

Abstract
While telomerase (hTERT) activity is absent from normal somatic cells, reactivation of hTERT expression is a hallmark of cancer cells. Telomerase activity is required for avoiding replicative senescence and supports immortalization of cellular proliferation. Only a minority of cancer cells rely on a telomerase-independent process known as alternative lengthening of telomeres, ALT, to sustain cancer cell proliferation. Multiple genetic, epigenetic, and viral mechanisms have been found to de-regulate telomerase gene expression, thereby increasing the risk of cellular transformation. Here, we review the different strategies used by the Human T-cell leukemia virus type 1, HTLV-I, to activate hTERT expression and stimulate its enzymatic activity in virally infected CD4 T cells. The implications of hTERT reactivation in HTLV-I pathogenesis and disease treatment are discussed.
Keywords: Telomerase, telomere; hTERT; Shelterin; HTLV-I; ATL; Tax; HBZ; NF-κB
Introduction
HTLV-I pathogenesis
Infection with the human retrovirus HTLV-I is associated with the development of an aggressive form of T-cell leukemia known as adult T-cell leukemia/lymphoma (ATLL)[1–3]. The diagnosis of ATLL is usually made on cytological examination of “cerebriform” or “flower cells” (activated lymphocytes with convoluted nuclei and basophilic cytoplasm) and the clonal integration of HTLV-I provirus in the host genome. The virus induces a lifelong infection, and while most HTLV-I-infected individuals remain asymptomatic carriers, 1 to 5% of infected patients have a lifelong risk of developing leukemia. ATLL has a broad clinical spectrum and has been divided into four clinically distinct entities (acute, chronic, smoldering, and lymphoma) that differ in their presentation, disease progression, and response to treatment[4]. While there are no karyotypic abnormalities specifically associated with the development of ATLL, cytogenetic analyses of leukemic cells from HTLV-I-infected patients revealed higher frequency for trisomy 3, 7 and 21 involvement of chromosomes 6 and 14 and loss of chromosome Y[5–7]. Loss of heterozygosity on chromosome 6q has been described in half of acute/lymphoma ATLL[8]. The tumor suppressor genes p53, RB, and p16INK are also frequently inactivated in ATLL cells. Treatment of ATLL remains disappointing, with marginal improvements in the overall survival of patients and a projected 4-year survival rate at 5%[4]. There are several factors associated with the poor prognosis of ATLL, including high serum thymidine kinase levels[9], high serum soluble interleukin-2 receptor (IL-2R) levels[10], and high serum β2 microglobulin levels[11]. The mechanism used by HTLV-I to transform human T cells and induce leukemia is still poorly understood. It is believed that the virus induces chronic T-cell proliferation resulting in accumulation of genetic defects and deregulated cellular growth[12]. The absence of a proofreading mechanism and the ensuing high error rates of reverse transcriptase enzymes have been associated with the diversity detected in retroviruses. However, HTLV-I is surprising in that the genetic variability detected between different isolates is extremely low. Therefore, it has been suggested that HTLV-I provirus replication occurs through the high-fidelity process of cellular DNA replication during cell division[13].
The viral oncoprotein Tax is thought to play a central role in the transformation process as demonstrated in various transgenic models and the transformation of fibroblasts[14–17]. Tax expression is also sufficient for immortalization of human primary T cells[18]. Tax has been shown to up-regulate the expression of IL-2 and the IL-2Rα chain[19,20], as well as IL-15 and the IL-15Rα chain[21,22], suggesting an autocrine/paracrine mechanism could be responsible for the proliferation of ATLL cells during the early stages of infection[23]. The transition from IL-2-dependent, HTLV-I-immortalized to IL-2-independent, transformed T-cells has been associated with constitutive activation of the JAK/STAT signaling pathways[24] and loss of the SRC homology 2 (SH2)-containing tyrosine phosphatase-1 (SHP-1)[25]. Tax is unusual in its ability to interact with components of the IKK complex and induce constitutive activation of the NF-κB pathway, which results in up-regulation of numerous pro-survival genes[26–28]. In addition, Tax has been shown to inhibit numerous cell cycle checkpoints to stimulate cellular growth[29–32]. In concert with an increased cellular proliferation, Tax can alter DNA repair pathways and chromosome stability, thereby increasing the mutation rate and possibly promoting genetic rearrangements in infected cells[33–37].
Despite the ability of Tax to promote transformation, a common and striking feature of ATLL cells is the absence of detectable Tax expression. Nearly half of all ATLL patients no longer express detectable tax RNA[38]. This suggests that Tax expression may not be absolutely required in fully transformed cells[39]. However, these ATLL cells retain phenotypic characteristics of Tax-expressing cells. In contrast, ATLL cells always retain expression of the viral HBZ transcript, encoded by the reverse strand of the viral 3′ LTR[40,41]. Similar to Tax, HBZ has been shown to promote the growth of T-cell lines[42]. However, only Tax has been shown to immortalize T cells. As described below, both Tax and HBZ play important roles in the reactivation and maintenance of hTERT expression in HTLV-I-transformed cells.
Telomeres and telomerase regulate a balance between senescence and genomic instability
Telomeres help to preserve genome integrity by preventing chromosome fusion and by regulating the onset of senescence[43–45]. Since DNA polymerase is unable to replicate the very ends of linear DNA, each cell division is associated with progressive erosion of telomeres. This limits the proliferative potential of somatic cells. Telomeres are capped by the shelterin complex that binds and covers telomeric ends. In mammalian cells, the shelterin complex is composed of telomeric repeat binding factors 1 and 2 (TRF1 and TRF2), TRF1 interacting nuclear factor 2 (TIN2), protector of telomeres 1 (POT1), repressor/activator protein 1 (RAP1), and a negative regulator of telomere length, PTOP/PIP1/TINT1[43–46]. The shelterin complex delivers signals regarding the length of telomeres that either allow or prevent telomerase from gaining access to the telomere ends[46,47]. The shelterin complex also forms sub-complexes that determine the structure of the telomeric ends and inhibits the activation of DNA damage pathways[48,49]. Critically short telomeres[50], and telomeres unprotected by the shelterin complex, are recognized and processed as double-strand DNA breaks (DSB) and become sites for recruitment of factors involved in the DNA damage response (DDR)[51,53]. These sites, referred to as telomere dysfunction-induced foci (TIF), stimulate the ATM/ATR kinase[54,55] and activate Chk2-mediated phosphorylation of p53 and p53-dependent transcription. This leads to senescence or apoptosis[56–60]. Initiation of senescence is regulated by the p16INK4a/RB-dependent pathway and a p53-dependent DDR pathway[61,62]. Most cancer cells avoid senescence by disruption of tumor suppressors and activation of telomerase. Telomerase is a holoenzyme that consists of several subunits, including hTR (human telomerase RNA) and hTERT (human telomerase reverse transcriptase)[63,64]. hTERT is a cellular reverse transcriptase that can extend telomeric ends[65] and is the rate-limiting factor for telomerase activity[66]. In cancer cells, reactivation of hTERT expression allows indefinite proliferation[67,68]. The hTERT promoter is tightly regulated and upheld in a repressive state in normal somatic cells[69,70]. The ability of telomerase to extend telomere length is subject to complicated controls[71], such as transcriptional[72,73], posttranscriptional, epigenetic[73,74], and access to the telomeres regulated by components of the shelterin complex[75].
Dysfunctional telomeres induced by progressive telomere shortening have been reported to lead to genomic instability at multiple levels. Cells with critically short telomeres that failed to enter permanent cell cycle arrest activate the non-homologous end joining (NHEJ) pathway, resulting in chromosome end fusions and subsequent breakage-fusion bridge cycles, eventually leading to a pro-cancer genotype[62,76–78]. Telomere attrition increases genomic instability and cumulative risks of cancer progression. Interbreeding of telomerase-deficient mice led to critically short telomeres in late generations, which severely affect the onset of tumorigenesis[79]. In mTerc−/− p53+/−mice, a significantly accelerated rate of tumor formation was observed and these animals suffered tumor types rarely seen in p53−/− telomere-competent mice[80]. These studies suggest that dysfunctional telomeres can drive the initiation of tumors in the absence of functional apoptosis or senescence checkpoints. Additional studies using conditionally deleted POT1 mice revealed highly unstable chromosomes, suggesting that POT1 is normally required to suppress genomic instability by preventing the formation of dysfunctional telomeres. In these animals, loss of POT1 activated a DDR pathway that resulted in cellular senescence[81]. In contrast, in p53 null cells, POT1-induced genomic instability supported malignant transformation and cancer[82,83]. These studies confirmed that dysfunctional telomeres can either suppress cancer by initiating p53-dependent cellular senescence (tumor suppressor activity), or promote cancer by fueling genomic instability in the absence of p53 (oncogenic activity).
Mechanisms Associated with Reactivation of hTERT Expression in HTLV-I Leukemic Cells
Transcriptional Regulation of hTERT Expression
In somatic cells the hTERT promoter is transcriptionally silenced to ensure a finite number of divisions and thereby prevent the accumulation of genetic defects and potential transforming events[84]. In most transformed cells hTERT expression is reactivated to support unlimited replication. Several transcriptional factors and signaling pathways that are frequently activated in tumor cells, c-Myc, SP1, USF, STAT3, PI3K, and NFAT, can positively stimulate hTERT promoter expression[85] (Table 1). In contrast, Mad, histone deacetylases, E2F1, TAK1, WT1, p53, Smad3, and Menin signaling negatively regulate hTERT promoter expression[85]. Multiple studies have confirmed that HTLV-I-infected cells in vitro and in vivo have elevated hTERT expression and hTERT activity[86–92]. Since HTLV-I cells are immortalized or transformed, it is possible that these cells acquire high telomerase activity through in vivo genetic alterations. This theory was disproved when it was demonstrated that the HTLV-I virus could directly induce endogenous telomerase activity[86]. In this study, gamma-irradiated HTLV-I-infected cells that produced infectious virus were co-cultured with non-infected, normal PBMCs. Due to the gamma-irradiation, the HTLV-I producer cells died. However, telomerase expression and activity remained high. This demonstrated that direct transmission of the HTLV-I virus to primary cells could cause activation of the endogenous hTERT gene.
Table 1.
| Effect on hTERT | Expression/Activity in HTLV cells | Known HTLV Activators/Repressors | Tested Effect(s) on hTERT promoter | |
|---|---|---|---|---|
| c-myc | + | + | Tax (+) | Tax increases c-myc binding to hTERT promoter |
| SP1 | + | − | − | Tax increases SP1 binding to hTERT promoter HBZ/JunD heterodimers interact with SP1 on hTERT promoter |
| USF | + | Tax (+) | USF binding to hTERT promoter not affected in Tax-expressing cells. | |
| STAT3 | + | + | − | Unknown (IL-2 signaling increases hTERT expression in HTLV cells) |
| PI3K | + | + | Tax (+) | Tax/IL-2-expression increase activity |
| NFAT | + | + | p12(I) | Unknown |
| Mad | − | UN | − | Max binding to hTERT promoter not affected in HTLV-/Tax-expressing cells; c-myc/Max may replace Mad1/Max on promoter |
| HDACs | − | − | HBZ (−) | |
| E2F1 | − | + | Tax (+) | Unknown |
| TAK1 | − | UN | Tax (+) | Unknown |
| p53 | − | +(inactive) | Tax (−) | p53 binding to hTERT promoter not affected in HTLV-/Tax-expressing cells |
| Smad3 | − | + | Tax (−) HBZ (+) | Unknown |
| Menin | − | + | HBZ (−) | HBZ/JunD/menin/p300 complex relieves JunD/menin inhibition of hTERT promoter |
| Tal1 | − | UN | Tax (+) HBZ (−) | Tal1 decreases hTERT mRNA/HBZ promotes proteosomal degradation of Tal1. |
The HTLV-I virus encodes several proteins through alternative splicing. Among these, the Tax gene is one of the most intensely studied due to its high oncogenic potential. The Tax oncoprotein of HTLV is a strong transcriptional co-regulator of cellular and viral genes[93]. Studies have demonstrated elevated hTERT expression in Tax-expressing cell lines[94] and that Tax can differentially modulate the hTERT promoter depending on the activation status and/or cell cycle progression of the cells[86,95]. Initially, Tax regulation of hTERT caused controversy in the field, since conflicting studies showed positive or negative Tax-mediated effects on hTERT expression[86,96]. However, several studies now confirm that Tax does indeed increase hTERT expression, and the discrepancy in regulation was due to whether prior stimulation was added to the cells of interest. In transient transfection assays, Tax repressed the hTERT promoter upon specific mitogenic stimulation[96]. However, in the absence of mitogenic stimulation, Tax had a general positive regulatory effect on hTERT endogenous gene expression[33,86]. This was confirmed in further studies demonstrating that Tax regulation of the hTERT promoter is cell cycle dependent, with Tax positively influencing the hTERT promoter in resting T cells, but negatively regulating the hTERT promoter in activated T cells[95]. This differential regulation of the hTERT promoter was due to the recruitment of different transcriptional regulators to a 43-bp region of the hTERT promoter. Tax was able to recruit E2F to this region of the hTERT promoter to positively influence telomerase expression. In addition, chromatin immunoprecipitation assays have demonstrated increased binding of two positive telomerase regulators, c-Myc and Sp1, to the endogenous hTERT promoter in HTLV-I- and Tax-expressing T cells[86]. Tax is also known to stimulate c-myc and Sp1 transcription through NF-κB activation[97–99]. Though it remains to be confirmed, this Tax-mediated increase in c-myc may favor the formation of c-myc/Max over Mad1/Max complexes. Mad1/Max is a known repressor of the hTERT promoter, and therefore the displacement of this complex would favor telomerase expression[100]. Together, these findings suggest a model of Tax-induced leukemogenesis whereby in active, proliferating T-cells Tax initially inhibits full hTERT expression. These events result in a transient, genetically unstable state for the cells, in which acquisition of chromosomal abnormalities are achieved leading to a transformed state. Tax then allows these cells to proliferate, expand, and be maintained by increasing telomerase expression[101].
In addition to Tax, another viral protein expressed in ATLL samples, HBZ, was found to stimulate hTERT transcription[102]. HBZ can increase hTERT expression in a JunD and SP-1-dependent manner while HBZ and c-Jun complexes exert a negative effect[102]. In addition, HBZ can antagonize Menin-mediated recruitment of HDAC and inhibition of JunD by interacting with and recruiting p300 instead to the hTERT promoter[103]. An additional negative regulator of the hTERT promoter, the T-cell acute lymphoblastic leukemia 1 protein (Tal1), was also found to be repressed by HBZ[104]. HBZ was able to degrade Tal1 through the proteosome, thereby preventing Tal1 from decreasing hTERT mRNA. HBZ has been shown to be expressed at the RNA level in most ATLL patients but it is unclear if the protein is also expressed at significant levels in these cells[42]. This suggests that in ATLL patients, where Tax expression is low to undetectable, ATLL cells can still retain high telomerase RNA levels through the concerted actions of HBZ and/or the elevated transcriptional and post-transcriptional effects of increased NF-κB activity.
Post-Transcriptional Regulation of hTERT Expression
Although transcriptional reactivation of hTERT is important, some studies demonstrated that transcriptional regulation of hTERT alone does not determine telomerase activity in human CD4 T lymphocytes[105,106]. The catalytic subunit of telomerase has been shown to be reversibly phosphorylated at specific serine, threonine, and tyrosine residues, leading to post-transcriptional control[107]. In fact, telomerase activity is positively regulated at the post-transcriptional level by AKT, the p65 subunit of NF-κB, SHP2, 14-3-3 protein, and PKC-mediated phosphorylation[108–111].
The PI3K-AKT-NF-κB signaling cascade can regulate hTERT transcriptionally and post-transcriptionally and plays an important role in HTLV-I leukemogenesis. In late stages of ATLL disease, Tax expression decreases and is only retained in about half of patients, however constitutive activation of NF-κB is always observed in ATLL cells regardless of their status for Tax. We have previously shown that in the absence of Tax, signaling through the IL-2Rα chain, over-expressed at the surface of ATLL cells[112], is sufficient for potent transcriptional and post-transcriptional activation of hTERT through the PI3K and AKT pathways. Studies shows that IL-2 signaling in HTLV-I cells results in cytoplasmic retention of the Wilm’s tumor suppressor protein (WT1)[94]. It is possible that elevated IL-2 activity leads to a PI3K-PKA-mediated phosphorylation of WT1, sequestering the protein in the cytoplasm where it can no longer bind to and negatively regulate the hTERT promoter. Studies have also found that AKT can directly phosphorylate hTERT and regulate its activity, while NF-κB can influence the nuclear/cytoplasmic distribution of hTERT[108,113]. The direct effect of these proteins on hTERT in HTLV-I cells still remains to be studied. Given the high levels of AKT and NF-κB in HTLV-I cells, it can be hypothesized that these pathways could be very important in maintaining the high telomerase activity seen in these cells. Another cellular protein with positive effects on telomerase activity is the protein kinase C (PKC) family of proteins[114]. PKC can phosphorylate hTERT, but the contribution of PKC in HTLV-I regulation of hTERT is unknown.
Aside from NF-κB, several other cellular proteins are believed to be involved in telomerase re-localization. Though not required for telomerase activity, the 14-3-3 proteins have been found to bind to hTERT and influence its nuclear/cytoplasmic distribution[115]. The SH2-containing protein tyrosine phosphatase, SHP-2, is also involved in telomerase localization by regulating tyrosine 707 phosphorylation and maintenance of hTERT in the nucleus[116]. Elevated SHP-2 expression also led to increased nuclear telomerase activity in endothelial cells. The nuclear/cytoplasmic distribution of telomerase has not been studied in HTLV-I cells, however, as stated above, NF-κB is constitutively active in ATLL cells and SHP-2 was found to be expressed in an HTLV-I line. Currently the expression of 14-3-3 proteins in HTLV-I cells is unknown.
In addition to positive hTERT regulators, several negative, post-transcriptional regulators have recently been discovered. Phosphatase PP2A has been shown to exert a negative control on telomerase activity by directly de-phosphorylating hTERT and/or indirectly de-phosphorylating AKT[117]. Tax has been shown to inhibit PP2A[118] and drug inhibitors (okadaic acid) or knock-down of PP2A caused cell death and cell cycle arrest in HTLV cells. What effect PP2A inhibition has on telomerase activity has not been studied in HTLV-I cells. Another negative regulator, c-Abl, is able to tyrosine phosphorylate hTERT, leading to telomerase inhibition and telomere shortening[110]. This may play a role in the DNA damage response, as cells exposed to ionizing radiation induce c-Abl phosphorylation of hTERT. The roles of c-Abl in HTLV-I pathogenesis and transformation are unknown.
Finally, control of telomerase in HTLV-I cells may also occur through epigenetic microRNA (miRNA)-mediated effects or somatic mutations in the telomerase promoter. For example, studies have found that hypermethylation of the upstream of the transcription start site (UTSS) region in cancer was associated with higher telomerase activity[119]. In pediatric brain cancers, methylation of this region correlated with tumor progression and an overall worse prognosis. Glioblastoma tumors were also found to harbor hTERT promoter mutations that correlated with increased hTERT expression[120]. The majority of research on telomerase regulation in ATLL has been focused on transcriptional regulation at the hTERT promoter. While a direct role for post-transcriptional regulation of telomerase in HTLV-I has not been studied, it is highly likely that such a means of telomerase regulation exists in HTLV-I cells (Table 2). The viral Tax protein has been shown to stimulate the activity of AKT and PKC downstream pathways[121,122]. Similarly, Tax has been shown to inhibit PP2A[118]. Hence it is expected, although not demonstrated, that Tax may alter post-transcriptional activity of hTERT. In addition, several microRNAs have been shown to be de-regulated in ATLL cells. A comprehensive look on microRNA-mediated regulation of telomerase in HTLV-I cells needs to be examined.
Table 2.
| Effect on hTERT | Expression/Activity in HTLV cells | Known HTLV Activators/Repressors | Tested Effect(s) on hTERT promoter | |
|---|---|---|---|---|
| AKT | + (phosphorylates/Expression) |
+ | Tax (+) | The PI3K/AKT pathway was shown to positively influence telomerase activity in HTLV-I cells; unknown direct effects. |
| NFκB (p65) | + (Localization/Expression) |
+ | Tax (+)HBZ (−) | NF-κB mediated SP1 and c-myc activation of hTERT promoter; unknown direct effects. |
| PKC | + (phosphorylates/Expression) |
+ | Tax (+) | Unknown |
| 14-3-3 | + (Localization) |
UN | UN | Unknown |
| SHP2 | + (Localization) |
+/UN | UN | Unknown |
| Abl | − (phosphorylates) |
UN | UN | Unknown |
| PPA | − (De-phosphorylates) |
UN | Tax (−) | Not tested; Inhibition of PPA leads to loss of cell viability in HTLV-I cells. |
Importance of telomerase regulation in ATLL disease and therapy
Telomere loss in cancer cells is believed to occur because of insufficient levels of telomerase, or due to a crisis originating from a combination of replication stress and a deficiency of double-strand break repair in telomeric regions[123]. Telomere attrition is an important driving factor in promoting the genetic alterations needed to establish a transformed state. Preventing telomere loss could prevent the genetic instability found in cancer and deter tumor progression. Several studies have found a correlation between telomerase activity and the progression of ATLL disease from the chronic to the acute stage[91,92]. It was found that ATLL leukemic cells have shorter telomere lengths than their non-infected counterpart. Also, acute ATLL patients have shorter telomeres when compared to asymptomatic patients. These studies have found that the median survival period of acute ATL patients harboring higher telomerase activity and shorter telomere lengths was significantly shorter than acute ATL patients with lower telomerase activity and normal telomeres (0.47 years vs 4.1 years). This suggests that the level of telomerase activity and the length of the telomere could therefore be used as a prognostic marker for ATL disease.
Though activated T cells express high levels of telomerase, this activity is transient and decreases once the stimulus is removed. In fact, non-stimulated, normal T cells have low to undetectable telomerase activity. The fact that ATLL cells demonstrate persistent, continuously high levels of telomerase activity suggests that high telomerase activity is required to maintain the transformed state. Several lines of evidence point to the fact that elevated telomerase activity is required for ATLL pathogenesis. First, telomerase activity correlates with proviral loads, which are known to correlate with disease progression[124,125]. In addition, after integration of the HTLV-I virus, infected cells undergo mitotic division for several years. Studies demonstrate that ATLL cells are derived from the clonal expansion of a single clone[126]. Given the long latency period of ATLL, it is understandable that these cells must gain a proliferative advantage. Reactivation of telomerase would allow these cells to continue to proliferate indefinitely. (Figure 1)
Figure 1.
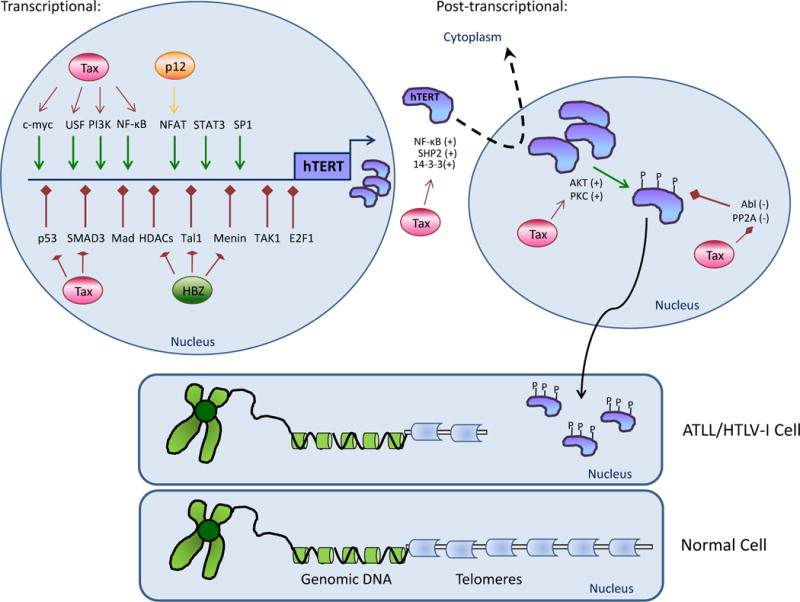
Telomerase regulation in ATLL cells.
Several transcriptional and post-transcriptional mechanisms cooperate to positively regulate telomerase activity in ATLL cells. In the nucleus, tight transcriptional control of the hTERT gene can be achieved through Tax (Red circle)-mediated increases in the positive regulators (green arrows), c-myc, USF, PI3K, or NF-κB. In addition, the viral protein p12 (yellow circle) can positively influence NFAT activity. Several negative regulators of hTERT expression (red arrows) are inhibited through Tax (p53 and Smad3) or HBZ (HDACs, Tal1, and menin) (green circle). The newly synthesized hTERT protein can be post-transcriptionally phosphorylated by positive regulators, AKT or PKC, or by the negative regulators, Abl and PP2A. Tax expression can affect both regulators. Tax could also influence the nuclear/cytoplasmic distribution of telomerase (dotted arrow) by positively regulating NF-κB, SHP-2, or 14-3-3 proteins. The concerted actions of HTLV-I proteins cause telomerase expression and activity to be increased in ATLL cells. These cells have higher telomerase levels and shorter telomeres (grey blocks) than their non-infected counterparts.
The importance of telomerase activity in HTLV-I pathogenesis is further supported by the fact that treatment with a telomerase inhibitor causes disease remission in ATLL patients who harbor a functional p53[86]. In this study, when HTLV-I cells were treated with Azidothymidine/zidovudine (AZT), which inhibits telomerase activity, telomere lengths were progressively shortened. These shorter telomeres promoted activation of the DDR-pathway. Telomere attrition resulted in formation of TIFs and an ATM response, leading to replicative senescence of HTLV-I-transformed cells with wild-type p53. In these long term AZT-treated cells, two critical regulators of p53 – MDM2 and MDMX– were eliminated. T cells undergoing AZT-mediated senescence also had elevated p21WAF, p16ink and decreased p-GSK3β expression.
ATLL cells are examples of a growing list of cancers that demonstrate short telomeres with re-activation of telomerase. Among leukemias, T-cell prolymphocytic leukemia (T-PLL), chronic myeloid leukemia (CML), and chronic lymphocytic leukemia also display short telomeres and high telomerase activity[127–129]. T-PLL cells were sensitive to the telomerase inhibitor, BIBR1532, which did not affect normal T cells. This same inhibitor was also able to inhibit proliferation of pre-B acute lymphoblastic leukemia cells[130]. Progress has recently been seen in the treatment of ATL patients with a combination of AZT and interferon-alpha (IFN-α)[131]. A high response rate was seen with minimal side effects in ATLL patients. The fact that zidovudine/IFN-α is now used in first-line therapy for treating leukemic sub-types of ATL demonstrates the importance of telomerase in ATLL disease[132,133].
Acknowledgments
This work was funded by NIH grant CA106258 to C.N. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Authors declare no conflict of interest.
References
- 1.Poiesz BJ, Ruscetti FW, Gazdar AF, et al. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA. 1980;77(12):7415–7419. doi: 10.1073/pnas.77.12.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshida M, Seiki M, Yamaguchi K, et al. Monoclonal integration of human T-cell leukemia provirus in all primary tumors of adult T-cell leukemia suggests causative role of human T-cell leukemia virus in the disease. Proc Natl Acad Sci USA. 1984;81(8):2534–2537. doi: 10.1073/pnas.81.8.2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uchiyama T, Yodoi J, Sagawa K, et al. Adult T-cell leukemia: clinical and hematologic features of 16 cases. Blood. 1977;50(3):481–492. [PubMed] [Google Scholar]
- 4.Tsukasaki K, Hermine O, Bazarbachi A, et al. Definition, prognostic factors, treatment, and response criteria of adult T-cell leukemia-lymphoma: a proposal from an international consensus meeting. J Clin Oncol. 2009;27(3):453–459. doi: 10.1200/JCO.2008.18.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Itoyama T, Chaganti RS, Yamada Y, et al. Cytogenetic analysis and clinical significance in adult T-cell leukemia/lymphoma: a study of 50 cases from the human T-cell leukemia virus type-1 endemic area, Nagasaki. Blood. 2001;97(11):3612–3620. doi: 10.1182/blood.v97.11.3612. [DOI] [PubMed] [Google Scholar]
- 6.Smith SD, Morgan R, Link MP, et al. Cytogenetic and immunophenotypic analysis of cell lines established from patients with T cell leukemia/lymphoma. Blood. 1986;67(3):650–656. [PubMed] [Google Scholar]
- 7.Shimoyama M, Abe T, Miyamoto K, et al. Chromosome aberrations and clinical features of adult T cell leukemia-lymphoma not associated with human T cell leukemia virus type I. Blood. 1987;69(4):984–989. [PubMed] [Google Scholar]
- 8.Hatta Y, Yamada Y, Tomonaga M, et al. Detailed deletion mapping of the long arm of chromosome 6 in adult T-cell leukemia. Blood. 1999;93(2):613–616. [PubMed] [Google Scholar]
- 9.Sadamori N, Ichiba M, Mine M, et al. Clinical significance of serum thymidine kinase in adult T-cell leukaemia and acute myeloid leukaemia. Br J Haematol. 1995;90(1):100–105. doi: 10.1111/j.1365-2141.1995.tb03386.x. [DOI] [PubMed] [Google Scholar]
- 10.Kamihira S, Atogami S, Sohda H, et al. Significance of soluble interleukin-2 receptor levels for evaluation of the progression of adult T-cell leukemia. Cancer. 1994;73(11):2753–2758. doi: 10.1002/1097-0142(19940601)73:11<2753::aid-cncr2820731117>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 11.Sadamori N, Mine M, Hakariya S, et al. Clinical significance of beta 2-microglobulin in serum of adult T cell leukemia. Leukemia. 1995;9(4):594–597. [PubMed] [Google Scholar]
- 12.Taylor G. Molecular aspects of HTLV-I infection and adult T-cell leukaemia/lymphoma. J Clin Pathol. 2007;60(12):1392–1396. doi: 10.1136/jcp.2007.052662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wattel E, Cavrois M, Gessain A, et al. Clonal expansion of infected cells: a way of life for HTLV-I. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;13(Suppl 1):S92–S99. doi: 10.1097/00042560-199600001-00016. [DOI] [PubMed] [Google Scholar]
- 14.Hasegawa H, Sawa H, Lewis MJ, et al. Thymus-derived leukemia-lymphoma in mice transgenic for the Tax gene of human T-lymphotropic virus type I. Nat Med. 2006;12(4):466–472. doi: 10.1038/nm1389. [DOI] [PubMed] [Google Scholar]
- 15.Grossman WJ, Ratner L. Cytokine expression and tumorigenicity of large granular lymphocytic leukemia cells from mice transgenic for the tax gene of human T-cell leukemia virus type I. Blood. 1997;90(2):783–794. [PubMed] [Google Scholar]
- 16.Nerenberg MI. An HTLV-I transgenic mouse model: role of the tax gene in pathogenesis in multiple organ systems. Curr Top Microbiol Immunol. 1990;160:121–128. doi: 10.1007/978-3-642-75267-4_7. [DOI] [PubMed] [Google Scholar]
- 17.Green JE, Hinrichs SH, Vogel J, et al. Exocrinopathy resembling Sjogren’s syndrome in HTLV-1 tax transgenic mice. Nature. 1989;341(6237):72–74. doi: 10.1038/341072a0. [DOI] [PubMed] [Google Scholar]
- 18.Pise-Masison CA, Jeong SJ, Brady JN. Human T cell leukemia virus type 1: the role of Tax in leukemogenesis. Arch Immunol Ther Exp. 2005;53(4):283–296. [PubMed] [Google Scholar]
- 19.Ruben S, Poteat H, Tan TH, et al. Cellular transcription factors and regulation of IL-2 receptor gene expression by HTLV-I tax gene product. Science. 1988;241(4861):89–92. doi: 10.1126/science.2838905. [DOI] [PubMed] [Google Scholar]
- 20.Good L, Maggirwar SB, Sun SC. Activation of the IL-2 gene promoter by HTLV-I tax involves induction of NF-AT complexes bound to the CD28-responsive element. EMBO J. 1996;15(14):3744–3750. [PMC free article] [PubMed] [Google Scholar]
- 21.Mariner JM, Lantz V, Waldmann TA, et al. Human T cell lymphotropic virus type I Tax activates IL-15R alpha gene expression through an NF-kappa B site. J Immunol. 2001;166(4):2602–2609. doi: 10.4049/jimmunol.166.4.2602. [DOI] [PubMed] [Google Scholar]
- 22.Waldmann TA. The promiscuous IL-2/IL-15 receptor: a target for immunotherapy of HTLV-I-associated disorders. J Acquir Immune Defic Syndr Hum Retrovirol. 1996;13(Suppl 1):S179–S185. doi: 10.1097/00042560-199600001-00027. [DOI] [PubMed] [Google Scholar]
- 23.Russell SJ. Interleukin-2 and T cell malignancies: an autocrine loop with a twist. Leukemia. 1989;3(10):755–757. [PubMed] [Google Scholar]
- 24.Migone TS, Lin JX, Cereseto A, et al. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995;269(5220):79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- 25.Migone TS, Cacalano NA, Taylor N, et al. Recruitment of SH2-containing protein tyrosine phosphatase SHP-1 to the interleukin 2 receptor; loss of SHP-1 expression in human T-lymphotropic virus type I-transformed T cells. Proc Natl Acad Sci USA. 1998;95(7):3845–3850. doi: 10.1073/pnas.95.7.3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao G. NF-kappaB activation: Tax sumoylation is out, but what about Tax ubiquitination? ( Retrovirology. 2012;9:78. doi: 10.1186/1742-4690-9-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun SC, Yamaoka S. Activation of NF-kappaB by HTLV-I and implications for cell transformation. Oncogene. 2005;24(39):5952–5964. doi: 10.1038/sj.onc.1208969. [DOI] [PubMed] [Google Scholar]
- 28.Harhaj EW, Harhaj NS. Mechanisms of persistent NF-kappaB activation by HTLV-I tax. IUBMB Life. 2005;57(2):83–91. doi: 10.1080/15216540500078715. [DOI] [PubMed] [Google Scholar]
- 29.Yamada Y, Kamihira S. Inactivation of tumor suppressor genes and the progression of adult T-cell leukemia-lymphoma. Leuk Lymphoma. 2005;46(11):1553–1559. doi: 10.1080/10428190500244217. [DOI] [PubMed] [Google Scholar]
- 30.Yoshida M. Multiple viral strategies of HTLV-1 for dysregulation of cell growth control. Annu Rev Immunol. 2001;19:475–496. doi: 10.1146/annurev.immunol.19.1.475. [DOI] [PubMed] [Google Scholar]
- 31.Pise-Masison CA, Mahieux R, Radonovich M, et al. Insights into the molecular mechanism of p53 inhibition by HTLV type 1 Tax. AIDS Res Hum Retroviruses. 2000;16(16):1669–1675. doi: 10.1089/08892220050193128. [DOI] [PubMed] [Google Scholar]
- 32.Marriott SJ, Semmes OJ. Impact of HTLV-I Tax on cell cycle progression and the cellular DNA damage repair response. Oncogene. 2005;24(39):5986–5995. doi: 10.1038/sj.onc.1208976. [DOI] [PubMed] [Google Scholar]
- 33.Bellon M, Baydoun HH, Yao Y, et al. HTLV-I Tax-dependent and -independent events associated with immortalization of human primary T lymphocytes. Blood. 2010;115(12):2441–2448. doi: 10.1182/blood-2009-08-241117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peloponese JM, Haller K, Miyazato A, et al. Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoprotein. Proc Natl Acad Sci USA. 2005;102(52):18974–18979. doi: 10.1073/pnas.0506659103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pise-Masison CA, Brady JN. Setting the stage for transformation: HTLV-1 Tax inhibition of p53 function. Front Biosci. 2005;10:919–930. doi: 10.2741/1586. [DOI] [PubMed] [Google Scholar]
- 36.Wycuff DR, Marriott SJ. The HTLV-I Tax oncoprotein: hyper-tasking at the molecular level. Front Biosci. 2005;10:620–642. doi: 10.2741/1558. [DOI] [PubMed] [Google Scholar]
- 37.Gatza ML, Watt JC, Marriott SJ. Cellular transformation by the HTLV-I Tax protein, a jack-of-all-trades. Oncogene. 2003;22(33):5141–5149. doi: 10.1038/sj.onc.1206549. [DOI] [PubMed] [Google Scholar]
- 38.Ko NL, Taylor JM, Bellon M, et al. PA28gamma is a novel corepressor of HTLV-1 replication and controls viral latency. Blood. 2013;121(5):791–800. doi: 10.1182/blood-2012-03-420414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miyazaki M, Yasunaga J, Taniguchi Y, et al. Preferential selection of human T-cell leukemia virus type 1 provirus lacking the 5′ long terminal repeat during oncogenesis. J Virol. 2007;81(11):5714–5723. doi: 10.1128/JVI.02511-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Satou Y, Matsuoka M. Implication of the HTLV-I bZIP factor gene in the leukemogenesis of adult T-cell leukemia. Int J Hematol. 2007;86(2):107–112. doi: 10.1532/IJH97.07103. [DOI] [PubMed] [Google Scholar]
- 41.Mesnard JM, Barbeau B, Devaux C. HBZ, a new important player in the mystery of adult T-cell leukemia. Blood. 2006;108(13):3979–3982. doi: 10.1182/blood-2006-03-007732. [DOI] [PubMed] [Google Scholar]
- 42.Satou Y, Yasunaga J, Yoshida M, et al. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA. 2006;103(3):720–725. doi: 10.1073/pnas.0507631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 44.Greider CW. Telomeres. Curr Opin Cell Biol. 1991;3(3):444–451. doi: 10.1016/0955-0674(91)90072-7. [DOI] [PubMed] [Google Scholar]
- 45.O’Sullivan RJ, Karlseder J. Telomeres: protecting chromosomes against genome instability. Nat Rev Mol Cell Biol. 2010;11(3):171–181. doi: 10.1038/nrm2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19(18):2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 47.Kendellen MF, Barrientos KS, Counter CM. POT1 association with TRF2 regulates telomere length. Mol Cell Biol. 2009;29(20):5611–5619. doi: 10.1128/MCB.00286-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Deng Y, Guo X, Ferguson DO, et al. Multiple roles for MRE11 at uncapped telomeres. Nature. 2009;460(7257):914–918. doi: 10.1038/nature08196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448(7157):1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 50.Capper R, Britt-Compton B, Tankimanova M, et al. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21(19):2495–2508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.d’Adda di, Fagagna F, Reaper PM, Clay-Farrace L, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426(6963):194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 52.Karlseder J, Smogorzewska A, Lange dT. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295(5564):2446–2449. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- 53.Karlseder J. Telomere repeat binding factors: keeping the ends in check. Cancer Lett. 2003;194(2):189–197. doi: 10.1016/s0304-3835(02)00706-1. [DOI] [PubMed] [Google Scholar]
- 54.Doksani Y, de Lange T. The Role of Double-Strand Break Repair Pathways at Functional and Dysfunctional Telomeres. Cold Spring Harb Perspect Biol. 2014;6(12) doi: 10.1101/cshperspect.a016576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karlseder J, Hoke K, Mirzoeva OK, et al. The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PLoS Biol. 2004;2(8):E240. doi: 10.1371/journal.pbio.0020240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shieh SY, Ikeda M, Taya Y, et al. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91(3):325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 57.Chehab NH, Malikzay A, Appel M, et al. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev. 2000;14(3):278–288. [PMC free article] [PubMed] [Google Scholar]
- 58.Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer. 2008;8(6):450–458. doi: 10.1038/nrc2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cosme-Blanco W, Chang S. Dual roles of telomere dysfunction in initiation and suppression of tumorigenesis. Exp Cell Res. 2008;314(9):1973–1979. doi: 10.1016/j.yexcr.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cosme-Blanco W, Shen MF, Lazar AJ, et al. Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53-dependent cellular senescence. EMBO Rep. 2007;8(5):497–503. doi: 10.1038/sj.embor.7400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Y, Sharpless N, Chang S. p16(INK4a) protects against dysfunctional telomere-induced ATR-dependent DNA damage responses. J Clin Invest. 2013;123(10):4489–4501. doi: 10.1172/JCI69574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cosme-Blanco W, Shen MF, Lazar AJ, et al. Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53-dependent cellular senescence. EMBO Rep. 2007;8(5):497–503. doi: 10.1038/sj.embor.7400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cairney CJ, Keith WN. Telomerase redefined: integrated regulation of hTR and hTERT for telomere maintenance and telomerase activity. Biochimie. 2008;90(1):13–23. doi: 10.1016/j.biochi.2007.07.025. [DOI] [PubMed] [Google Scholar]
- 64.Counter CM. The roles of telomeres and telomerase in cell life span. Mutat Res. 1996;366(1):45–63. doi: 10.1016/s0165-1110(96)90006-8. [DOI] [PubMed] [Google Scholar]
- 65.Meyerson M, Counter CM, Eaton EN, et al. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell. 1997;90(4):785–795. doi: 10.1016/s0092-8674(00)80538-3. [DOI] [PubMed] [Google Scholar]
- 66.Hukezalie KR, Wong JM. Structure-function relationship and biogenesis regulation of the human telomerase holoenzyme. FEBS J. 2013;280(14):3194–3204. doi: 10.1111/febs.12272. [DOI] [PubMed] [Google Scholar]
- 67.Shay JW, Wright WE. Telomerase activity in human cancer. Curr Opin Oncol. 1996;8(1):66–71. doi: 10.1097/00001622-199601000-00012. [DOI] [PubMed] [Google Scholar]
- 68.Rhyu MS. Telomeres, telomerase, and immortality. J Natl Cancer Inst. 1995;87(12):884–894. doi: 10.1093/jnci/87.12.884. [DOI] [PubMed] [Google Scholar]
- 69.Horikawa I, Michishita E, Barrett JC. Regulation of hTERT transcription: a target of cellular and viral mechanisms for immortalization and carcinogenesis. Cytotechnology. 2004;45(1–2):23–32. doi: 10.1007/s10616-004-5122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horikawa I, Cable PL, Afshari C, et al. Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res. 1999;59(4):826–830. [PubMed] [Google Scholar]
- 71.Stern JL, Bryan TM. Telomerase recruitment to telomeres. Cytogenet. Genome Res. 2008;122(3–4):243–254. doi: 10.1159/000167810. [DOI] [PubMed] [Google Scholar]
- 72.Kyo S, Takakura M, Fujiwara T, et al. Understanding and exploiting hTERT promoter regulation for diagnosis and treatment of human cancers. Cancer Sci. 2008;99(8):1528–1538. doi: 10.1111/j.1349-7006.2008.00878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daniel M, Peek GW, Tollefsbol TO. Regulation of the human catalytic subunit of telomerase (hTERT) Gene. 2012;498(2):135–146. doi: 10.1016/j.gene.2012.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhu J, Zhao Y, Wang S. Chromatin and epigenetic regulation of the telomerase reverse transcriptase gene. Protein Cell. 2010;1(1):22–32. doi: 10.1007/s13238-010-0014-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang Y, Chen LY, Han X, et al. Phosphorylation of TPP1 regulates cell cycle-dependent telomerase recruitment. Proc Natl Acad Sci USA. 2013;110(14):5457–5462. doi: 10.1073/pnas.1217733110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De lange T. Telomere-related genome instability in cancer. Cold Spring Harb Symp Quant Biol. 2005;70:197–204. doi: 10.1101/sqb.2005.70.032. [DOI] [PubMed] [Google Scholar]
- 77.Deng Y, Chan SS, Chang S. Telomere dysfunction and tumour suppression: the senescence connection. Nat Rev Cancer. 2008;8(6):450–458. doi: 10.1038/nrc2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo X, Deng Y, Lin Y, et al. Dysfunctional telomeres activate an ATM-ATR-dependent DNA damage response to suppress tumorigenesis. EMBO J. 2007;26(22):4709–4719. doi: 10.1038/sj.emboj.7601893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blasco MA, Lee HW, Hande MP, et al. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91(1):25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 80.Artandi SE, Chang S, Lee SL, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406(6796):641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 81.Chang S. Cancer chromosomes going to POT1. Nat Genet. 2013;45(5):473–475. doi: 10.1038/ng.2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.He H, Multani AS, Cosme-Blanco W, et al. POT1b protects telomeres from end-to-end chromosomal fusions and aberrant homologous recombination. EMBO J. 2006;25(21):5180–5190. doi: 10.1038/sj.emboj.7601294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu L, Multani AS, He H, et al. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell. 2006;126(1):49–62. doi: 10.1016/j.cell.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 84.Counter CM, Meyerson M, Eaton EN, et al. Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene. 1998;16(9):1217–1222. doi: 10.1038/sj.onc.1201882. [DOI] [PubMed] [Google Scholar]
- 85.Bellon M, Nicot C. Regulation of telomerase and telomeres: human tumor viruses take control. J Natl Cancer Inst. 2008;100(2):98–108. doi: 10.1093/jnci/djm269. [DOI] [PubMed] [Google Scholar]
- 86.Sinha-Datta U, Horikawa I, Michishita E, et al. Transcriptional activation of hTERT through the NF-kappaB pathway in HTLV-I-transformed cells. Blood. 2004;104(8):2523–2531. doi: 10.1182/blood-2003-12-4251. [DOI] [PubMed] [Google Scholar]
- 87.Bellon M, Datta A, Brown M, et al. Increased expression of telo-mere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int J Cancer. 2006;119(9):2090–2097. doi: 10.1002/ijc.22026. [DOI] [PubMed] [Google Scholar]
- 88.Datta A, Bellon M, Sinha-Datta U, et al. Persistent inhibition of telomerase reprograms adult T-cell leukemia to p53-dependent senescence. Blood. 2006;108(3):1021–1029. doi: 10.1182/blood-2006-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Franzese O, Balestrieri E, Comandini A, et al. Telomerase activity of human peripheral blood mononuclear cells in the course of HTLV type 1 infection in vitro. AIDS Res Hum Retroviruses. 2002;18(4):249–251. doi: 10.1089/088922202753472810. [DOI] [PubMed] [Google Scholar]
- 90.Tsumuki H, Nakazawa M, Hasunuma T, et al. Infection of synoviocytes with HTLV-I induces telomerase activity. Rheumatol Int. 2001;20(5):175–179. doi: 10.1007/s002960100111. [DOI] [PubMed] [Google Scholar]
- 91.Kubuki Y, Suzuki M, Sasaki H, et al. Telomerase activity and telomere length as prognostic factors of adult T-cell leukemia. Leuk Lymphoma. 2005;46(3):393–399. doi: 10.1080/10428190400018349. [DOI] [PubMed] [Google Scholar]
- 92.Uchida N, Otsuka T, Arima F, et al. Correlation of telomerase activity with development and progression of adult T-cell leukemia. Leuk Res. 1999;23(3):311–316. doi: 10.1016/s0145-2126(98)00170-2. [DOI] [PubMed] [Google Scholar]
- 93.Currer R, Van DR, Jaworski E, et al. HTLV tax: a fascinating multifunctional co-regulator of viral and cellular pathways. Front Microbiol. 2012;3:406. doi: 10.3389/fmicb.2012.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bellon M, Nicot C. Central role of PI3K in transcriptional activation of hTERT in HTLV-I-infected cells. Blood. 2008;112(7):2946–2955. doi: 10.1182/blood-2008-01-134692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hara T, Matsumura-Arioka Y, Ohtani K, et al. Role of human T-cell leukemia virus type I Tax in expression of the human telomerase reverse transcriptase (hTERT) gene in human T-cells. Cancer Sci. 2008;99(6):1155–1163. doi: 10.1111/j.1349-7006.2008.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gabet AS, Mortreux F, Charneau P, et al. Inactivation of hTERT transcription by Tax. Oncogene. 2003;22(24):3734–3741. doi: 10.1038/sj.onc.1206468. [DOI] [PubMed] [Google Scholar]
- 97.Trejo SR, Fahl WE, Ratner L. The tax protein of human T-cell leukemia virus type 1 mediates the transactivation of the c-sis/platelet-derived growth factor-B promoter through interactions with the zinc finger transcription factors Sp1 and NGFI-A/Egr-1. J Biol Chem. 1997;272(43):27411–27421. doi: 10.1074/jbc.272.43.27411. [DOI] [PubMed] [Google Scholar]
- 98.Dittmer J, Pise-Masison CA, Clemens KE, et al. Interaction of human T-cell lymphotropic virus type I Tax, Ets1, and Sp1 in transactivation of the PTHrP P2 promoter. J Biol Chem. 1997;272(8):4953–4958. doi: 10.1074/jbc.272.8.4953. [DOI] [PubMed] [Google Scholar]
- 99.Duyao MP, Kessler DJ, Spicer DB, et al. Transactivation of the c-myc gene by HTLV-1 tax is mediated by NFkB. Curr Top Microbiol Immunol. 1992;182:421–424. doi: 10.1007/978-3-642-77633-5_53. [DOI] [PubMed] [Google Scholar]
- 100.Kyo S, Takakura M, Taira T, et al. Sp1 cooperates with c-Myc to activate transcription of the human telomerase reverse transcriptase gene (hTERT) Nucleic Acids Res. 2000;28(3):669–677. doi: 10.1093/nar/28.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hara T, Matsumura-Arioka Y, Ohtani K, et al. Role of human T-cell leukemia virus type I Tax in expression of the human telomerase reverse transcriptase (hTERT) gene in human T-cells. Cancer Sci. 2008;99(6):1155–1163. doi: 10.1111/j.1349-7006.2008.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kuhlmann AS, Villaudy J, Gazzolo L, et al. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT) Retrovirology. 2007;4:92. doi: 10.1186/1742-4690-4-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Borowiak M, Kuhlmann AS, Girard S, et al. HTLV-1 bZIP factor impedes the menin tumor suppressor and upregulates JunD-mediated transcription of the hTERT gene. Carcinogenesis. 2013;34(11):2664–2672. doi: 10.1093/carcin/bgt221. [DOI] [PubMed] [Google Scholar]
- 104.Terme JM, Mocquet V, Kuhlmann AS, et al. Inhibition of the hTERT promoter by the proto-oncogenic protein TAL1. Leukemia. 2009;23(11):2081–2089. doi: 10.1038/leu.2009.131. [DOI] [PubMed] [Google Scholar]
- 105.Liu K, Hodes RJ, Weng N. Cutting edge: telomerase activation in human T lymphocytes does not require increase in telomerase reverse transcriptase (hTERT) protein but is associated with hTERT phosphory-lation and nuclear translocation. J Immunol. 2001;166(8):4826–4830. doi: 10.4049/jimmunol.166.8.4826. [DOI] [PubMed] [Google Scholar]
- 106.Liu K, Schoonmaker MM, Levine BL, et al. Constitutive and regulated expression of telomerase reverse transcriptase (hTERT) in human lymphocytes. Proc Natl Acad Sci USA. 1999;96(9):5147–5152. doi: 10.1073/pnas.96.9.5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cong YS, Wright WE, Shay JW. Human telomerase and its regulation. Microbiol Mol Biol Rev. 2002;66(3):407–425. doi: 10.1128/MMBR.66.3.407-425.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kang SS, Kwon T, Kwon DY, et al. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem. 1999;274(19):13085–13090. doi: 10.1074/jbc.274.19.13085. [DOI] [PubMed] [Google Scholar]
- 109.Chang JT, Lu YC, Chen YJ, et al. hTERT phosphorylation by PKC is essential for telomerase holoprotein integrity and enzyme activity in head neck cancer cells. Br J Cancer. 2006;94(6):870–878. doi: 10.1038/sj.bjc.6603008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kharbanda S, Kumar V, Dhar S, et al. Regulation of the hTERT telomerase catalytic subunit by the c-Abl tyrosine kinase. Curr Biol. 2000;10(10):568–575. doi: 10.1016/s0960-9822(00)00483-8. [DOI] [PubMed] [Google Scholar]
- 111.Li H, Zhao L, Yang Z, et al. Telomerase is controlled by protein kinase Calpha in human breast cancer cells. J Biol Chem. 1998;273(50):33436–33442. doi: 10.1074/jbc.273.50.33436. [DOI] [PubMed] [Google Scholar]
- 112.Uchiyama T, Hori T, Tsudo M, et al. Interleukin-2 receptor (Tac antigen) expressed on adult T cell leukemia cells. J Clin Invest. 1985;76(2):446–453. doi: 10.1172/JCI111992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Akiyama M, Hideshima T, Hayashi T, et al. Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res. 2003;63(1):18–21. [PubMed] [Google Scholar]
- 114.Chang JT, Lu YC, Chen YJ, et al. hTERT phosphorylation by PKC is essential for telomerase holoprotein integrity and enzyme activity in head neck cancer cells. Br J Cancer. 2006;94(6):870–878. doi: 10.1038/sj.bjc.6603008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Seimiya H, Sawada H, Muramatsu Y, et al. Involvement of 14-3-3 proteins in nuclear localization of telomerase. EMBO J. 2000;19(11):2652–2661. doi: 10.1093/emboj/19.11.2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jakob S, Schroeder P, Lukosz M, et al. Nuclear protein tyrosine phosphatase Shp-2 is one important negative regulator of nuclear export of telomerase reverse transcriptase. J Biol Chem. 2008;283(48):33155–33161. doi: 10.1074/jbc.M805138200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li H, Zhao LL, Funder JW, et al. Protein phosphatase 2A inhibits nuclear telomerase activity in human breast cancer cells. J Biol Chem. 1997;272(27):16729–16732. doi: 10.1074/jbc.272.27.16729. [DOI] [PubMed] [Google Scholar]
- 118.Fu DX, Kuo YL, Liu BY, et al. Human T-lymphotropic virus type I tax activates I-kappa B kinase by inhibiting I-kappa B kinase-associated serine/threonine protein phosphatase 2A. J Biol Chem. 2003;278(3):1487–1493. doi: 10.1074/jbc.M210631200. [DOI] [PubMed] [Google Scholar]
- 119.Castelo-Branco P, Choufani S, Mack S, et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: an integrative genomic and molecular study. Lancet Oncol. 2013;14(6):534–542. doi: 10.1016/S1470-2045(13)70110-4. [DOI] [PubMed] [Google Scholar]
- 120.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lindholm PF, Tamami M, Makowski J, et al. Human T-cell lymphotropic virus type 1 Tax1 activation of NF-kappa B: involvement of the protein kinase C pathway. J Virol. 1996;70(4):2525–2532. doi: 10.1128/jvi.70.4.2525-2532.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu Y, Wang Y, Yamakuchi M, et al. Phosphoinositide-3 kinase-PKB/Akt pathway activation is involved in fibroblast Rat-1 transformation by human T-cell leukemia virus type I tax. Oncogene. 2001;20(20):2514–2526. doi: 10.1038/sj.onc.1204364. [DOI] [PubMed] [Google Scholar]
- 123.Murnane JP. Telomere dysfunction and chromosome instability. Mutat Res. 2012;730(1‐2):28–36. doi: 10.1016/j.mrfmmm.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kamihira S, Dateki N, Sugahara K, et al. Significance of HTLV-1 proviral load quantification by real-time PCR as a surrogate marker for HTLV-1-infected cell count. Clin Lab Haematol. 2003;25(2):111–117. doi: 10.1046/j.1365-2257.2003.00503.x. [DOI] [PubMed] [Google Scholar]
- 125.Iwanaga M, Watanabe T, Utsunomiya A, et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood. 2010;116(8):1211–1219. doi: 10.1182/blood-2009-12-257410. [DOI] [PubMed] [Google Scholar]
- 126.Etoh K, Tamiya S, Yamaguchi K, et al. Persistent clonal proliferation of human T-lymphotropic virus type I-infected cells in vivo. Cancer Res. 1997;57(21):4862–4867. [PubMed] [Google Scholar]
- 127.Roth A, Durig J, Himmelreich H, et al. Short telomeres and high telomerase activity in T-cell prolymphocytic leukemia. Leukemia. 2007;21(12):2456–2462. doi: 10.1038/sj.leu.2404968. [DOI] [PubMed] [Google Scholar]
- 128.Keller G, Brassat U, Braig M, et al. Telomeres and telomerase in chronic myeloid leukaemia: impact for pathogenesis, disease progression and targeted therapy. Hematol Oncol. 2009;27(3):123–129. doi: 10.1002/hon.901. [DOI] [PubMed] [Google Scholar]
- 129.Sellmann L, de BD, Bartels M, et al. Telomeres and prognosis in patients with chronic lymphocytic leukaemia. Int J Hematol. 2011;93(1):74–82. doi: 10.1007/s12185-010-0750-2. [DOI] [PubMed] [Google Scholar]
- 130.Bashash D, Ghaffari SH, Mirzaee R, et al. Telomerase inhibition by non-nucleosidic compound BIBR1532 causes rapid cell death in pre-B acute lymphoblastic leukemia cells. Leuk Lymphoma. 2013;54(3):561–568. doi: 10.3109/10428194.2012.704034. [DOI] [PubMed] [Google Scholar]
- 131.Bazarbachi A, Nasr R, El-Sabban ME, et al. Evidence against a direct cytotoxic effect of alpha interferon and zidovudine in HTLV-I associated adult T cell leukemia/lymphoma. Leukemia. 2000;14(4):716–721. doi: 10.1038/sj.leu.2401742. [DOI] [PubMed] [Google Scholar]
- 132.Kchour G, Makhoul NJ, Mahmoudi M, et al. Zidovudine and interferon-alpha treatment induces a high response rate and reduces HTLV-1 proviral load and VEGF plasma levels in patients with adult T-cell leukemia from North East Iran. Leuk Lymphoma. 2007;48(2):330–336. doi: 10.1080/10428190601071717. [DOI] [PubMed] [Google Scholar]
- 133.Bazarbachi A, Hermine O. Treatment of adult T-cell leukaemia/lymphoma: current strategy and future perspectives. Virus Res. 2001;78(1–2):79–92. doi: 10.1016/s0168-1702(01)00286-6. [DOI] [PubMed] [Google Scholar]