

Abstract
We have developed a thin layer, multiplexed biosensing platform that features two working-electrode arrays for detecting small molecules, nucleic acid sequences, and DNA-binding proteins. DNA duplexes are patterned onto the primary electrode array, while a secondary electrode array is used both to initiate DNA monolayer formation, and for electrochemical readout via DNA-mediated charge transport (DNA CT) chemistry. Electrochemical reduction of Cu(phendione)22+ (phendione is 1,10-phenanthroline-5,6-dione) at the secondary electrodes induces covalent attachment via click chemistry of ethynyl-labeled DNA probe duplexes onto the primary electrodes that have been treated with azide-terminated alkythiols. Electrochemical impedance spectroscopy and cyclic voltammetry confirm that catalyst activation at the secondary electrode is essential to maintain the integrity of the DNA monolayer. Electrochemical readout of DNA CT processes that occur at the primary electrode is accomplished at the secondary electrode. The two-electrode system enables the platform to function as a collector-generator using either ferrocyanide or ferricyanide as mediators with methylene blue and DNA charge transport. Electrochemical measurements at the secondary electrode eliminate the need for large background corrections. The resulting sensitivity of this platform enables the reliable and simultaneous detection of femtomoles of the transcription factors TATA-binding protein and CopG on a single multiplexed device.
INTRODUCTION
Analytical methods for reliable biomolecule detection are becoming increasingly important with the continued discovery of disease-related biomarkers. Electrochemical nucleic acid-based assays,1–10 particularly those that utilize DNA-mediated charge transport (DNA CT)11,12 are especially promising for sensing platforms. Devices based on DNA CT effectively report on the integrity of the π-stacked DNA bases; perturbations to the proper stacking, resulting from lesions, single nucleotide polymorphisms, or protein binding events that affect the base stack, attenuate the electrochemical signal. DNA CT has been employed successfully in the detection of a variety of biomolecules, including DNA fragments, chemically modified DNA, and DNA-binding proteins, many of which are not specifically detectable using alternative sensing platforms.11,13
Substrates for DNA-based electrochemical systems typically are prepared by self-assembling thiolated DNA duplexes onto gold electrodes, followed by backfilling with an alkylthiol to passivate any remaining surface-exposed gold.14–16 One major challenge with this methodology is the limited control over monolayer composition, both in the total amount of DNA assembled and its dispersion within the monolayer.17–19 For biosensing applications, which rely on direct interactions between target biomolecules and the DNA duplexes attached to the electrode surface, adequate spacing between the DNA duplexes is critical to provide the target sufficient access. Inevitably, clustering occurs with thiolated DNA. Recently, we demonstrated the utility of applying mixed alkylthiol monolayers doped with variable amounts of azide-terminated functional groups to gain more control over monolayer formation.20 Subsequent coupling via conjugation of cyclooctyne-labeled DNA yields surfaces containing evenly dispersed DNA with coverages that mirror the mole fraction of azide in the underlying film. The resulting monolayers allow greater access of DNA-binding proteins to individual helices within the films, permitting devices with greater sensitivity to these biomolecules.
While tethering DNA to surfaces with cyclooctyne provides a strong foundation for more controlled monolayer formation, ideally, DNA probe molecules would feature a simple terminal alkyne group to avoid additional synthetic steps. The well known Huisgen 1,3-dipolar cycloaddition (“click” reaction)21 catalyzed by copper(I) has been used previously to form homogenous monolayers using terminal alkyne-labeled probe molecules.22,23 Indeed, because of the instability of copper(I) in aqueous solution and its reactivity with DNA,24–26 electrochemical methods to generate copper(I) in situ from copper(II) precursors have been developed, and the coupling of alkyne-labeled oligonucleotides to azide-terminated surfaces via electrochemically induced click chemistry has been reported.27–30
We have now employed a two-electrode platform in which simple alkyne-labeled duplexes are coupled to azide-terminated surfaces by copper(I) species generated in situ at a secondary working electrode positioned over the alkylthiol monolayer.31 Our attempts to fabricate surfaces suitable for DNA CT using published methods with a single working electrode were unsuccessful: reliable electrochemical readout of DNA-mediated chemistry was hampered by interference from the irreversible products of copper(II) reduction at the modified electrochemical surface (likely adsorption of copper films onto the electrode surface). Our new method31 allows for the attachment of multiple DNA sequences onto a single electrode, with tight control over the probe-molecule spacing.32 A multiplexed version of this methodology has enabled the sensitive detection of DNA methyltransferase activity directly from human tissue samples.33
Here we report the full characterization of this multiplexed, two working-electrode platform. In addition to minimizing undesirable copper byproducts at DNA-modified surfaces, we have found that readout of electrocatalytically generated reporter molecules at the secondary electrode greatly enhances the sensitivity and specificity of DNA CT assays. Notably, this mode of detection eliminates large background signals, and hence the requirement for sophisticated data processing. This platform enables detection of single base mismatches as well as the selective and specific detection of two transcription factors, TATA binding protein (TBP) and CopG, with sensitivities significantly greater than those achieved using single working-electrode platforms.
EXPERIMENTAL
Design of Experimental Platform
The multiplexed, two-electrode array was fabricated from two ¼” Teflon blocks separated by a Teflon gasket of various thicknesses (Figure 1). Gold wires (1-mm diameter) were then inserted into holes drilled into the Teflon to form complementary 5 × 3 electrode arrays on each block. Each pair of complementary electrodes was 5 mm from its nearest neighbor, providing the opportunity to isolate each pair into individual wells. The electrodes were sealed into the Teflon using superglue. The top array featured additional holes (1.5-mm diameter) to provide the reference and auxiliary electrodes access to the working solution. Spacers that separated the individual wells were constructed from 1.5 mm thick Teflon.
Figure 1.

Schematic diagram of the thin-layer, two-electrode sensing platform. The multiplexed platform contains two 5 × 3 arrays of 1-mm diameter gold electrodes embedded in Teflon blocks, separated by a Teflon spacer to form a solution well. The top (secondary) array contains four holes into which reference and auxiliary electrodes can be inserted. The bottom (primary) array features the same number and positioning of electrodes as the top array.
DNA Attachment to Alkanethiol Monolayers
A 10 mM aqueous Cu(phendione)22+ (phendione=1,10-phenanthroline-5,6-dione) solution was prepared by combining one equivalent of CuSO4 (10 μmol, 15.9 mg) with two equivalents of phendione (20 μmol, 42.0 mg) in 10 mL of deionized H2O. ESI-MS: 580.2 (calc: 580.0). The complex was additionally isolated as the PF6− salt. Prior to application to the electrode surface, the complex was diluted to a final concentration of 1 mM in Tris buffer (10 mM Tris, 100 mM KCl, 2.5 mM MgCl2, 1 mM CaCl2, pH 7.6). The catalyst solution was combined with 5′-labeled ethynyl DNA (final concentration of 25 μM), and a constant potential of −350 mV v. AgCl/Ag was applied to the sensing (top) electrode array to reduce the Cu(II) and initiate the coupling of the DNA to the azide-terminated monolayers. The potential was applied for 15 minutes. Multiple sequences of DNA were attached to the array through the sequential activation of different secondary electrodes. For example, well matched and mismatched DNA were attached to the same array through the preliminary activation of secondary electrodes 1 – 9 in the presence of well matched DNA, followed by rinsing of the platform and subsequent activation of secondary electrodes 10 – 15 in the presence of DNA containing a single-base mismatch.
Characterization of DNA-modified Monolayers
All electrochemical experiments were performed on a CH Instruments 760E bipotentiostat. For electrochemical impedance spectroscopy experiments, 400 μM potassium ferricyanide in phosphate buffer (5 mM phosphate, 50 mM NaCl, pH 7.0) was used. For all other experiments, electrochemistry was conducted in Tris buffer (10 mM Tris, 100 mM KCl, 2.5 mM MgCl2, 1 mM CaCl2, pH 7.6) with 4 μM methylene blue and 300 μM potassium ferricyanide; for experiments with covalent Nile Blue, methylene blue was omitted. For mismatch discrimination and protein binding experiments, constant potential amperometry was used, with potential applied for 90 s. The primary electrode was held at −400 mV v. AgCl/Ag, and the secondary electrode was held at 350 mV v. AgCl/Ag.
RESULTS AND DISCUSSION
We have designed and fabricated an addressable, multiplexed biosensing array that features two sets of complementary electrodes separated by a thin film (Figure 1). The bottom electrodes, which comprise the primary array, are modified with covalently bound DNA sequences dispersed within a mixed alkylthiol monolayer, while the top electrodes, which form the secondary array, are unmodified and are used both for activating a DNA-coupling catalyst and for the electrochemical readout. An overview of the steps required for the primary electrode-array modification, DNA coupling, and electrocatalytic detection using this two-electrode sensing platform are summarized in Scheme 1.
Scheme 1.
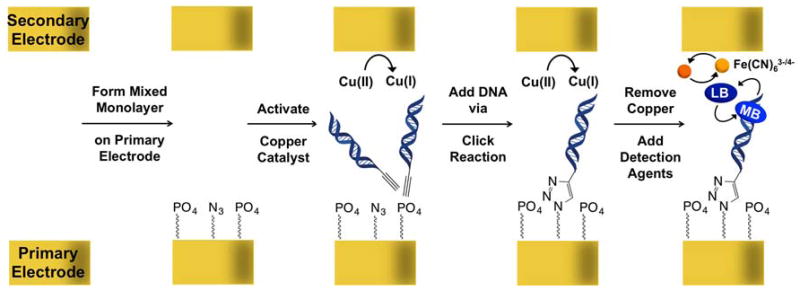
General strategy for monolayer formation and detection. First, a mixed alkanethiol monolayer is formed on the primary electrode array containing phosphate and azide head groups. Subsequently, a copper catalyst for click chemistry is activated at the secondary electrode to enable the click reaction to proceed between the azides on the surface and alkyne-modified DNA. Finally, all copper is removed and methylene blue and ferricyanide are added for electrochemical detection.
Electrochemical Response of the Coupling Catalyst
The electrochemical initiation of azide/alkyne coupling via copper(II) reduction has been explored previously.34 While several chelating ligands for copper(I) click chemistry have been reported, our experiments have focused on the bipyridyl derivative 1,10-phenanthroline-5,6-dione (phendione), as this ligand is commercially available and yields a water-soluble complex. The cyclic voltammogram (CV) of [Cu(phendione)2][SO4] in Tris buffer shows several copper-centered reductions between + 0.20 and −0.40 V vs. AgCl/Ag (Figure S1). Importantly, these processes are only partially chemically reversible. The CV also shows a coupled oxidative response with a shape characteristic of anodic stripping, suggesting that the electrochemical reactions result in the adsorption of at least some copper-containing species onto the electrode surface. The deposition of copper following electrochemical reduction is further supported by the formation of a visible, black surface film following the application of potentials in the range of −0.30 to −0.40 V vs. AgCl/Ag.
Formation of DNA Monolayers with Activation from Primary vs. Secondary Electrodes
DNA-modified surfaces were prepared on the primary array using a two-step process that involved: (i) the self-assembly of a mixed alkylthiol monolayer containing 50% azide and 50% phosphate head groups onto the primary electrodes, followed by (ii) the electrochemical reduction of Cu(phendione)22+ at the top (secondary) electrodes in the presence of alkyne-labeled DNA duplexes. Electrogenerated copper(I) induces covalent attachment of the DNA via alkyne/azide coupling (Scheme 1). Based on previous studies, an underlying 1:1 ratio of azide-to-phosphate head groups provides adequate spacing between the individual helices for substrate access within the DNA monolayer, while still maintaining a sufficient concentration of DNA on the surface for reliable detection.20 Importantly, before attachment of the DNA duplexes, the azide/phosphate-modified primary electrodes showed no electrochemical response in the presence of up to 8 μM methylene blue (MB) and/or 500 μM Fe(CN)63−.
The catalytic precursor, Cu(phendione)22+ (1 mM), was activated by applying a potential of −350 mV (vs. AgCl/Ag) to the secondary electrodes for 15 minutes. After rinsing the system with Tris buffer, cyclic voltammetry at the primary electrodes revealed reproducible and featureless background currents, characteristic of DNA-modified electrodes. No signals attributable to copper-reduction byproducts were observed. By contrast, if the Cu(phendione)22+ activation was carried out at the primary (bottom) electrodes instead, the resulting cyclic voltammograms exhibited large, sloping, and irreproducibly “bumpy” signals, even after multiple rinsings. This observation suggests partial (and variable) passivation of the primary-electrode surfaces due to the irreversible copper(II) electrochemistry.
Electrochemical impedance spectroscopy (EIS)36 was also performed to evaluate the effect of copper-film deposition on the electrochemical properties of DNA-modified electrodes. Data were collected at DNA-modified surfaces prepared via Cu(phendione)22+ activation at both the primary and secondary electrodes, and compared to analogous data gathered at bare gold and at electrodes modified with only the underlying mixed alkylthiol monolayers. EIS was conducted at the Fe(CN)63−/4− redox couple, following literature protocols for the label-free detection of DNA.35, 36 Nyquist plots (Figure S2) constructed from measurements recorded at both bare gold surfaces and electrodes modified only with the underlying alkylthiols display a small impedance arc, consistent with low surface capacitance and a response dominated by the diffusion of Fe(CN)63−/4− species to the surface.36–39 Alternatively, the conjugation of DNA duplexes onto the primary gold monolayers provides an electrostatic barrier that effectively passivates the negatively charged Fe(CN)63−/4− complexes from the electrode surface; the corresponding Nyquist plots therefore show a significantly larger capacitive arc. Notably, the plots from DNA monolayers formed via catalyst activation at the primary electrodes show the largest (and least reproducible) arcs, indicating higher electron-transfer resistance through these films than through the analogous DNA monolayers prepared via activation from the secondary array (Figure S2). These results are fully consistent with the cyclic voltammetry of Cu(phendione)22+, and with the visible appearance of black deposits on whichever array was used to activate the copper(II). Importantly, when the secondary electrodes were used for activation, the precipitate could be easily removed by polishing the array prior to detection, without perturbing the DNA monolayers on the primary-electrode surfaces.
Electrochemical Readout at the Primary Electrode
In order to amplify the electrochemical readout from DNA CT-based sensors, we previously developed an electrocatalytic cycle using MB and Fe(CN)63− in solution.12 The cycle begins with the DNA-mediated electrochemical reduction of intercalated MB. The product of that reaction, leucomethylene blue (LB), has a lower affinity for DNA and dissociates from the π-stack. LB then reduces Fe(CN)63− to Fe(CN)64− in solution, thereby regenerating MB that re-intercalates into the film to begin the cycle again. Under certain experimental conditions, 39 the measured current is limited only by the diffusion of ferricyanide in solution, providing dramatically larger signals than those obtained by the direct surface electrochemistry of MB at the DNA-modified electrode.
In order to evaluate the utility of this electrocatalytic cycle in our two-electrode platform, we prepared a series of primary electrodes that contained either well-matched (WM) or mismatched (MM) 18-base-pair DNA duplexes (see SI for sequences). We then monitored the thin-film MB/Fe(CN)63− electrochemical response at both the primary and secondary electrode arrays. As DNA CT is sharply attenuated by lesions within the π-stack, the ability to discriminate between WM and MM monolayers provides strong evidence for a DNA-mediated process.
Figure 2 shows representative results when the current is measured at the primary electrode. Because of the low surface coverage of DNA, the relatively large background currents, and the restricted diffusion of Fe(CN)63− owing to the proximity of the secondary electrode, the cyclic voltammograms do not show the characteristic electrocatalytic wave shapes observed in the absence of the secondary array. Nevertheless, integrating the measured current traces reveals a marginally higher net-charge passed through the well- vs. mismatched-DNA films. (As also illustrated in Figure 2, if instead the DNA films are formed via copper(II) activation at the primary electrodes, the background signals are even larger, and the differences in charge passed through well- vs. mismatched DNA are correspondingly smaller.) Thus, while mismatch discrimination can be accomplished using electrochemical readout at the primary electrodes, the signal amplification afforded by the addition of Fe(CN)63− is essentially only stoichiometric, so the MB/Fe(CN)63− signals remain very small relative to the much larger background currents. As with other systems where a single electrode is used both for target capture and electrochemical readout, reliable mismatch detection at the primary array would require extensive background subtraction of the large non-Faradaic currents that dominate the electrochemical response in order to discern the subtle differences in the (much smaller) Faradaic signals that occur at well- vs. mismatched sequences.
Figure 2.
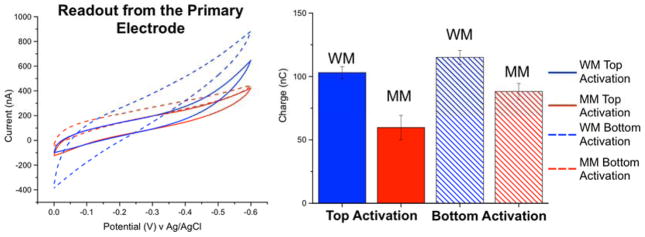
(Left) Cyclic voltammetry (ν = 100 mV/s) of 4 μM MB/300 μM [K]3[Fe(CN)6] in Tris buffer, using the two-electrode multiplexed platform with a 127-μm spacer separating the primary- and secondary-electrode arrays. Data were recorded at four separate primary electrodes featuring an underlying 50/50 azide/phosphate monolayer, covalently modified with either well-matched (blue) or mismatched (red) alkyne-labeled DNA duplexes (18-mers). The solid traces were obtained at electrodes prepared by Cu(phendione)22+ activation at the corresponding secondary electrodes, while the dashed traces were obtained by activating Cu(phendione)22+ directly at the primary electrodes. (Right) Charges obtained by integrating the cyclic voltammograms. Electrodes prepared by catalyst activation at the secondary electrode display modest, though significantly better mismatch discrimination.
Electrochemical Readout at the Secondary Electrode
In contrast, using a secondary array for electrochemical readout offers an inherently more sensitive detection platform, with virtually no interference from background currents. Because the secondary-electrode array effectually traps solution-bourn analytes within the thin layer created between the primary and secondary electrodes, holding the secondary electrodes at potentials positive of the Fe(CN)63−/4− couple should continually replenish Fe(CN)63− in the vicinity of the DNA films by re-oxidizing Fe(CN)64− as soon as it is generated during the electrocatalytic cycle. This would enable multiple rounds of turnover and a correspondingly higher level of amplification. Although the rate of MB/Fe(CN)63− electrocatalysis is limited by the DNA-binding dynamics of MB under the experimental conditions used in this study, 39 the two-electrode platform offers, in principle, the possibility of secondary-electrode readout currents that are larger than would be possible using passive diffusion.
Indeed, as demonstrated in Figure 3, poising the secondary electrode at +0.35 V (i.e., 0.1 V positive of the Fe(CN)63−/4− couple) during the electrocatalytic assay enables the system to function in this “collector-generator” mode: upon reduction of Fe(CN)63− in solution by the LB produced via DNA-mediated reduction of MB at the primary electrode, the resulting Fe(CN)64− is immediately re-oxidized at the secondary electrode to generate a large, “turn-on” readout current. In contrast, if no DNA CT occurs at the primary electrode, no current is generated at the secondary electrode, negating the need for any background correction. Because of the thin layer created by sandwiching the primary and secondary arrays together, the platform can function as a collector-generator when starting either with Fe(CN)64− (Figure 3b), as was done previously using scanning electrochemical microscopy, or with Fe(CN)63− (Figure 3a), which is rapidly converted to Fe(CN)64− via DNA CT at the primary electrode surface.
Figure 3.

Detection strategy (left) and constant-potential amperometry assay (right) for DNA CT using the two-electrode detection platform. Electrochemical readout is carried out at a single, addressable electrode in the secondary array held at +0.35 V. In (a), the thin layer between the primary and secondary electrode arrays contains 4 μM MB and 300 μM Fe(CN)63−. Because the applied potential is positive of the Fe(CN)63−/4− redox couple, there is no initial current (shown in red) at the secondary electrode. 15 seconds into the experiment, the entire primary electrode array (current shown in black) is activated at a potential of −0.40 V. This initiates the MB/Fe(CN)63− electrocatalytic cycle, and current begins to flow at the secondary electrode due to the re-oxidation of Fe(CN)64− generated at the primary array. For sensing applications, readout at the secondary electrode is measured only after a steady-state current is achieved (~ 90 s) to eliminate any complications arising from double-layer charging effects. The lower panel (b) shows the analogous experiment, except that the thin-layer solution initially contains 4 μM MB and 300 μM Fe(CN)64−. When the secondary electrode is turned on at time zero, there is an initial current (shown in green) due to the oxidation of Fe(CN)64− to Fe(CN)63−. This current rapidly approaches zero as the Fe(CN)64− is converted to Fe(CN)63− within the thin layer. At the 15-s mark, the primary array is turned on, and the assay proceeds as in (a).
Note that for this platform to function as a collector-generator, the distance between the two sets of electrodes must be sufficiently close to prevent the Fe(CN)63−/4− species from diffusing away. In scanning electrochemical microscopy (SECM), the optimal distance between the tip and the electrode surface is determined by an approach curve.40,41 Here, we have experimentally determined the optimal distance between our two sets of working electrodes by carrying out the analysis using various thicknesses of the Teflon spacer that separates the two arrays (Figure S3). Empirically, the spacer that simultaneously yielded the largest readout currents at the secondary electrodes while providing the greatest level of mismatch discrimination was 127 μm thick, which we therefore used for all experiments. It may be the case that if the spacer is too small, the electrode arrays are too close to one another, and our signals are no longer DNA-mediated. This is likely the case with the 50 μm spacer, with which we do not see mismatch discrimination.
Single-Base Mismatch Detection with Non-covalent and Covalent Redox Probes
To evaluate the ability of this platform to detect single-base mismatches, we prepared a primary-electrode array that featured the side-by-side conjugation of both well- and mismatched alkyne-labeled DNA duplexes at adjacent electrode sites. DNA addressing was readily accomplished by activating the copper(II) coupling catalyst only at specific (complementary) secondary electrodes for each DNA sequence used. As shown in Figure 4, electrochemical readout was then carried out using constant-potential amperometry at the secondary electrodes, in the presence of MB/Fe(CN)63−.33 The differences in readout currents generated by well- vs. mismatched duplexes are both large and robust. (For comparison, if the monolayers are instead addressed by copper(II) activation at the primary electrodes, the resulting readout currents are small, and mismatch discrimination is poor; this further highlights the importance of catalyst activation from the secondary electrode.) These signal differences obviate the need for dramatic background subtractions, essentially providing an on/off sensor for mismatch detection.
Figure 4.
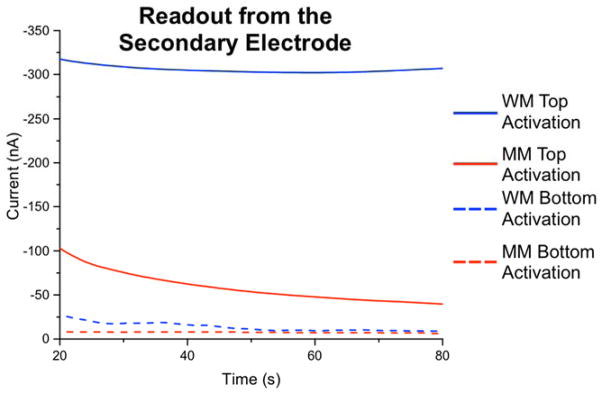
Mismatch detection with electrochemical readout at the secondary electrode. Constant-potential amperometry was conducted at the secondary electrodes with an applied potential of +0.35 V, while the primary-electrodes were held at −0.40 V in the presence of 4 μM MB and 300 μM Fe(CN)63−. The blue traces represent currents measured at readout electrodes complementary to well-matched DNA sequences (18-mers) on the primary array, while the red traces represent the analogous currents generated at electrodes complementary to mismatched duplexes. For comparison (dashed lines), the identical assay was carried out on a separate array in which the DNA duplexes were conjugated via copper(II) activation directly at the primary electrodes; clearly, greater signal differential between well- vs, mismatched sequences occurs when monolayers are formed by catalyst activation at the secondary electrode. In these experiments, all primary-array electrodes were first modified with an underlying 50/50 azide/phosphate monolayer before DNA conjugation.
We also investigated the possibility of replacing MB with a covalent redox probe, which may be necessary for some biomolecule detection applications. Nile Blue, a covalent reporter molecule that is electronically conjugated to the DNA π-stack,48 was previously found to mediate the electrocatalytic reduction of Fe(CN)63− in solution. As shown in Figure 5, replacing MB with NB in the two-electrode platform still allows for robust mismatch discrimination, despite the fact that the NB is covalently tethered to DNA. Based on electrochemical readout at the secondary electrodes, a 60±10% decrease in the current upon incorporation of a single-base mismatch occurs with the covalent Nile blue redox probe, as compared to an 80±10% decrease with noncovalent MB. This difference in mismatch discrimination is likely due to the higher binding stoichiometry of methylene blue to DNA, as well as its enhanced mobility within the film. It is therefore unsurprising that MB yields larger signals and a correspondingly larger differential. This result is consistent with previous single-electrode studies, in which larger differentials are observed with free probes than with covalent probes.48,49
Figure 5.

Comparison of mismatch discrimination recorded with the two-electrode platform using methylene blue vs. Nile blue redox reporters. (a) Electrochemical readout recorded at a secondary electrode during the electrocatalytic reduction of 300 μM Fe(CN)63− in the presence of 4 μM MB at primary electrodes modified with well-matched DNA duplexes (blue), and mismatched DNA duplexes (red). (b) The same assay, but with Nile blue covalently bound to the DNA probe sequences substituting for MB. Percent signal changes were calculated from the amperometry data recorded 90 seconds after initiation of the electrochemical readout.
Detection of DNA-binding Proteins
To establish the relevance of this platform for biomolecule detection, DNA-binding protein detection was investigated using two transcription factors, TBP and CopG. Both proteins bind to specific sequences of DNA, kinking the duplex to a large degree, and are ideal targets for DNA CT-based assays. The transcription factor TBP (TATA-binding protein, a subunit of the eukaryotic TFIID transcription factor) was previously employed as a measure of sensitivity for a DNA CT-based protein-detection platform.50,51 TBP kinks DNA by over 80° when bound to its TATA target sequence, 52 destacking the DNA bases and disrupting DNA CT.20 Likewise, the transcription repressor CopG, which binds DNA as a tetramer at an ACGTxxxxxACGT site, bends the helix up to 120° and also has a nano-molar binding affinity.53–55
To determine detection limits using the two-electrode assay, each protein was titrated individually into the two-electrode platform, and electrochemical readout at the secondary-electrode during MB/Fe(CN)63− electrocatalysis was used to monitor protein binding. The titration data, shown in Figure 6, indicate a greater than 30% decrease in the readout signals associated with both proteins at concentrations as low as 10 nM; based on the small volumes required to fill the thin layer between the two electrode arrays, this translates to less than 50 femtomoles of protein. From these titration curves, dissociation constants can be calculated for protein binding at the DNA-modified surfaces, using a cooperative binding model (the Hill model).20 TBP has a surface KD of 14±2 nM, and CopG has a surface KD of 17±4 nM. Both TBP and CopG are therefore detectable at concentrations near their solution KD’s (3.3 nM and 10 nM, respectively). 50
Figure 6.
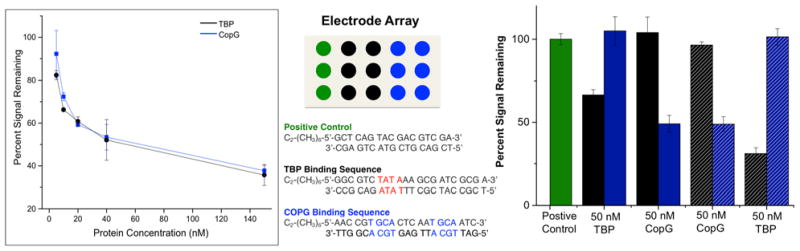
Titration of transcription factors TATA-binding protein (TBP) and CopG on two-electrode array. Each protein was titrated onto an array in concentrations ranging from 0 nM to 150 nM. Shown (left) are the percent electrochemical signal remaining from constant potential amperometry plotted as a function of protein concentration. The proteins caused significant signal decreases on the arrays in very low nanomolar concentrations. All electrochemistry was conducted in Tris buffer (10 mM Tris, 100 mM KCl, 2.5 mM MgCl2, 1 mM CaCl2, pH 7.6) with 4 μM methylene blue and 300 μM K3[Fe(CN)6]. Constant potential amperometry at the secondary electrode was conducted with an applied potential of 350 mV v. AgCl/Ag to the secondary electrode and −400 mV v. AgCl/Ag to the primary electrode for 90 seconds. Specific detection of transcription factors TBP and CopG on a single array is also shown (right). Arrays are formed with six electrodes modified with DNA containing specific binding sites for each of the transcription factors TBP and CopG, as well as three electrodes containing positive control DNA that does not contain the binding site for either protein. A single protein was added to the surface at a 50 nM concentration, and the signal decrease for each sequence was monitored. The second protein was then added, and signal decreases monitored. TBP detection followed by CopG detection is shown with solid bars, while CopG detection followed by TBP detection is shown with dashed bars. The TBP binding sequence is represented by black, both in terms of the location of the electrodes modified with this sequence on the electrode array (center, black circles) and in terms of the percent signal remaining for this sequence (right, black bars). Independent of which protein was added first, signal decreases were only observed on the sequences to which the specific protein binds.
Notably, the multiplexed nature of this sensing platform enables the specific detection of multiple proteins simultaneously, using a single primary-electrode array. To illustrate this capability, each transcription factor was added to a primary array that featured: (i) three electrodes modified with a non-binding DNA sequence; (ii) six electrodes modified with DNA containing a TBP binding site; and (iii) six electrodes modified with DNA containing a CopG binding site. The assay itself was carried out by introducing one protein (either TBP or CopG) first onto the platform, then measuring the secondary-electrode readout at addresses that correspond to the locations of the DNA-binding sequences, as well as at the locations of the non-binding sequences (the positive controls). Any signal attenuation was measured relative to the readout current at the positive-control addresses. The surface was subsequently rinsed, the second protein was added, and the readout signal attenuation was again measured. In each case, signal attenuation occurred only at secondary electrodes that were complementary to the primary addresses which featured the binding sequence of the protein added. This observation verifies that changes are due only to the specific binding of each protein. We note that greater variability between protein-detection experiments is observed in the dual protein detection experiments, as compared to the individual titrations, likely because of the added normalization of these data to a non-binding positive control sequence. This additional normalization is necessary for combined protein experiments to further ensure that signal decreases are due to specific binding of only one of the proteins.
CONCLUSIONS
We have described a highly sensitive, multiplexed, two-working-electrode platform for DNA CT-based electrochemical detection. Using in situ electrochemical activation of a copper(II) catalyst for Huisgen 1,3-dipolar coupling, this platform enables the addressing of low-density DNA monolayers onto specific sites within a single, primary sensing array. Catalyst activation at a secondary electrode is essential to maintain the integrity of the DNA, as shown by cyclic voltammetry, EIS, and constant-potential amperometry. Detection and electrochemical readout from the secondary-electrode array is similarly necessary to provide high sensitivity without large background signals. Operating in collector-generator mode using the well-known MB/Fe(CN)63− electrocatalytic cycle for DNA-modified surfaces, this platform provides a “signal-on” detection assay for DNA CT, with virtually no background currents whatsoever. Notably, this platform allows the specific and simultaneous detection of femtomoles of the transcription factors, TBP and CopG in a single electrochemical assay. The new multiplexed, two-electrode detection platform significantly broadens the scope and applications for detection using DNA CT.
Supplementary Material
Acknowledgments
We are grateful to the NIH (GM61077) for their financial support of this research.
Footnotes
Supporting Information Available: Additional experimental details as well as a CV of the copper precatalyst, impedance spectrometry data of the DNA monolayers, and spacer height optimization data are available.
References
- 1.Wang J. Electrochemical nucleic acid biosensors. Analytica Chimica Acta. 2002;469:63–71. [Google Scholar]
- 2.Liu J, Cao Z, Lu Y. Functional Nucleic Acid Sensors. Chem Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lam JCF, Aguirre S, Li Y. Nucleic acids as detection tools. Chem Biol of Nucl Acids. 2010:401–431. [Google Scholar]
- 4.Gorodetsky AA, Buzzeo MC, Barton JK. DNA-mediated Electrochemistry. Bioconjugate Chem. 2008;19:2285–2296. doi: 10.1021/bc8003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang H, Hui A, Pampalakis G, Soleymani L, Liu FF, Sargent EH, Kelley SO. Direct, Electronic MicroRNA Detection Reveals Differential Expression Profiles in 30 Minutes. Angew Chem. 2009;48:8461–8464. doi: 10.1002/anie.200902577. [DOI] [PubMed] [Google Scholar]
- 6.Soleymani L, Fang Z, Sun X, Yang H, Taft BJ, Sargent EH, Kelley SO. Nanostructuring of Patterned Microelectrodes To Enhance the Sensitivity of Electrochemical Nucleic Acids Detection. Angew Chem Int Ed. 2009;48:8457–8460. doi: 10.1002/anie.200902439. [DOI] [PubMed] [Google Scholar]
- 7.Kelley SO, Mirkin CA, Walt DR, Ismagilov RF, Toner M, Sargent EH. Advancing the Speed, Sensitivity and Accuracy of Biomolecular Detection. Nat Nanotech. 2014;9:969–980. doi: 10.1038/nnano.2014.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Das J, Cederquist KB, Lee P, Sargent EH, Kelley SO. An Ultrasensitive, Universal Detector Based on Neutralizer Displacement. Nat Chem. 2012;4:642–648. doi: 10.1038/nchem.1367. [DOI] [PubMed] [Google Scholar]
- 9.Gooding JJ. Electrochemical DNA Hybridization Biosensors. Electroanalysis. 2002;14:1149–1156. [Google Scholar]
- 10.Pei H, Lu N, Wen Y, Song S, Liu Y, Yan H, Fan C. A DNA Nanostructure-based Biomolecular Probe Carrier Platform for Electrochemical Biosensing. Adv Mater. 2010;22:4754–4758. doi: 10.1002/adma.201002767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drummond TG, Hill MG, Barton JK. Electrochemical DNA Sensors. Nat Biotechnol. 2003;21:1192–1199. doi: 10.1038/nbt873. [DOI] [PubMed] [Google Scholar]
- 12.Furst AL, Hill MG, Barton JK. Electrocatalysis in DNA Sensors. Polyhedron. 2014;84:150–159. doi: 10.1016/j.poly.2014.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muren NB, Olmon ED, Barton JK. Solution, Surface, and Single Molecule Platforms for the Study of DNA-mediated Charge Transport. Phys Chem Chem Phys. 2012;14:13754–13771. doi: 10.1039/c2cp41602f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kelley SO, Jackson NM, Hill MG, Barton JK. Long-Range Electron Transfer Through DNA Films. Angewandte Chemie. 1999 doi: 10.1002/(SICI)1521-3773(19990401)38:7<941::AID-ANIE941>3.0.CO;2-7. Germ. Ed. 111, 991; Intl. Ed. 38, 941. [DOI] [PubMed] [Google Scholar]
- 15.Levicky R, Herne TM, Tarlov MJ, Satija SK. Using Self-Assembly to Control the Structure of DNA Monolayers on Gold: A Neutron Reflectivity Study. J Am Chem Soc. 1998;120:9787–9792. [Google Scholar]
- 16.Abi A, Ferapontova EE. Unmediated by DNA electron transfer in redox-labeled DNA duplexes end-tethered to gold electrodes. J Am Chem Soc. 2012;134:14499–14507. doi: 10.1021/ja304864w. [DOI] [PubMed] [Google Scholar]
- 17.Murphy JN, Cheng AKH, Yu HZ, Bizzotto D. On the Nature of DNA Self-Assembled Monolayers on Au: Measuring Surface Heterogeneity with Electrochemical in Situ Fluorescence Microscopy. J Am Chem Soc. 2009;131:4042–4050. doi: 10.1021/ja808696p. [DOI] [PubMed] [Google Scholar]
- 18.Ricci F, Lai RF, Heeger AJ, Plaxco KW, Sumner JW. Effect of Molecular Crowding on the Response of the Electrochemical DNA Sensor. Langmuir. 2007;23:6827–6834. doi: 10.1021/la700328r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sam M, Boon EM, Barton JK, Hill MG, Spain EM. Morphology of 15-mer Duplexes Tethered to Au(111) using Scanning Probe Microscopy. Langmuir. 2001;17:5727–5730. [Google Scholar]
- 20.Furst AL, Hill MG, Barton JK. DNA-Modified Electrodes Fabricated Using Copper-Free Click Chemistry for Enhanced Protein Detection. Langmuir. 2013;29:16141–16149. doi: 10.1021/la403262v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huisgen R, Grashey R, Sauer J. Cycloaddition reactions of alkenes. In: Patai S, editor. The Alkenes. Vol. 1. John Wiley & Sons, Ltd; Chichester, UK: 1964. [Google Scholar]
- 22.Collman JP, Devaraj NK, Chidsey CED. “Clicking” Functionality onto Electrode Surfaces. Langmuir. 2004;20:1051–1053. doi: 10.1021/la0362977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerasimov JY, Lai RY. Design and characterization of an electrochemical peptide-based sensor fabricated via ‘click’ chemistry. Chem Commun. 2011;47:8688–8690. doi: 10.1039/c1cc12783g. [DOI] [PubMed] [Google Scholar]
- 24.Shuman MS, Woodward GP., Jr Stability constants of copper-organic chelates in aquatic samples. Environ Sci Technol. 1977;11:809–813. [Google Scholar]
- 25.Meijler MM, Zelenko O, Sigman DS. Chemical Mechanism of DNA Scission by (1,10-Phenanthroline)copper. Carbonyl Oxygen of 5-Methylenefuranone Is Derived from Water. J Am Chem Soc. 1997;119:1135–1136. [Google Scholar]
- 26.Kazakov SA, Astashkina TG, Mamaev SV, Vlassov VV. Site-specific cleavage of single-stranded DNAs at unique sites by a copper-dependent redox reaction. Nature. 1988;335:186–188. doi: 10.1038/335186a0. [DOI] [PubMed] [Google Scholar]
- 27.Devaraj NK, Miller GP, Ebima W, Kakaradov B, Collman JP, Kool ET, Chidsey ED. Chemoselective Covalent Coupling of Oligonucleotide Probes to Self-Assembled Monolayers. J Am Chem Soc. 2005;127:8600–8601. doi: 10.1021/ja051462l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Devaraj NK, Dinolfo PH, Chidsey CED, Collman JP. Selective Functionalization of Independently Addressed Microelectrodes by Electrochemical Activation and Deactivation of a Coupling Catalyst. J Am Chem Soc. 2006;128:1794–1795. doi: 10.1021/ja058380h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Canete SJ, Lai RY. Fabrication of an electrochemical DNA sensor array via potential-assisted ‘click’ chemistry. Chem Commun. 2010;46:3941–3943. doi: 10.1039/c002371j. [DOI] [PubMed] [Google Scholar]
- 30.Ripert M, Farre C, Chaix C. Selective functionalization of Au electrodes by electrochemical activation of the ‘click’ reaction catalyst. Electrochimica Acta. 2013;91:82–89. [Google Scholar]
- 31.Furst AL, Landfield S, Hill MG, Barton JK. Electrochemical Patterning and Detection of DNA Arrays on a Two-Electrode Platform. J Am Chem Soc. 2013;135:19099–19102. doi: 10.1021/ja410902j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quinton D, Maringa A, Griveau S, Nyokong T, Beioui F. Surface patterning using scanning electrochemical microscopy to locally trigger a “click” chemistry reaction. Electrochem Commun. 2013;31:112–115. [Google Scholar]
- 33.Furst AL, Muren NB, Hill MG, Barton JK. Label-free electrochemical detection of human methyltransferase from tumors. Proc Natl Acad Sci USA. 2014;111:14985–14989. doi: 10.1073/pnas.1417351111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hong V, Presolski SI, Ma C, Finn MG. Analysis and Optimization of Copper-Catalyzed Azide-Alkyne Cycloaddition for Bioconjugation. Angew Chem Int Ed. 2009;48:9879–9883. doi: 10.1002/anie.200905087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Janek RP, Fawcett WR. Impedance Spectroscopy of Self-Assembled Monolayers on Au(111): Sodium Ferrocyanide Charge Transfer at Modified Electrodes. Langmuir. 1998;14:3011–3018. [Google Scholar]
- 36.Ceres DM, Udit AK, Hill HD, Hill MG, Barton JK. Differential Ionic Permeation of DNA-Modified Electrodes. J Phys Chem B. 2007;111:663–668. doi: 10.1021/jp066324c. [DOI] [PubMed] [Google Scholar]
- 37.Bard AJ, Faulkner LR. Fundamental and Applications. 2. John Wiley & Sons, Inc; New York: 2001. Electrochemical Methods. [Google Scholar]
- 38.Kafka J, Pänke O, Abendroth B, Lisdat F. A label-free DNA sensor based on impedance spectroscopy. Electrochimica Acta. 2008;53:7467–7474. [Google Scholar]
- 39.Boon EM, Barton JK, Bhaghat V, Nerissian M, Wang W, Hill MG. Reduction of Ferricyanide by Methylene Blue at a DNA-Modified Rotating-Disk Electrode. Langmuir. 2003;19:9255–9259. [Google Scholar]
- 40.Fernandez JL, Bard AJ. Scanning Electrochemical Microscopy. 47. Imaging Electrocatalytic Activity for Oxygen Reduction in an Acidic Medium by the Tip Generation-Substrate Collection Mode. Anal Chem. 2003;75:2967–2974. doi: 10.1021/ac0340354. [DOI] [PubMed] [Google Scholar]
- 41.Martin RD, Unwin PR. Theory and Experiment for the Substrate Generation/Tip Collection Mode of the Scanning Electrochemical Microscope: Application as an Approach for the Measuring the Diffusion Coefficient Ratio of a Redox Couple. Anal Chem. 1998;70:276–284. [Google Scholar]
- 42.Whitworth AL, Mandler D, Unwin PR. Theory of scanning electrochemical microscopy (SECM) as a probe of surface conductivity. Phys Chem Chem Phys. 2005;7:356–365. [Google Scholar]
- 43.Turcu F, Schulte A, Hartwich G, Schuhmann W. Label-free electrochemical recognition of DNA hybridization by means of modulation of the feedback current in SECM. Angew Chem Int Ed. 2004;43:3482–3485. doi: 10.1002/anie.200454228. [DOI] [PubMed] [Google Scholar]
- 44.Wierzbinski E, Arndt J, Hammond W, Slowinski K. In Situ Electrochemical Distance Tunneling Spectroscopy of ds-DNA Molecules. Langmuir. 2006;22:2426–2429. doi: 10.1021/la053224+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu B, Bard AJ, Li CZ, Kraatz HB. Scanning Electrochemical Microscopy. 51. Studies of Self-Assembled Monolayers of DNA in the Absence and Presence of Metal Ions. J Phys Chem B. 2005;109:5193–5198. doi: 10.1021/jp045050v. [DOI] [PubMed] [Google Scholar]
- 46.Wain AJ, Feimeng Z. Scanning electrochemical microscopy imaging of DNA microarrays using methylene blue as a redox-active intercalator. Langmuir. 2008;24:5155–5160. doi: 10.1021/la703922v. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Zhou F. Scanning electrochemical microscopic imaging of surface-confined DNA probes and their hybridization via guanine oxidation. J Electroanalytical Chem. 2002;537:95–102. [Google Scholar]
- 48.Kelley SO, Boon EM, Barton JK, Jackson NM, Hill MG. Single-base mismatch detection based on charge transduction through DNA. Nucleic Acids Res. 1999;27:4830–4837. doi: 10.1093/nar/27.24.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pheeney CG, Barton JK. DNA electrochemistry with tethered methylene blue. Langmuir. 2012;28:7063–7070. doi: 10.1021/la300566x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorodetsky AA, Ebrahim A, Barton JK. Electrical Detection of TATA Binding Protein at DNA-Modified Microelectrodes. J Am Chem Soc. 2008;130:2924–2925. doi: 10.1021/ja7106756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boon EM, Salas JW, Barton JK. An electrical probe of protein-DNA interactions on DNA-modified surfaces. Nature Biotechnology. 2002;20:282–287. doi: 10.1038/nbt0302-282. [DOI] [PubMed] [Google Scholar]
- 52.Hahn S, Bruatowski S, Sharp PA, Guarente L. Yeast TATA-binding protein TFIID binds to TATA elements with both consensus and nonconsensus DNA sequences. Proc Natl Acad Sci USA. 1989;86:5718–5722. doi: 10.1073/pnas.86.15.5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.del Solar G, de la Campa AG, Perez-Martin J, Choli T, Espinosa M. Purification and characterization of RepA, a protein involved in the copy number control of plasmid pLS1. Nucleic Acids Res. 1989;17:2405–2420. doi: 10.1093/nar/17.7.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomis-Ruth FX, Sola M, Acebo P, Parraga A, Guasch A, Eritja R, Gonzalez A, Espinosa M, del Solar G, Coll M. The structure of plasmid-encoded transcriptional repressor CopG unliganded and bound to its operator. EMBO Journal. 1998;17:7404–7415. doi: 10.1093/emboj/17.24.7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Del Solar G, Albericio F, Eritja R, Espinosa M. Chemical synthesis of a fully active transcriptional repressor protein. Proc Natl Acad Sci, USA. 1994;91:5178–5182. doi: 10.1073/pnas.91.11.5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.