority of any of these methods to one another.16 In a review published in 2009, the 5-year survival rates were 40 to 67% and 50 to 65% in single embolization and chemoembolization, respectively.42 In a series including 148 patients who were treated with radioembolization (Yttrium-90 resin microspheres) the median survival time was 70 months, the rate of complete response was 2.75, the rate of partial response was 60.5%, and the progression-free diseases was reported to be 22.7%.43
Radiofrequency ablation (RFA)
RFA is used as a symptom-relieving or supportive treatment in nonresectable liver metastases. It may be performed via the relieving of small lesions during open or laparoscopic liver tumor resections or via the percutaneous route alone. The ablation of the tumoral tissue is provided by the application of inserting high temperatures into the tumor via an RFA needle in the guidance of ultrasound. In a series including 89 patients with neuroendocrine liver metastasis, the symptoms were relieved in 97% of the patients via laparoscopic RFA. Although the recent hepatic lesion development and extrahepatic metastasis was about 60%, the disease-free survival was 1.6 years and total survival was 6 years.44
Peptide Receptor Radionuclide Therapy (PRRT)
PRRT is based on the systemic transmission of radioisotope labeled synthetic somatostatin analogue (Lutetium-177 or Yttrium-90) to the metastatic cells within the whole body.45 The number of somatostatin receptors should be high in the tumor cells in order to be able to use this method. Although it is not counted in the primary therapies, PRRT is recommended when the systemic medical therapy is unsuccessful in the diffuse diseases with extrahepatic metastasis.36 In a cohort study where Yttrium-90 DOTATOC and consequent Yttrium-90 DOTATOC and Lutetium-177 DOTATOC treatments were performed on 2 separate groups of 486 patients, consequent radioisotope usage was shown to provide a longer survival rate (5.51 versus 3.96 years).46
Medical Treatment
Somatostatin analogue therapy
Long-term somatostatin analogue therapy is an antiproliferative therapy used in patients with a slow progressive PNET and a low rate of liver involvement (less than 50%) in order to stop/slow down the progression of the disease. In a randomized, double-blind, placebo controlled study on patients with GEP-NET, the somatostatin analogue octreotide-LAR delayed the tumor progression compared to the placebo (14.3 months versus 6 months, hazard ratio = 0.34), and following 6 months of therapy, progression-free disease was observed in 66.7% of the therapy group compared to 37.2% of the placebo group.47 Since the somatostatin analogue is well tolerated, it may be primarily used primarily; however, chemotherapy is recommended for nonfunctional PNET patients with well-differentiated tumors and a Ki-67 index of >15%.48
Interferon
Similarly, interferon therapy is used to stop or stabilize the progression of the disease and may be used in metastatic nonfunctional PNETs. However, frequent side effects limit the wide and long-term usage of this therapy.31 In a randomized study performed on 80 patients with metastatic GEP-NET, the somatostatin analogue lanreotide, interferon-α or the combination was applied to the patients, and the stable disease was found to be between 18 to 28% in all groups.48
Chemotherapy
Cytotoxic chemotherapy may be used in PNETs with diffuse liver metastasis, in patients with rapid local progression, in poorly-differentiated patients, in well-differentiated but high proliferative Ki-67 index patients, or in patients not responding to the somatostatin analogue therapy.31 In a retrospective examination of 84 patients with advanced-stage PNET, the response to the triple therapy of streptozocin, fluorouracil, and doxorubicin was 39%; progression-free survival was 18 months, and general survival was 37 months.49 In another phase II study on advanced grade PNET patients with dacarbazine, the objective response rate was found to be 34%.50
A recent ENETS consensus guideline has summarized the chemotherapeutic approach in GEP-NETs with hepatic and distant metastases.51
VEGF inhibitor
VEGF is a growth factor that plays a role in angiogenesis. The progression-free survival was found to be improved in advanced-stage, well-differentiated PNETs with VEGF inhibitor sunitinib (11.5 months versus 5.5 months); however, the clinical study was ceased due to serious adverse events and death observed in the placebo arm.52 Since there is no sufficient data on issues such as long-term resistance or side effects yet, it is not currently recommended as a first-step therapy.51
MTOR inhibitor
mTOR (mammalian target of rapamycin) is a serine/treonine protein kinase molecule that has a role in the tyrosine kinase pathway. The mTOR inhibitor everolimus was compared to a placebo in a phase III study including patients with advanced, low and moderate stage PNET. In this study, where the best possible supportive treatment was performed on both groups, everolimus provided a significantly improved survival rate (11 months versus 4.6 months) and was well tolerated.53 However, since everolimus is a recent agent like sunitinib, it is not recommended as a first-step therapy. There is the suggestion that it be used as a second- or third-step therapy if no response to other therapies is observed.51
Conclusion
Gastroenteropancreatic neuroendocrine tumors are rare diseases and there is quite a controversy concerning the diagnosis of the disease. Although we tried to review the literature to have a wide understanding about the disease, more data are needed to have a better prognosis with treatment modalities.
References
- 1.Lawrence B, Gustafsson BI, Chan A, Svejda B, Kidd M, Modlin IM. The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40(1):1–18. doi: 10.1016/j.ecl.2010.12.005. vii. [DOI] [PubMed] [Google Scholar]
- 2.Mignon M. Natural history of neuroendocrine enteropancreatic tumors. Digestion. 2000;62(Suppl 1):51–58. doi: 10.1159/000051856. [DOI] [PubMed] [Google Scholar]
- 3.Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci. 1991;36(7):933–942. doi: 10.1007/BF01297144. [DOI] [PubMed] [Google Scholar]
- 4.Halfdanarson TR, Rabe KG, Rubin J, Petersen GM. Pancreatic neuroendocrine tumors (PNETs): incidence, prognosis and recent trend toward improved survival. Ann Oncol. 2008;19(10):1727–1733. doi: 10.1093/annonc/mdn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pape UF, Berndt U, Muller-Nordhorn J, Bohmig M, Roll S, Koch M. Prognostic factors of long-term outcome in gastroenteropancreatic neuroendocrine tumours. Endocr Relat Cancer. 2008;15(4):1083–1097. doi: 10.1677/ERC-08-0017. [DOI] [PubMed] [Google Scholar]
- 6.Soga J. Carcinoids of the pancreas: an analysis of 156 cases. Cancer. 2005;104(6):1180–1187. doi: 10.1002/cncr.21291. [DOI] [PubMed] [Google Scholar]
- 7.Falconi M, Plockinger U, Kwekkeboom DJ, Manfredi R, Korner M, Kvols L. Well-differentiated pancreatic nonfunctioning tumors/carcinoma. Neuroendocrinology. 2006;84(3):196–211. doi: 10.1159/000098012. [DOI] [PubMed] [Google Scholar]
- 8.Bilimoria KY, Tomlinson JS, Merkow RP, Stewart AK, Ko CY, Talamonti MS. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: analysis of 9,821 patients. J Gastrointest Surg. 2007;11(11):1460–1467. doi: 10.1007/s11605-007-0263-3. discussion 1467–1469. [DOI] [PubMed] [Google Scholar]
- 9.Fitzgerald TL, Hickner ZJ, Schmitz M, Kort EJ. Changing incidence of pancreatic neoplasms: a 16-year review of statewide tumor registry. Pancreas. 2008;37(2):134–138. doi: 10.1097/MPA.0b013e318163a329. [DOI] [PubMed] [Google Scholar]
- 10.Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. 2008;113(7 Suppl):1807–1843. doi: 10.1002/cncr.23648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bosman FT. WHO Classification of Tumor of the Digestive System. Lyon, France: IARC Press; 2010. [Google Scholar]
- 12.Rindi G, Kloppel G, Alhman H, Caplin M, Couvelard A, de Herder WW. TNM staging of foregut (neuro)endocrine tumors: a consensus proposal including a grading system. Virchows Arch. 2006;449(4):395–401. doi: 10.1007/s00428-006-0250-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A. AJCC Cancer Staging Manual (7th ed) New York: Springer; 2010. [Google Scholar]
- 14.Pape UF, Jann H, Muller-Nordhorn J, Bockelbrink A, Berndt U, Willich SN. Prognostic relevance of a novel TNM classification system for upper gastroenteropancreatic neuroendocrine tumors. Cancer. 2008;113(2):256–265. doi: 10.1002/cncr.23549. [DOI] [PubMed] [Google Scholar]
- 15.Strosberg JR, Cheema A, Weber J, Han G, Coppola D, Kvols LK. Prognostic validity of a novel American Joint Committee on Cancer Staging Classification for pancreatic neuroendocrine tumors. J Clin Oncol. 2011;29(22):3044–3049. doi: 10.1200/JCO.2011.35.1817. [DOI] [PubMed] [Google Scholar]
- 16.Muniraj T, Vignesh S, Shetty S, Thiruvengadam S, Aslanian HR. Pancreatic neuroendocrine tumors. Dis Mon. 2013;59(1):5–19. doi: 10.1016/j.disamonth.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 17.Legmann P, Vignaux O, Dousset B, Baraza AJ, Palazzo L, Dumontier I. Pancreatic tumors: comparison of dual-phase helical CT and endoscopic sonography. AJR Am J Roentgenol. 1998;170(5):1315–1322. doi: 10.2214/ajr.170.5.9574609. [DOI] [PubMed] [Google Scholar]
- 18.Heller MT, Shah AB. Imaging of neuroendocrine tumors. Radiol Clin North Am. 2011;49(3):529–548. doi: 10.1016/j.rcl.2011.02.011. vii. [DOI] [PubMed] [Google Scholar]
- 19.Thoeni RF, Mueller-Lisse UG, Chan R, Do NK, Shyn PB. Detection of small, functional islet cell tumors in the pancreas: selection of MR imaging sequences for optimal sensitivity. Radiology. 2000;214(2):483–490. doi: 10.1148/radiology.214.2.r00fe32483. [DOI] [PubMed] [Google Scholar]
- 20.Dromain C, de Baere T, Lumbroso J, Caillet H, Laplanche A, Boige V. Detection of liver metastases from endocrine tumors: a prospective comparison of somatostatin receptor scintigraphy, computed tomography, and magnetic resonance imaging. J Clin Oncol. 2005;23(1):70–78. doi: 10.1200/JCO.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 21.Rockall AG, Reznek RH. Imaging of neuroendocrine tumours (CT/MR/US) Best Pract Res Clin Endocrinol Metab. 2007;21(1):43–68. doi: 10.1016/j.beem.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Langer P, Kann PH, Fendrich V, Richter G, Diehl S, Rothmund M. Prospective evaluation of imaging procedures for the detection of pancreaticoduodenal endocrine tumors in patients with multiple endocrine neoplasia type 1. World J Surg. 2004;28(12):1317–1322. doi: 10.1007/s00268-004-7642-7. [DOI] [PubMed] [Google Scholar]
- 23.Patel KK, Kim MK. Neuroendocrine tumors of the pancreas: endoscopic diagnosis. Curr Opin Gastroenterol. 2008;24(5):638–642. doi: 10.1097/MOG.0b013e32830bf7fb. [DOI] [PubMed] [Google Scholar]
- 24.de Herder WW, Kwekkeboom DJ, Feelders RA, van Aken MO, Lamberts SW, van der Lely AJ. Somatostatin receptor imaging for neuroendocrine tumors. Pituitary. 2006;9(3):243–248. doi: 10.1007/s11102-006-0270-5. [DOI] [PubMed] [Google Scholar]
- 25.Lebtahi R, Cadiot G, Sarda L, Daou D, Faraggi M, Petegnief Y. Clinical impact of somatostatin receptor scintigraphy in the management of patients with neuroendocrine gastroenteropancreatic tumors. J Nucl Med. 1997;38(6):853–858. [PubMed] [Google Scholar]
- 26.Schillaci O, Corleto VD, Annibale B, Scopinaro F, Delle Fave G. Single photon emission computed tomography procedure improves accuracy of somatostatin receptor scintigraphy in gastro-entero pancreatic tumours. Ital J Gastroenterol Hepatol. 1999;31(Suppl 2):S186–S189. [PubMed] [Google Scholar]
- 27.Lawrence B, Gustafsson BI, Kidd M, Pavel M, Svejda B, Modlin IM. The clinical relevance of chromogranin A as a biomarker for gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am. 2011;40(1):111–134. doi: 10.1016/j.ecl.2010.12.001. viii. [DOI] [PubMed] [Google Scholar]
- 28.Bajetta E, Ferrari L, Martinetti A, Celio L, Procopio G, Artale S. Chromogranin A, neuron specific enolase, carcinoembryonic antigen, and hydroxyindole acetic acid evaluation in patients with neuroendocrine tumors. Cancer. 1999;86(5):858–865. doi: 10.1002/(sici)1097-0142(19990901)86:5<858::aid-cncr23>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 29.Panzuto F, Severi C, Cannizzaro R, Falconi M, Angeletti S, Pasquali A. Utility of combined use of plasma levels of chromogranin A and pancreatic polypeptide in the diagnosis of gastrointestinal and pancreatic endocrine tumors. J Endocrinol Invest. 2004;27(1):6–11. doi: 10.1007/BF03350903. [DOI] [PubMed] [Google Scholar]
- 30.Hellman P, Andersson M, Rastad J, Juhlin C, Karacagil S, Eriksson B. Surgical strategy for large or malignant endocrine pancreatic tumors. World J Surg. 2000;24(11):1353–1360. doi: 10.1007/s002680010224. [DOI] [PubMed] [Google Scholar]
- 31.Minter RM, Simeone DM. Contemporary management of nonfunctioning pancreatic neuroendocrine tumors. J Gastrointest Surg. 2012;16(2):435–446. doi: 10.1007/s11605-011-1693-5. [DOI] [PubMed] [Google Scholar]
- 32.Hill JS, McPhee JT, McDade TP, Zhou Z, Sullivan ME, Whalen GF. Pancreatic neuroendocrine tumors: the impact of surgical resection on survival. Cancer. 2009;115(4):741–751. doi: 10.1002/cncr.24065. [DOI] [PubMed] [Google Scholar]
- 33.Haynes AB, Deshpande V, Ingkakul T, Vagefi PA, Szymonifka J, Thayer SP. Implications of incidentally discovered, nonfunctioning pancreatic endocrine tumors: short-term and long-term patient outcomes. Arch Surg. 2011;146(5):534–538. doi: 10.1001/archsurg.2011.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee LC, Grant CS, Salomao DR, Fletcher JG, Takahashi N, Fidler JL. Small, nonfunctioning, asymptomatic pancreatic neuroendocrine tumors (PNETs): role for nonoperative management. Surgery. 2012;152(6):965–974. doi: 10.1016/j.surg.2012.08.038. [DOI] [PubMed] [Google Scholar]
- 35.Bettini R, Partelli S, Boninsegna L, Capelli P, Crippa S, Pederzoli P. Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery. 2011;150(1):75–82. doi: 10.1016/j.surg.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 36.Falconi M, Bartsch DK, Eriksson B, Kloppel G, Lopes JM, O'Connor JM. ENETS Consensus Guidelines for the management of patients with digestive neuroendocrine neoplasms of the digestive system: well-differentiated pancreatic non-functioning tumors. Neuroendocrinology. 2012;95(2):120–134. doi: 10.1159/000335587. [DOI] [PubMed] [Google Scholar]
- 37.Cusati D, Zhang L, Harmsen WS, Hu A, Farnell MB, Nagorney DM. Metastatic nonfunctioning pancreatic neuroendocrine carcinoma to liver: surgical treatment and outcomes. J Am Coll Surg. 2012;215(1):117–124. doi: 10.1016/j.jamcollsurg.2012.05.002. discussion 124–125. [DOI] [PubMed] [Google Scholar]
- 38.De Jong MC, Farnell MB, Sclabas G, Cunningham SC, Cameron JL, Geschwind JF. Liver-directed therapy for hepatic metastases in patients undergoing pancreaticoduodenectomy: a dual-center analysis. Ann Surg. 2010;252(1):142–148. doi: 10.1097/SLA.0b013e3181dbb7a7. [DOI] [PubMed] [Google Scholar]
- 39.Kianmanesh R, Sauvanet A, Hentic O, Couvelard A, Levy P, Vilgrain V. Two-step surgery for synchronous bilobar liver metastases from digestive endocrine tumors: a safe approach for radical resection. Ann Surg. 2008;247(4):659–665. doi: 10.1097/SLA.0b013e31816a7061. [DOI] [PubMed] [Google Scholar]
- 40.Pascher A, Klupp J, Neuhaus P. Endocrine tumours of the gastrointestinal tract. Transplantation in the management of metastatic endocrine tumours. Best Pract Res Clin Gastroenterol. 2005;19(4):637–648. doi: 10.1016/j.bpg.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 41.Yao KA, Talamonti MS, Nemcek A, Angelos P, Chrisman H, Skarda J. Indications and results of liver resection and hepatic chemoembolization for metastatic gastrointestinal neuroendocrine tumors. Surgery. 2001;130(4):677–682. doi: 10.1067/msy.2001.117377. discussion 682–685. [DOI] [PubMed] [Google Scholar]
- 42.Vogl TJ, Naguib NN, Zangos S, Eichler K, Hedayati A, Nour-Eldin NE. Liver metastases of neuroendocrine carcinomas: interventional treatment via transarterial embolization, chemoembolization and thermal ablation. Eur J Radiol. 2009;72(3):517–528. doi: 10.1016/j.ejrad.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Kennedy AS, Dezarn WA, McNeillie P, Coldwell D, Nutting C, Carter D. Radioembolization for unresectable neuroendocrine hepatic metastases using resin 90Y-microspheres: early results in 148 patients. Am J Clin Oncol. 2008;31(3):271–279. doi: 10.1097/COC.0b013e31815e4557. [DOI] [PubMed] [Google Scholar]
- 44.Akyildiz HY, Mitchell J, Milas M, Siperstein A, Berber E. Laparoscopic radiofrequency thermal ablation of neuroendocrine hepatic metastases: long-term follow-up. Surgery. 2010;148(6):1288–1293. doi: 10.1016/j.surg.2010.09.014. discussion 1293. [DOI] [PubMed] [Google Scholar]
- 45.Kwekkeboom DJ, Kam BL, van Essen M, Teunissen JJ, van Eijck CH, Valkema R. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr Relat Cancer. 2010;17(1):R53–R73. doi: 10.1677/ERC-09-0078. [DOI] [PubMed] [Google Scholar]
- 46.Villard L, Romer A, Marincek N, Brunner P, Koller MT, Schindler C. Cohort study of somatostatin-based radiopeptide therapy with [(90)Y-DOTA]-TOC versus [(90)Y-DOTA]-TOC plus [(177)Lu-DOTA]-TOC in neuroendocrine cancers. J Clin Oncol. 2012;30(10):1100–1106. doi: 10.1200/JCO.2011.37.2151. [DOI] [PubMed] [Google Scholar]
- 47.Rinke A, Muller HH, Schade-Brittinger C, Klose KJ, Barth P, Wied M. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: a report from the PROMID Study Group. J Clin Oncol. 2009;27(28):4656–4663. doi: 10.1200/JCO.2009.22.8510. [DOI] [PubMed] [Google Scholar]
- 48.Faiss S, Pape UF, Bohmig M, Dorffel Y, Mansmann U, Golder W. Prospective, randomized, multicenter trial on the antiproliferative effect of lanreotide, interferon alfa, and their combination for therapy of metastatic neuroendocrine gastroenteropancreatic tumors–the International Lanreotide and Interferon Alfa Study Group. J Clin Oncol. 2003;21(14):2689–2696. doi: 10.1200/JCO.2003.12.142. [DOI] [PubMed] [Google Scholar]
- 49.Kouvaraki MA, Ajani JA, Hoff P, Wolff R, Evans DB, Lozano R. Fluorouracil, doxorubicin, and streptozocin in the treatment of patients with locally advanced and metastatic pancreatic endocrine carcinomas. J Clin Oncol. 2004;22(23):4762–4771. doi: 10.1200/JCO.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 50.Ramanathan RK, Cnaan A, Hahn RG, Carbone PP, Haller DG. Phase II trial of dacarbazine (DTIC) in advanced pancreatic islet cell carcinoma. Study of the Eastern Cooperative Oncology Group-E6282. Ann Oncol. 2001;12(8):1139–1143. doi: 10.1023/a:1011632713360. [DOI] [PubMed] [Google Scholar]
- 51.Pavel M, Baudin E, Couvelard A, Krenning E, Oberg K, Steinmuller T. ENETS Consensus Guidelines for the management of patients with liver and other distant metastases from neuroendocrine neoplasms of foregut, midgut, hindgut, and unknown primary. Neuroendocrinology. 2012;95(2):157–176. doi: 10.1159/000335597. [DOI] [PubMed] [Google Scholar]
- 52.Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 53.Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]